

Abstract
Background
Plastome sequences for 18 species of the PACMAD grasses (subfamilies Panicoideae, Aristidoideae, Chloridoideae, Micrairoideae, Arundinoideae, Danthonioideae) were analyzed phylogenomically. Next generation sequencing methods were used to provide complete plastome sequences for 12 species. Sanger sequencing was performed to determine the plastome of one species, Hakonechloa macra, to provide a reference for annotation. These analyses were conducted to resolve deep subfamilial relationships within the clade. Divergence estimates were assessed to determine potential factors that led to the rapid radiation of this lineage and its dominance of warmer open habitats.
Results
New plastomes were completely sequenced and characterized for 13 PACMAD species. An autapomorphic ~1140 bp deletion was found in Hakonechloa macra putatively pseudogenizing rpl14 and eliminating rpl16 from this plastome. Phylogenomic analyses support Panicoideae as the sister group to the ACMAD clade. Complete plastome sequences provide greater support at deep nodes within the PACMAD clade. The initial diversification of PACMAD subfamilies was estimated to occur at 32.4 mya.
Conclusions
Phylogenomic analyses of complete plastomes provides resolution for deep relationships of PACMAD grasses. The divergence estimate of 32.4 mya at the crown node of the PACMAD clade coincides with the Eocene-Oligocene Transition (EOT). The Eocene was a period of global cooling and drying, which led to forest fragmentation and the expansion of open habitats now dominated by these grasses. Understanding how these grasses are related and determining a cause for their rapid radiation allows for future predictions of grassland distribution in the face of a changing global climate.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-015-0563-9) contains supplementary material, which is available to authorized users.
Keywords: Complete plastome, Divergence estimates, PACMAD Clade, Panicoideae, Phylogenomics, Rapid radiation
Background
Poaceae have been the subject of numerous phylogenetic studies due to their economic and ecological importance, as well as their dominance in major terrestrial biomes [1–5]. The current phylogenetic classification of Poaceae includes a deep grade of three lineages: Anomochlooideae, Pharoideae, and Puelioideae, as well as the crown grasses represented by the BEP (Bambusoideae, Ehrhartoideae, Pooideae) [2] and PACMAD (Panicoideae, Aristidoideae, Chloridoideae, Micrairoideae, Arundinoideae, Danthonioideae) [3] clades. The PACMAD clade is of particular interest in this study because despite its paramount economic and ecological importance, phylogenetic relationships among its major lineages remain uncertain.
The sister group to the BEP clade has been variously defined as the PACC, PACCAD, PACCMAD, or PACMAD clade with different constituent subfamilies. A previous study [1] utilized the plastid gene sequence ndhF, which supported a monophyletic PACC (Panicoideae, Arundinoideae, Chloridoideae, Centothecoideae) clade, as well as indicating the polyphyletic nature of Arundinoideae. Subsequent work by the Grass Phylogeny Working Group (GPWG) [2] addressed weak support within and among the grass subfamilies by making use of informative characters in seven molecular datasets along with a morphological dataset. For comparative purposes we will refer to their results for three plastid sequences (ndhF, rbcL, trnK/matK), and not their eight-dataset analysis, as these did not differ in subfamilial arrangement, or provide further resolution.
The GPWG also increased taxon sampling over those of previous phylogenetic studies to include representatives of 62 genera, 30 of which fell within a group described under the newly established PACCAD (Panicoideae, Arundinoideae, Chloridoideae, Centothecoideae, Aristidoideae, Danthonioideae) clade [2]. Three taxa that nested within the PACCAD clade (Eriachne, Gynerium, and Micraira) were classified as incertae sedis (of uncertain placement). Arundinoideae were also found to lack unifying morphological or molecular synapomorphies to establish it as monophyletic. The genera classified as incertae sedis were analyzed further in a separate study with other representatives from Eriachne and Micraira through the use of 69 structural characters as well as ndhF and rpl16 plastid sequences [6]. Their reinstatement of Micrairoideae as a distinct subfamily changed the PACCAD acronym to PACCMAD. With increased taxon sampling across Panicoideae and Centothecoideae in a subsequent study [7], it was concluded that Centothecoideae were paraphyletic with Panicoideae “…and the name should not have phylogenetic implications” (p. 1738). This study defined the constituent subfamilies of the PACMAD clade and established a backbone phylogenetic hypothesis against which deeper phylogenetic relationships could be explored.
The second GPWG constructed the most detailed grass phylogeny to date [4]. One of their major goals was to determine the number of C4 photosynthesis origins across the PACMAD clade. They analyzed 452 PACMAD species, encompassing two thirds of the genera within the clade using the same plastid markers from the previous GPWG study (rbcL, ndhF, trnK/matK). Multiple phylogenetic analyses and an increase in taxonomic sampling provided support for Aristidoideae as the sister subfamily to the rest of the clade. However, the relationship between Panicoideae and the CMAD (Chloridoideae, Micrairoideae, Arundinoideae, Danthonioideae) clade was only weakly supported (bootstrap (bs) value: 61 %, posterior probability (pp): 0.99), as well as the relationship between the MA (Micrairoideae, Arundinoideae) and DC (Danthonioideae, Chloridoideae) clades (bs value: 51 %, pp: 0.98). Furthermore, the arundinoids were only weakly supported as monophyletic.
Deep divergence time estimates of PACMAD grasses have been relatively few. This is partly because of the paucity of confidently dated grass fossils for use as calibration points at specific nodes [8, 9]. The fossils used for calibration include pollen, phytoliths, and spikelets [8, 10, 11]. Another contributing factor is the lack of a well-supported topology at the subfamilial level, especially for deep relationships within the PACMAD clade, which requires sufficient molecular sequences. Previous divergence estimates of the PACMAD clade are highly variable and have been examined in the stem Aristidoideae (28.8 to 61.1 mya), crown PACMAD (38 to 61.1 mya), and stem Panicoideae (26 to 42.1 mya) [8, 10–12]. These four studies used a relatively small number of molecular markers in their phylogenetic analyses and the lack of informative characters likely caused the topologies to vary.
Phylogenomic studies using complete plastomes from Poaceae have provided strong support for the relationships within and among other subfamilies [13–15]. This study addresses the weak support in previous research for deep nodes in the PACMAD clade by utilizing complete plastomes. A plastome from one arundinoid species (Hakonechloa macra) was sequenced using Sanger technology to provide a reference for annotation, and 12 additional complete plastomes were determined by next generation sequencing (NGS) methods for PACMAD taxa. Complete plastomes were analyzed phylogenomically and divergence dates estimated to seek potential selective causes for the PACMAD radiation. The analyses presented here utilize more phylogenetically informative characters and well supported phylogenomic relationships to provide a greater accuracy of divergence estimates through the use of complete plastomes.
Mitochondrial and plastome sequences may produce incongruent gene trees due to incomplete lineage sorting, recombination events, or potential elevated rates of substitution in grasses [16, 17]. Mitochondrial sequences were here explored with the goal of increasing maternally inherited character sampling among representative taxa. Mitochondrial sequence data were extracted and analyzed, as a source of potentially conserved characters, which have proven useful in determining subfamilial relationships in combination with plastome sequences [18–20].
Results
Outgroup selection, plastome
The PACMAD topology based on plastome data remained largely consistent across likelihood analyses conducted with different outgroups. In analyses of all but one outgroup taxon, Panicoideae were sister to the remaining PACMAD taxa. When the single taxon Oryza sativa was selected as the outgroup, Aristidoideae were sister to the remaining PACMAD taxa with a bs value of 56 %. Note that the use of O. rufipogon as an outgroup did not alter the topology in this way.
Outgroup selection greatly influenced support values for the position of Aristidoideae (Additional file 1: Fig. S1). Considering only single-taxon outgroups, the choice of Puelia olyriformis generated a bootstrap support (bs) value of 67 % for the PACMAD node. The use of the somewhat more closely related ehrhartoid species, Oryza rufipogon, increased the bs value to 76 %. When Bambusa oldhamii was used as the outgroup this node had a bs value of 80 %, but Rhynchoryza subulata provided the greatest support, a bs value of 99 %. Bambusa oldhamii was selected as the outgroup for the mitochondrial analysis since mitochondrial data were available for this species.
Plastome characterization
The 13 new plastomes were largely conserved in gene content and organization. The short single copy (SSC) regions had ranges of 11,771 to 14,756 bp in length, long single copy regions (LSC) from 78,798 to 82,525 bp, and inverted repeat regions (IR) from 20,103 to 22,730 bp (Table 1). A unique deletion of ~1140 bp was found in the rpl14 and rpl16 region of Hakonechloa macra. This deletion eliminated all of rpl16 as well as the first 70 bp of rpl14 and the noncoding sequence between them. The deletion is found ~450 bp downstream of rpl8 and ~100 bp upstream of rps3.
Table 1.
Lengths of plastome subregions
| Species | SSC | LSC | IR | %AT |
|---|---|---|---|---|
| Aristida purpurea | 12540 | 80437 | 22723 | 62.5 |
| Centotheca lappacea | 12553 | 81451 | 22730 | 61.4 |
| Coleataenia prionitis | 12551 | 81886 | 22730 | 61.3 |
| Panicum virgatum | 12562 | 81659 | 22699 | 61.4 |
| Thysanolaena maxima | 12466 | 82173 | 22729 | 61.3 |
| Zea mays | 11771 | 82352 | 22748 | 61.5 |
| Elytrophorus spicatus | 12543 | 81221 | 21291 | 61.6 |
| Hakonechloa macra | 12743 | 80977 | 20103 | 61.3 |
| Monachather paradoxus | 12626 | 82525 | 21230 | 61.4 |
| Phragmites australis | 14756 | 82327 | 21268 | 61.3 |
| Eriachne stipacea | 12573 | 79849 | 21162 | 61.4 |
| Isachne distichophylla | 12650 | 81006 | 21063 | 61.5 |
| Micraira spiciformis | 12721 | 80073 | 21262 | 61.4 |
| Chaetobromus dregeanus | 12083 | 78798 | 20922 | 61.6 |
| Chionochloa macra | 12518 | 80888 | 21157 | 61.6 |
| Danthonia californica | 12346 | 79647 | 21099 | 61.6 |
| Neyraudia reynaudiana | 12695 | 80616 | 21028 | 61.6 |
| Rhynchoryza subulata | 12594 | 82029 | 20840 | 61.0 |
All lengths are reported in base pairs. The percent AT richness of each plastome is also reported. SSC: short single-copy; LSC: long single-copy; IR: inverted-repeat
Plastome phylogenomic analyses
The maximum likelihood (ML) analysis produced a tree with − lnL = 274737.67. The tree had mean terminal branch lengths (0.009) more than 2.5 times greater than the mean of the internal branch lengths (0.0035). In the topology generated from the ML analysis, Panicoideae (six species) are resolved as the sister subfamily to the rest of the PACMAD clade (Fig. 1). The next subfamily to diverge is Aristidoideae, which is united with the CMAD clade with a bs value of 77 %. Chloridoideae is supported as sister to Danthonioideae with a bs value of 100 %. The sister relationship between Micrairoideae and Arundinoideae is also supported with a bs value of 100 %. Elytrophorus spicatus was embedded within the Arundinoideae and resolved as sister to the clade of Hakonechloa macra and Phragmites australis with maximum bs support. The chloridoid/danthonioid clade is supported with a bs value of 100 as sister to the arundinoid/micrairoid clade. Although the ML topology retrieved here was well-supported, the Shimodaira-Hasegawa (SH) test failed to reject the alternative hypothesis of Aristidoideae sister to the PCMAD clade (p < 0.151).The Bayesian inference (BI) topology was identical to the ML topology. The position of Aristidoideae and all other nodes in the topology were supported with posterior probability (pp) values of 1.0. The same data matrix under parsimony analysis had 10,779 parsimony informative sites. The maximum parsimony (MP) analysis produced a single tree of length 33,593 steps. The MP analysis had an ensemble consistency index of 0.5764 and retention index of 0.7911. The divergence order for the MP analysis varied slightly from the ML and BI analyses with Aristidoideae sister to the rest of the PACMAD subfamilies, followed by the Panicoideae. Bootstrap support values at each node were 100 with the exception of: the crown Arundinoideae (bs value = 65 %), crown DC (bs value = 99 %), crown CMAD (bs value = 98 %), and crown Panicoideae (bs value = 86 %). The sister relationship of two outgroup taxa, Triticum aestivum and Aegilops geniculata, was supported with a bs value of 99 % in the MP analysis. The partitioned analysis, using only 76 protein coding sequences, produced identical topological results with similar bootstrap support and will not be further considered here.
Fig. 1.
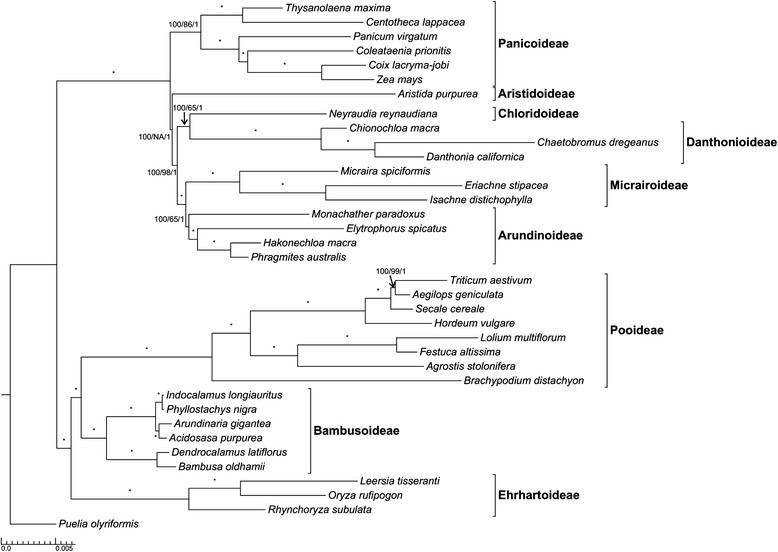
Maximum likelihood phylogram produced from a complete plastome analysis of 18 species of PACMAD grasses. Each node that was fully supported with a ML bs value = 100, MP bs value = 100 and a pp = 100 is labelled with *, except where noted (ML bs/MP bs value/pp). MP bootstrap values are marked NA in the one case where the topology differed
Mitochondrial analysis
The mitochondrial analysis produced a tree with -lnL = 17041.15 (Fig. 2). Panicoideae were monophyletic, but with relatively weak support (bs value = 79 %). The sister relationships of T. maxima and Centotheca lappacea as well as Z. mays and Coleataenia prionitis were each supported with bs values of 98 %. A bs value of 89 % supported Danthonioideae as monophyletic. The sister relationship of Danthonioideae and Chloridoideae was retained, but with little support (bs value: 56 %). Arundinoideae, as sampled here, were characterized as monophyletic, but with bs value < 50 %.
Fig. 2.
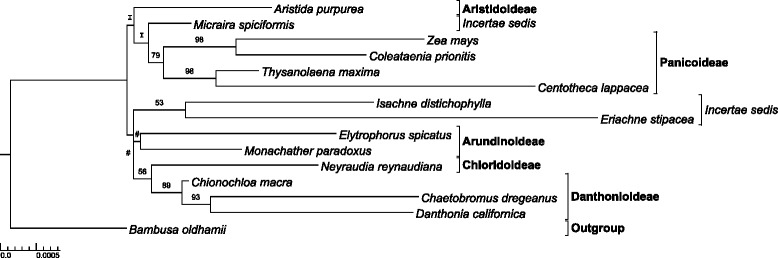
Maximum likelihood phylogram produced from analysis of assembled and aligned mitochondrial matR and seven intron sequences from 15 species. Branch lengths are proportional to the substitution rate along the branch. Bambusa oldhamii was selected as the outgroup. Bs values >50 and <100 are noted. Nodes labelled with # denote bs values <50. Each node marked “I” was incongruent with our ML plastome analysis topology (Fig. 1)
The deep mitochondrial topology differed greatly from that of the plastome analysis (Fig. 2). Arundinoideae were sister to the Chloridoideae/Danthonioideae clade with a bs value < 50 % unlike the relationships in GPWG II [4] or this paper (Fig. 1). Micrairoideae were polyphyletic and all bs values placing these three species were lower than in the plastid ML tree. The deep nodes of the mitochondrial topology were substantially incongruent to those from the plastome analyses and were not used here in further analyses of the deep PACMAD nodes.
Divergence date estimation
Divergence dates were estimated under two calibration scenarios (Table 2). The estimated divergence date at the crown PACMAD node was 32.44 [11.9, 50.6] mya and 32.74 [17.0, 45.2] mya for calibration sets one and two respectively, indicating that the use of the controversial phytolith in the outgroup had little impact on the estimated age of this node. The Aristidoideae divergence of 31.19 [12.8, 46.6] mya for the first set and 20.46 [10.6, 25.6] mya for the second set was the most variable between the two. In set two, the addition of another calibration point caused the stem and crown arundinoid divergences to decrease while the divergence dates of the crown micrairoid, danthonioid, panicoid and stem chloridoid lineages increased (Table 2). The crown panicoid divergence date of 20.24 [7.9, 36.8] mya for set one and 23.61[8.2, 36.8] mya for set two is also of note. Our estimated date for the crown Panicoideae is consistent with recent divergence estimates of Andropogoneae [21].
Table 2.
Divergence estimations for two BEAST analyses
| Calibrations | Crown PACMAD | Arist | MADC | Arund/Micr | Chlor | Danth | Panic | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 | (Chloridoideae: 14–19 mya) | Age | 32.44 | 31.19 | 25.77 | 22.58 | 13.48 | 7.48 | 20.24 |
| (Crown Panicoideae: 7–8 mya) | 95 % HPD Lower | 11.89 | 12.75 | 10.33 | 9.80 | 7.63 | 1.80 | 7.93 | |
| 95 % HPD Upper | 50.55 | 46.57 | 38.26 | 34.33 | 19.58 | 15.60 | 36.82 | ||
| 2 | (Chloridoideae: 14–19 mya) | Age | 32.74 | 20.46 | 19.08 | 17.31 | 14.43 | 12.99 | 23.61 |
| (Crown Panicoideae: 7–8 mya) | 95 % HPD Lower | 16.99 | 10.55 | 9.51 | 7.26 | 5.74 | 2.86 | 8.21 | |
| (Crown BEP/PACMAD: 65–67 mya) | 95 % HPD Upper | 45.20 | 25.61 | 22.61 | 19.79 | 18.97 | 13.29 | 36.75 |
Respective fossil calibrations and estimated ages of each node are reported in mya. Arist: Aristidoideae; Arund/Micr: Arundinoideae + Micrairoideae; Chlor: Chloridoideae; Danth: Danthonioideae; Panic: Panicoideae
Discussion
Short, deep branches of the PACMAD clade in our analyses are consistent with rapid radiations early in this group. Regardless of the cause, deep branches in the PACMAD grasses are difficult to resolve. The full plastome phylogenomic analyses of the PACMAD clade with hundreds of informative characters offer a clear advantage to understanding the deep divergences in the group. Full plastome analysis and estimation of divergence times allows for speculation on the cause of this accelerated diversification.
Phylogenomic analysis
Phylogenetic analyses of rapid radiations in plant lineages tend to be challenging because long ingroup branches are connected only by short deep branches with relatively little phylogenetic information, which may hinder robust resolution of deep relationships [22]. When outgroups are on relatively long branches they can artifactually attract long ingroup branches and suggest erroneous relationships. The conflict between our ML and MP analyses is also suggestive of long-branch attraction to which parsimony is somewhat more susceptible [23].
With one exception, each ML analysis generated an identical ingroup topology. Long branch attraction tends to be indicative of homoplasious substitutions or possible elevated substitution rates, and may be responsible for the weakly supported result with O. sativa. This stands as a unique exception compared to all of the other outgroup combinations. Several outgroup taxa produced less support for the stem Aristidoideae. Outgroup taxa were selected for their relatively close relationships to the PACMAD grasses. Bambusa oldhamii, Oryza rufipogon, and Rhynchoryza subulata presented stronger support for this node than the more distantly related Puelia olyriformis. B. oldhamii exhibited a shorter terminal branch length than R. subulata and O. rufipogon [15], which was correlated with greater support at the stem Aristidoideae (80 %) that fell between those values when R. subulata (99 %) and O. rufipogon (76 %) were selected as outgroups. The inclusion of 18 outgroup taxa resulted in the addition of more phylogenetic information. Although support for the sister relationship of Panicoideae to the rest of the PACMAD clade was less than that of analyses with a single outgroup taxon (77 %), the larger outgroup allowed for greater confidence in the ingroup topology.
A significant past study on the systematics of Poaceae [4] provided weak support for the sister relationship of Panicoideae to the CMAD clade (bs value: 61 %) as well as between the micrairoid/arundinoid and danthonioid/chloridoid clades (bs values: 51 %). Although the taxon sampling of GPWG II included 452 PACMAD species, only three genetic markers of 600–800 base pairs (bp) each were analyzed. The phylogenomic methods here allow for an increase in the number of molecular markers by several orders of magnitude to provide additional informative sites and raise support values for the phylogeny.
In our most well-supported likelihood topology (Fig. 1), Panicoideae are sister to the other PACMAD grasses, and Aristidoideae are sister to the CMAD clade, but this relationship is not statistically different from the GPWG II topology by the SH test. The difficulty in discriminating between these two alternative topologies may be due to the rapid radiation of the PACMAD grasses. All subfamilies sampled with two or more species were recovered as monophyletic. Within Panicoideae, the two Andropogoneae (Coix lacryma-jobi and Zea mays) were sisters as were two species formerly characterized as centothecoids (Thysanolaena maxima and Centotheca lappacea) (bs values, both 100 %). Complete plastome sequence analyses were thus able to provide greater support for phylogenetic relationships and suggests that further sampling of complete plastomes from PACMAD taxa might be useful to address relationships at lower taxonomic levels. This analysis may be creating artifactual groups due to modest taxon sampling as compared to GPWG II [4], but greatly improves upon character sampling.
The ML topology of the mitochondrial data was substantially incongruent with our whole plastome phylogenies (Figs. 1, 2) and the relationships in the GPWGII analysis [4], especially at the deepest nodes. This may be due to the relatively low rate of substitution seen in plant mitochondrial genomes, or their tendency to recombine with fragments of other genomes/organisms that could link loci with different evolutionary histories [24]. Some subfamilies (Panicoideae, Danthonioideae) were retrieved with moderate support. Mitochondrial sequences may be more appropriately used as phylogenetic tools within subfamilies of PACMAD grasses.
Divergence estimates
Previous studies have set out to determine divergence dates for PACMAD grasses using many taxa, but with a relatively small number of molecular markers. The increase in molecular data in this study allows for a more accurate assessment of divergence dates at deep nodes due to the presence of a greater number of phylogenetically informative sites. The age of the BEP/PACMAD clade was recently estimated to be 54.9 (±7.0) mya [9]. This estimate suggests that the BEP and PACMAD clades diverged at the approximate time of the Paleocene-Eocene Thermal Maximum (PETM) (55–56 mya) and the transition to the Eocene era. The Eocene is characterized as a period of cooling and drying, which led to forest fragmentation and created new or more extensive habitats for open habitat and forest margin species [25].
The divergence of the PACMAD subfamilies, which has been formerly estimated to fall between 38 mya [12] and 45 mya [8], was resolved at an age of 32.4 [11.9, 50.6] (95 % HPD lower and upper, respectively) mya in this analysis. The rapid radiation of the PACMAD clade according to this somewhat younger estimate occurs along the Eocene-Oligocene transition (EOT). Throughout the Eocene there was a global cooling trend following Antarctic glaciation events [26] as well as declining atmospheric CO2 [12]. These climatic changes influenced habitat diversification across the globe, increasing open habitats and forest margins for plant colonizations following the EOT.
Aristidoideae are almost exclusively open habitat grasses and the most parsimonious interpretation for the ancestral condition for the subfamily is that it was also an open habitat lineage [2]. If, as suggested by GPWG II, the Aristidoideae are sister to the other PACMAD grasses, then the exploitation of open habitats long preceded the radiation of the PACMAD clade for which there is no corresponding explanatory hypothesis [4]. In the context of the overall grass phylogeny, PACMAD habitat shifts are more parsimoniously interpreted if the sister group to other PACMAD species has the ancestral habitat type. Note that the deeply diverging lineages Puelioideae, Pharoideae, and Anomochlooideae, are exclusively found on forest floors [2]. Panicoideae comprise genera that occur in shady habitats, open habitats, and mixed habitats [2]. This kind of habitat diversity among species of the subfamily sister to the rest of the PACMAD clade would be expected if climatic changes promoted the radiation and diversification of open habitat species at the time of the EOT. Species composition of these descendants, which are resolved here as the sister group to the rest of the PACMAD lineage, would be expected to fill both shade tolerant and open habitat niches, which is seen in contemporary Panicoideae [4]. Notably, in our analysis the lineages sister to the rest of Panicoideae are forest margin (Thysanolaena) and shade tolerant (Centotheca) species [27], consistent with the hypothesis that the Panicoideae initially occupied forests and forest margins, and then radiated, possibly multiple times, into open habitats.
Conclusions
Deep PACMAD relationships are here retrieved with greater support than in previous studies through the use of a phylogenomic approach. Our results support a PACMAD topology where Panicoideae is sister to the ACMAD clade allowing for further exploration of terminal relationships. It also offers a general phylogenomic approach for investigating rapidly radiating plant lineages.
Divergence estimates for the PACMAD clade provide insight into the putative role of climate changes leading to habitat diversification, which possibly triggered the rapid radiation of these grasses. The date of 32.4 mya for the initial radiation of the lineage is correlated with the EOT. Glaciation events and an overall global cooling trend throughout the Eocene led to environmental diversification via forest fragmentation and expansion of open habitats. These changes may have allowed grasses to rapidly speciate, hybridize, and ultimately dominate newly developed habitats.
Methods
Outgroup selection
Outgroup sampling with respect to taxon selection and number posed a challenge due to the potential sensitivity of the positions of Aristidoideae and Panicoideae as the sister group to the other PACMAD lineages. Initially, Oryza rufipogon (NC_022668) was selected as the outgroup, but support for Aristidoideae as sister to the CMAD clade was low (bs value 76 %). Another BEP representative, Bambusa oldhamii (NC_012927), was chosen as the outgroup and the topology remained consistent, but weak support was still seen at the stem Aristidoideae node (bs value 80 %). Up to 17 representative BEP taxa, as well as basal grade taxa (Anomochloa marantoidea, NC_014062; Pharus latifolius NC_021372; Puelia olyriformis, NC_023449), were combined in nine subsets to determine the effect of outgroup selection on ingroup topology and support. Ingroup topology proved to be stable across these analyses, although support for short deep branches was variable. Ultimately, the single outgroup species Rhynchoryza subulata (NC_016718; Ehrhartoideae) was found to produce results congruent to those of the largest outgroup species sets analyzed here.
Taxon sampling
Taxa were sampled based on subfamilial membership to obtain representation for all major groups of interest. Taxa from Panicoideae include representatives of five major tribes, Panicum virgatum (Paniceae) (NC_015990), Zea mays (Andropogoneae) (NC_001666), and Coleataenia prionitis (Paspaleae). Centotheca lappacea (Centotheceae) and Thysanolaena maxima (Thysanolaeneae) were also included from the formerly recognized Centothecoideae [1, 2, 6], which [7] classified as Panicoideae. Three representative Arundinoideae, Hakonechloa macra, Monachather paradoxus, and Phragmites australis, which represent three major clades of Arundinoideae as retrieved by GPWG II were included [4]. One member of an arundinoid genus classified as incertae sedis, Elytrophorus spicatus, was also included in an attempt to resolve this arundinoid relationship [28]. Three taxa from the subfamily Micrairoideae were also analyzed to provide representation for the micrairoid/arundinoid clade. Micraira spiciformis (Micraireae), Eriachne stipacea (Eriachneae), and Isachne distichophylla (Isachneae) were chosen to represent three of the four tribes of micrairoids. Four genera within Danthonioideae were also included to represent the danthonioid lineage as well as a representative species of Aristidoideae, Aristida purpurea (Aristideae). The only published plastome for a chloridoid species, Neyraudia reynaudiana, was also included in our analyses [29].
Mitochondrial sequence data for each taxon included in the plastome analysis that was sequenced using NGS methods were retrieved from the Illumina read files. Sequences from other taxa were retrieved from the NCBI database. Because of this, our mitochondrial sampling was limited to species that were sequenced using NGS and those represented in Genbank. For this portion of the study, the taxon set was limited to 15 species with B. oldhamii as the outgroup taxon.
DNA extraction and sequencing
Leaf tissue samples were obtained for each species of interest (sources listed in Table 3) and dried with silica gel desiccant. Liquid nitrogen was used to lyse cells during manual homogenization of plant tissue to maximize DNA yields. Extractions were performed using the DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol.
Table 3.
Species newly sequenced for this study
| Species | Subfamily | Voucher | GenBank Accession |
|---|---|---|---|
| Aristida purpurea | Aristidoideae | Saarela 605 (CAN) | KJ920224 |
| Centotheca lappacea | Panicoideae | Sanchez-Ken s.n. (ISC) | KJ920225 |
| Coleataenia prionitis | Panicoideae | Morrone 6195 (SI) | KJ920228 |
| Thysanolaena maxima | Panicoideae | Clark & Gallaher 1717 (ISC) | KJ920236 |
| Elytrophorus spicatus | Arundinoideae | Saarela 1685 (CAN) | KJ920230 |
| Monachather paradoxus | Arundinoideae | Saarela 1629 (CAN) | KJ920235 |
| Eriachne stipacea | Micrairoideae | Jacobs 8773 (NSW) | KJ920231 |
| Isachne distichophylla | Micrairoideae | Morden 1227 (HAW) | KJ920233 |
| Micraira spiciformis | Micrairoideae | Jacobs 8850 (NSW) | KJ920234 |
| Chaetobromus dregeanus | Danthonioideae | Barker 978 (PRE) | KJ920226 |
| Chionochloa macra | Danthonioideae | Buxton s.n. (CHR) | KJ920227 |
| Danthonia californica | Danthonioideae | Saarela 860 (CAN) | KJ920229 |
| Hakonechloa macra | Arundinoideae | Burke s.n. (DEK) | KJ920232 |
Subfamily, voucher information and Genbank accession are reported
The plastome of one species, Hakonechloa macra, was amplified and Sanger sequenced using grass-specific primers [30] and the general methods of [31]. Only one copy of the IR (inverted repeat) was sequenced as well as all four IR boundaries. The alternative methods of [32] were followed when amplifications failed, including the design and use of primers tailored to H. macra (Table 4). Amplifications were prepared for sequencing using the Wizard SV PCR Clean-up System (Promega, Madison, WI, USA). Sanger sequencing was performed at Macrogen, Seoul, South Korea. A complete plastome was manually assembled from the overlapping sequences and adjacent segments using Geneious Pro version 7.0.1 (Biomatters, Auckland, New Zealand). The other 12 plastomes not acquired through the NCBI nucleotide database were sequenced using NGS. Starting quantities of total genomic DNA from three DNA extracts (Danthonia californica, Elytrophorus spicatus, Monachather paradoxus) were determined with a Nanodrop 1000 (ThermoFisher Scientific, Wilmington, DE, USA) to be 1.5 μg each. DNAs were diluted to 2 ng/μl and sheared into 300 base pair (bp) fragments using a Bioruptor® sonicator (Diagenode, Denville, NJ, USA) in two 12 min periods, with inversion of tubes between them. DNA preparations were then purified and concentrated using the MinElute Extraction Kit (Qiagen Inc., Valencia, CA, USA). Single read libraries were prepared with the TruSeq low throughput protocol (gel method) following manufacturer instructions (Illumina, San Diego, CA, USA). Single-end sequencing was conducted on a HiSeq 2000 instrument (Illumina, San Diego, CA, USA) at Iowa State University, Ames, USA. Illumina reads were 99 bp in length.
Table 4.
Species-specific Primers Designed for Hakonechloa macra
| Primer Name | Sequence |
|---|---|
| 1FHma | CATTACCCACTTGTCCGACTGTTGC |
| 2RHma | CGATACTGGAACTCAGAGCATAGGAGG |
| 12RHma | GCATGATATTGGGGAATCTCCTTGC |
| 31FHma | GCTGCGTGTATAAGAGCCGAAATGGG |
| 31RHma | GGCAACGAGCATCCAAAACCAAAAG |
| 31FHma2 | GCCCGAAATGGAGTGGGTCCTTCC |
| 31RHma2 | GGTTTTGGATGCTCGTTGCCGCTAG |
| 39FHma | CCTTGGGGTAAAGAGTTTACACTGC |
| 99RHma | CCTGTAGTAGGGATCTGGTTCACTGC |
| 101FHma | GCGACCCCACAGGCTTGTACTTTCG |
| 101RHma | CTTCGTCTACCCACTTCTCTTTCAGG |
| 116FHma | GAACCTGGAGGAGTTAGGTATGTAGG |
| 116RHma | GGAGTCGGCAAATGGACTGAGAATG |
| 148FHma | GGCGTTTTTTTAGTGAGTCCTGG |
| 148RHma | CTCTTTGAGAGACCCCGCTTATTGC |
| 148FHma2 | GAGACCGACCCACTTCCTATCTAG |
| 169FHma | GCCGTTATTGTCGCAAGCATACGAC |
| 169RHma | GCAGAATTTCTCTAGTAGGAGCGTC |
| 169FHma2 | CTCTATGGTTTGGGGCTTTAGCAGG |
| 169RHma2 | CACTCGGATAAACGCAGCAACACGG |
| 184FHma | GCAACAGTCGGACAAGTGGGTAATG |
An improved NGS method was used for the remaining nine species. Total genomic DNA extracts for the remaining taxa were diluted to 2.5 ng/μl in 20 μl water. The Nextera Illumina library preparation kit (Illumina, San Diego, CA, USA) was used to prepare libraries for sequencing and the DNA Clean and Concentrator kit (Zymo Research, Irvine, CA, USA) was used for library sample purification. Single-end sequencing was performed with the HiSeq 2000 instrument at the Iowa State University Sequencing Facility as above.
The reads were first quality filtered using DynamicTrim v2.1 from the SolexaQA software package [33] with default settings, and then sequences less than 25 bp in length (default setting) were removed with LengthSort v2.1 in the same package. The quality of the reads was then assessed using FastQC v0.10.1 (www.bioinformatics.babraham.ac.uk/projects/fastqc/). The complete quality trimmed set of reads was used for assembly.
Plastome assembly and annotation
Plastome assembly was performed with entirely de novo methods. The Velvet software package [34] was run iteratively following previously established methods [26] to assemble reads into contiguous sequences (contigs). Contigs were scaffolded using the anchored conserved region extension (ACRE) method [29]. Conserved regions were identified using a grass family-wide alignment of plastomes. Any remaining gaps in the plastomes were resolved using contigs or reads by locating overlapping regions until the circular map was complete. Assembled plastomes were annotated in Geneious Pro by aligning them to a closely related and previously annotated reference plastome and transferring the annotations from the reference to the assembled plastome when the annotation shared a similarity of 70 % or above. Each coding sequence was then examined and necessary adjustments were made to preserve intron boundaries, reading frames, stop codons, or to identify pseudogenes. The IR boundaries were located using the methods of [13].
Plastome analysis
Fully assembled plastomes were aligned in Geneious Pro using the MAFFT plugin [35]. Characters for which gaps were introduced in one or more sequences by the alignment were removed to reduce regions of ambiguous homology and one of the IR sequences was removed to reduce overrepresentation of duplicated sequence. This alignment was 96,493 bp in length after IR and gap removal. An ML analysis was performed using RAxML-HPC2 on XSEDE [36] and was accessed using the CIPRES science gateway [37]. The tree with the highest likelihood was obtained. Default parameters were used under a GTR model with estimation of the 25 per site gamma rate categories and all other free model parameters. A rapid bootstrap analysis was performed with 1,000 pseudoreplicates. The specified outgroup included 18 BEP taxa (Additional file 1: Fig. S1). The same methods were used for other combinations of outgroup taxa. A consensus bootstrap tree was produced with the Consense function of the Phylip software package [38] on CIPRES (Fig. 1). Tree files were visualized and edited using FigTree v1.4.0. [39]. Several other likelihood analyses were conducted using the separate outgroup taxa, Puelia olyriformis, Bambusa oldhamii, and Oryza rufipogon, but these analyses provided low bootstrap support (bs) values for the stem Aristidoideae. The SH test [40] was performed to determine if the ML topology obtained here differed significantly from the GPWG II hypothesis. The SH test was performed using PAUP* v4.0b10 [41] to determine if the topology with Aristidoideae as the sister group to the other PACMAD subfamilies [4] could be rejected by the data analyzed here.
A BI analysis was performed using MrBayes 3.2.2 on XSEDE [42], which was accessed using the CIPRES science gateway. The Markov chain Monte Carlo (MCMC) analysis was run twice at 10,000,000 generations each. This was run twice with a burn-in of 5,000,000 generations. The analysis was run until completion, and the average standard deviation of split frequencies was < 0.00001.
Branch and bound maximum parsimony (MP) analyses were performed using PAUP* v4.0b10 [41]. An MP bootstrap analysis with 1000 pseudoreplicates, each with 10 random addition sequence replicates, was also performed.
A partitioned analysis was performed by concatenating 76 protein coding sequences from each full plastome. The partitioned matrix was analyzed using identical methods as the full plastome analysis.
Mitochondrial analysis
Mitochondrial sequences were assembled from the same NGS read files and analyzed in a likelihood framework using the same utilities as the plastome analysis. The matR gene sequence was chosen based on prior use in phylogenetic analyses [18]. Six relatively variable intron sequences [19] were also chosen: nad1 intron 2, nad4 intron 1, and nad7 introns 1, 2, 3, and 4. Each intron sequence had less than 99 % pairwise identity when aligned (Table 5). Mitochondrial sequences from Zea mays and Bambusa oldhamii were obtained directly from GenBank. A reference mapping analysis was performed for each species that was included in the NGS libraries by mapping each read file to each coding sequence of interest from the full mitochondrial sequence of Zea mays. The consensus sequences from each reference mapping were examined for missing nucleotide sites and for frameshift mutations, where applicable, to detect erroneous assemblies. The corresponding matR and intron sequence data (Table 5) were assembled, concatenated, and aligned for each species. ML analyses were performed on these data as before, but with B. oldhamii as the outgroup, to generate a ML tree (Fig. 2).
Table 5.
Mitochondrial sequence analysis
| Locus | Percent nucleotide identity |
|---|---|
| mat-r | 99.3 |
| cox2 intron | 91.1 |
| nad1 intron 2 | 97.4 |
| nad4 intron 1 | 98.1 |
| nad7 intron 1 | 95.5 |
| nad7 intron 2 | 98.3 |
| nad7 intron 3 | 98.9 |
| nad7 intron 4 | 98.0 |
Divergence estimates
Divergence dates were estimated using BEAST v2.1.2 [43] and parameters were set in BEAUTi. Preliminary divergence dates with different combinations of calibrations and different prior distributions were estimated. There are few fossils useful for calibrations of PACMAD grasses and dates are sometimes contradictory. Two divergence analyses were selected and presented here to show the maximum range of estimated dates. Both estimates were each given two different seed values and chain lengths of 10 million for a total of 20 million generations each. Priors were set to constrain relationships that preserved the topology generated by the ML plastome analysis with Rhynchoryza subulata as the outgroup. The substitution model utilized was GTR + G + I under an uncorrelated relaxed log-normal clock. For each of these a log-normal distribution was selected. Parametersolver v3.0 [44] was used to calculate the mean and variance of these log-normal prior distributions. Fossil estimates for calibration of each run differed to evaluate the effect of a potentially controversial fossil in the outgroup. A total of six fossils were used to calibrate the analyses. The first run was assessed using a lower bound at the stem Chloridoideae node of 14 mya calibrated with a fossil identified as a member of Chloridoideae, due to the shape of the stomatal subsidiary cells and silica bodies [45]. There has been some debate, however, on the taxonomic identity of this fossil to the genus Distichlis upon further microanatomical evaluation [46]. However, our use of this fossil here is as a calibration point at the crown Chloridoideae node. An upper bound of 19 mya was placed at the stem Chloridoideae node using a chloridoid phytolith fossil [47]. The core Panicoideae (Panicodae) [28] were also constrained for the first run with a lower bound of seven mya using a Setaria fossil, and upper bound of eight mya with a fossil assigned to Dichanthelium sp. [48]. The second BEAST run was conducted using the same calibrations as the first run, but with the addition of a lower bound on the crown BEP/PACMAD node of 65 mya and an upper bound of 67 mya using Oryzeae phytolith fossils [10]. The use of phytoliths as calibrations for estimation of divergence times in Poaceae is somewhat controversial [49]. Comparing two sets of analyses, with and without the phytoliths allows evaluation of the impact of this calibration on the divergence estimation. This calibration strategy incorporated taxonomically identified fossils for PACMAD grasses that are available. It also evaluated the effect of using the Oryzeae phytolith and associated cuticle fossils for the outgroup, which have been shown to produce date estimates that are incompatible with those from other data [9]. The tree and log files produced from the BEAST analysis were combined with Logcombiner v2.1.2 and convergence assessed using Tracer v1.6 [50]. FigTree v1.4.2 [39] was used to view and edit the combined tree file generated for each run of BEAST with a burn-in of 20 percent. Divergence estimates for nodes relevant to the deep relationships of PACMAD grasses are given (Table 2).
Availability of supporting data
The data set(s) supporting the results of this article are available in the TreeBase repository, http://purl.org/phylo/treebase/phylows/study/TB2:S17812. All nucleotide sequences were deposited in the NCBI GenBank repository. Accession numbers can be found in Table 3.
Acknowledgements
We thank K. A. Murrell, K. Zeno, and C. Jäeger for technical assistance and Y. Yin for allowing access to computing resources. We also thank L. Attigala for submitting orders to the ISU DNA facility. Special thanks to those who provided samples for use in this study; J. Saarela, G. Sanchez-Ken, O. Morrone (1957–2011), T. Gallaher, S. Jacobs (1946–2009), C. Morden, and N. Barker. This work was supported in part by the Plant Molecular Biology Center, the Department of Biological Sciences at Northern Illinois University and the National Science Foundation under Grant Numbers DEB-1120750 to LGC, DEB-1120856 to SAK and DEB-1120761 to MRD. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Abbreviations
- ACRE
anchored conserved region extension
- BEAST
Bayesian evolutionary analysis sampling trees
- BEAUTi
Bayesian evolutionary analysis utility
- BEP
Bambusoideae Ehrhartoideae Pooideae
- CIPRES
cyber infrastructure for phylogenetic research
- CMAD
Chloridoideae Micrairoideae Arundinoideae Danthonioideae
- DC
Danthonioideae Chloridoideae
- EOT
Eocene-Oligocene transition
- GPWG (II)
grass phylogeny working group (II)
- GTR + G + I
general time reversible plus gamma distribution plus proportion of invariant sites
- IR
inverted repeat
- LSC
long single copy
- MA
Micrairoideae Arundinoideae
- MAFFT
multiple alignment using fast fourier transform
- MCMC
Markov chain Monte Carlo
- ML
maximum likelihood
- MP
maximum parsimony
- NGS
next generation sequencing
- PAUP*
Phylogenetic Analysis Using Parsimony * and other methods
- PACC
Panicoideae Arundinoideae Chloridoideae Centothecoideae
- PACCAD
Panicoideae Arundinoideae Chloridoideae Centothecoideae Arundinoideae Danthonioideae
- PACCMAD
Panicoideae Arundinoideae Chloridoideae Centothecoideae Micrairoideae Arundinoideae Danthonioideae
- PACMAD
Panicoideae Arundinoideae Chloridoideae Micrairoideae Arundinoideae Danthonioideae
- PETM
Paleocene-Eocene thermal maximum
- SSC
short single copy
Additional file
Schematic outlining the effects of outgroup selection on subfamilial relationships within the PACMAD clade. The included outgroups for each estimation is shown along with the subfamily that is sister to the rest of the PACMAD clade. Additionally, the ML bootstrap support for this sister relationship is included. Figure S1. Outgroup taxon subsets chosen for phylogenomic analyses including total number and identities of outgroup species. Bootstrap values uniting all taxa sister to the deepest diverging clade, which is either Panicoideae or Aristidoideae, are shown. Generalized tree of phylogenetic relationships among Poaceae is indicated above.
Footnotes
Joseph L. Cotton, William P. Wysocki and Melvin R. Duvall contributed equally to this work.
Competing interests
The authors declare no competing interests in regards to the work presented here.
Authors’ contributions
MRD contributed to design of the study, drafting the manuscript, and facilitating interactions between co-authors. JLC performed data acquisition and analyses as well as drafting the manuscript. WPW assisted in drafting the manuscript, data analysis, and sequence assembly. CJP, PPE, and DMJ assisted in TruSeq library preparation. LGC and SAK also contributed to the design of the project and general methodological approach. All authors read and contributed written sections of the final manuscript.
Contributor Information
Joseph L. Cotton, Email: joebtcotton@gmail.com
William P. Wysocki, Email: wwysoc2@gmail.com
Lynn G. Clark, Email: lgclark@iastate.edu
Scot A. Kelchner, Email: kelchner@isu.edu
J Chris Pires, Email: piresjc@missouri.edu.
Patrick P. Edger, Email: pedger@gmail.com
Dustin Mayfield-Jones, Email: dmayfield-jones@danforthcenter.org.
Melvin R. Duvall, Email: mel-duvall@niu.edu
References
- 1.Clark LG, Zhang W, Wendel JF. A phylogeny of the grass family (Poaceae) based on ndhF sequence data. Syst Bot. 1995;436–460.
- 2.Grass Phylogeny Working Group I. Barker NP, Clark LG, Davis JI, Duvall MR, Guala GF, et al. Phylogeny and subfamilial classification of the grasses (Poaceae) Ann Mo Bot Gard. 2001;88:373–457. doi: 10.2307/3298585. [DOI] [Google Scholar]
- 3.Duvall MR, Davis JI, Clark LG, Noll JD, Goldman DH, Sánchez-Ken JG. Phylogeny of the grasses (Poaceae) revisited. Aliso: A Journal of Systematic and Evolutionary Botany. 2007;23(1):237–247. doi: 10.5642/aliso.20072301.18. [DOI] [Google Scholar]
- 4.Grass Phylogeny Working Group II. Aliscioni S, Bell HL, Besnard G, Christin PA, Columbus JT, et al. New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol. 2012;193:304–312. doi: 10.1111/j.1469-8137.2011.03972.x. [DOI] [PubMed] [Google Scholar]
- 5.Saarela JM, Graham SW. Inference of phylogenetic relationships among the subfamilies of grasses (Poaceae: Poales) using meso-scale exemplar-based sampling of the plastid genome. Botany. 2010;88(1):65–84. doi: 10.1139/B09-093. [DOI] [Google Scholar]
- 6.Sánchez-Ken JG, Clark LG. Phylogenetic relationships within the Centothecoideae + Panicoideae clade (Poaceae) based on ndhF and rpl16 intron sequences and structural data. Aliso: A Journal of Systematic and Evolutionary Botany. 2007;23(1):487–502. doi: 10.5642/aliso.20072301.38. [DOI] [Google Scholar]
- 7.Sánchez-Ken JG, Clark LG. Phylogeny and a new tribal classification of the Panicoideae s.l. (Poaceae) based on plastid and nuclear sequence data and structural data. Am J Bot. 2010;97(10):1732–1748. doi: 10.3732/ajb.1000024. [DOI] [PubMed] [Google Scholar]
- 8.Bouchenak-Khelladi Y, Verboom GA, Savolainen V, Hodkinson TR. Biogeography of the grasses (Poaceae): a phylogenetic approach to reveal evolutionary history in geographical space and geological time. Bot J Linnean Soc. 2010;162(4):543–557. doi: 10.1111/j.1095-8339.2010.01041.x. [DOI] [Google Scholar]
- 9.Christin PA, Spriggs E, Osborne CP, Stromberg CA, Salamin N, Edwards EJ. Molecular dating, evolutionary rates, and the age of the grasses. Syst Biol. 2013;63(2):153–65. doi: 10.1093/sysbio/syt072. [DOI] [PubMed] [Google Scholar]
- 10.Prasad V, Stromberg CA, Leache AD, Samant B, Patnaik R, Tang L, et al. Late Cretaceous origin of the rice tribe provides evidence for early diversification in Poaceae. Nat Commun. 2011;2:480. doi: 10.1038/ncomms1482. [DOI] [PubMed] [Google Scholar]
- 11.Vicentini A, Barber JC, Aliscioni SS, Giussani LM, Kellogg EA. The age of the grasses and clusters of origins of C4 photosynthesis. Glob Chang Biol. 2008;14(12):2963–2977. doi: 10.1111/j.1365-2486.2008.01688.x. [DOI] [Google Scholar]
- 12.Christin PA, Besnard G, Samaritani E, Duvall MR, Hodkinson TR, Savolainen V, et al. Oligocene CO2 Decline Promoted C4 Photosynthesis in Grasses. Current Biol. 2008;18(1):37–43. doi: 10.1016/j.cub.2007.11.058. [DOI] [PubMed] [Google Scholar]
- 13.Burke SV, Grennan CP, Duvall MR. Plastome sequences of two New World bamboos—Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)—extend phylogenomic understanding of Bambusoideae. Am J Bot. 2012;99(12):1951–1961. doi: 10.3732/ajb.1200365. [DOI] [PubMed] [Google Scholar]
- 14.Jones SS, Burke SV, Duvall MR. Phylogenomics, molecular evolution, and estimated ages of lineages from the deep phylogeny of Poaceae. Plant Syst and Evol. 2014;300(6):1421–1436. doi: 10.1007/s00606-013-0971-y. [DOI] [Google Scholar]
- 15.Wu ZQ, Ge S. The phylogeny of the BEP clade in grasses revisited: Evidence from the whole-genome sequences of chloroplasts. Mol Phylogenet Evol. 2012;62(1):573–578. doi: 10.1016/j.ympev.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Nyffeler R. The closest relatives of cacti: insights from phylogenetic analyses of chloroplast and mitochondrial sequences with special emphasis on relationships in the tribe Anacampseroteae. Am J Bot. 2007;94(1):89–101. doi: 10.3732/ajb.94.1.89. [DOI] [PubMed] [Google Scholar]
- 17.Sha LN, Xing F, Zhang HQ, Kang HY, Wang Y, Wang XL, et al. Phylogenetic relationships in Leymus (Triticeae; Poaceae): Evidence from chloroplast trnH‐psbA and mitochondrial coxII intron sequences. J Syst Evol. 2014;52(6):722–734. doi: 10.1111/jse.12097. [DOI] [Google Scholar]
- 18.Clifton SW, Minx P, Fauron CM, Gibson M, Allen JO, Sun H, et al. Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol. 2004;136(3):3486–3503. doi: 10.1104/pp.104.044602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo YL, Ge S. Molecular phylogeny of Oryzeae (Poaceae) based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes. Am J Bot. 2005;92(9):1548–1558. doi: 10.3732/ajb.92.9.1548. [DOI] [PubMed] [Google Scholar]
- 20.Freudenstein JV, Chase MW. Analysis of Mitochondrial nad1b-c Intron Sequences in Orchidaceae: Utility and Coding of Length-change Characters. Syst Bot. 2001;26(3):643–657. [Google Scholar]
- 21.Estep MC, McKain MR, Diaz DV, Zhong J, Hodge JG, Hodkinson TR, et al. Allopolyploidy, diversification, and the Miocene grassland expansion. PNAS. 2014;111(42):15149–15154. doi: 10.1073/pnas.1404177111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothfels CJ, Larsson A, Kuo LY, Korall P, Chiou WL, Pryer KM. Overcoming deep roots, fast rates, and short internodes to resolve the ancient rapid radiation of eupolypod II ferns. Syst Biol. 2012;61(3):490–509. doi: 10.1093/sysbio/sys001. [DOI] [PubMed] [Google Scholar]
- 23.Swofford DL, Waddell PJ, Huelsenbeck JP, Foster PG, Lewis PO, Rogers JS. Bias in phylogenetic estimation and its relevance to the choice between parsimony and likelihood methods. Systematic Biol. 2001;50(4):525–539. doi: 10.1080/106351501750435086. [DOI] [PubMed] [Google Scholar]
- 24.Jaramillo-Correa JP, Bousquet J. Mitochondrial genome recombination in the zone of contact between two hybridizing conifers. Genetics. 2005;171(4):1951–1962. doi: 10.1534/genetics.105.042770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellosi ES, Krause JM. Onset of the Middle Eocene global cooling and expansion of open-vegetation habitats in central Patagonia. Andean Geol. 2013;41(1):29–48. [Google Scholar]
- 26.Mudelsee M, Bickert T, Lear CH, Lohmann G. Cenozoic climate changes: a review based on time series analysis of marine benthic δ18O records. Rev Geophys. 2014;52(3):333–374. doi: 10.1002/2013RG000440. [DOI] [Google Scholar]
- 27.Watson L, Dallwitz MJ: 1992 onwards. The grass genera of the world: descriptions, illustrations, identification, and information retrieval; including synonyms, morphology, anatomy, physiology, phytochemistry, cytology, classification, pathogens, world and local distribution, and references. Version: 12th August 2014. http://delta-intkey.com
- 28.Soreng RJ, Davidse G, Peterson PM, Zuloaga FO, Judziewicz EJ, Filgueiras TS, Morrone O, Romaschenko K. A world-wide phylogenetic classification of Poaceae (Gramineae). Tropicos. 2014. etc. http://www.tropicos.org/Project/CNWG Accessed: 2015-01-07.
- 29.Wysocki WP, Clark LG, Kelchner SA, Burke SV, Pires JC, Edger PP, et al. A multi-step comparison of short-read full plastome sequence assembly methods in grasses. Taxon. 2014;63(4):899–910. [Google Scholar]
- 30.Leseberg CH, Duvall MR. The complete chloroplast genome of Coix lacryma-jobi and a comparative molecular evolutionary analysis of plastomes in cereals. J Mol Evol. 2009;69(4):311–318. doi: 10.1007/s00239-009-9275-9. [DOI] [PubMed] [Google Scholar]
- 31.Dhingra A, Folta KM. ASAP: amplification, sequencing & annotation of plastomes. BMC Genomics. 2005;6(1):176. doi: 10.1186/1471-2164-6-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris LM, Duvall MR. The chloroplast genome of Anomochloa marantoidea (Anomochlooideae; Poaceae) comprises a mixture of grass-like and unique features. Am J Bot. 2010;97(4):620–627. doi: 10.3732/ajb.0900226. [DOI] [PubMed] [Google Scholar]
- 33.Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC bioinformatics. 2010;11(1):485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katoh K, Kuma KI, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33(2):511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 37.Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. GCE. 2010;1–8.
- 38.Felsenstein J. PHYLIP (phylogeny inference package) Seattle: Department of Genome Sciences, University of Washington; 2005. [Google Scholar]
- 39.Rambaut A: FigTree v1.4.2. 2014. Available from http://tree.bio.ed.ac.uk/software/figtree/
- 40.Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16:1114–1116. doi: 10.1093/oxfordjournals.molbev.a026201. [DOI] [Google Scholar]
- 41.Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates. Massachusetts, USA: Sunderland; 2003. [Google Scholar]
- 42.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10(4) doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cook J, Wathen KJ, Nguyen H: Parametersolver v3.0. 2013, Available from https://biostatistics.mdanderson.org/SoftwareDownload/SingleSoftware.aspx?Software_Id=6
- 45.Dugas DP, Retallack GJ. Middle Miocene fossil grasses from Fort Ternan, Kenya. J Paleontol. 1993;67:113–128. [Google Scholar]
- 46.Bell HL, Columbus JT. Proposal for an expanded Distichlis (Poaceae, Chloridoideae): support from molecular, morphological, and anatomical characters. Syst Bot. 2008;33(3):536–551. doi: 10.1600/036364408785679879. [DOI] [Google Scholar]
- 47.Strömberg CA. Decoupled taxonomic radiation and ecological expansion of open-habitat grasses in the Cenozoic of North America. Proc Natl Acad Sci U S A. 2005;102(34):11980–11984. doi: 10.1073/pnas.0505700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomasson JR. Observations on the characteristics of the lemma and palea of the late Cenozoic grass Panicum elegans. Am J Bot. 1978;65:34–39. doi: 10.2307/2442550. [DOI] [Google Scholar]
- 49.Iles WD, Smith SY, Gandolfo MA, Graham SW. Monocot fossils suitable for molecular dating analyses. Bot J Linn Soc. 2015;178(3):346–374. doi: 10.1111/boj.12233. [DOI] [Google Scholar]
- 50.Rambaut A, Suchard MA, Xie D, Drummond AJ: Tracer v1.6. 2014. Available from http://beast.bio.ed.ac.uk/Tracer