

The crystal structure of a human dihydrouridine synthase, an enzyme associated with lung cancer, with 18% sequence identity to a T. maritima enzyme, has been determined at 1.9 Å resolution by molecular replacement after extensive molecular remodelling of the template.
Keywords: dihydrouridine synthase, X-ray crystallography, MR-Rosetta, tRNA modification, lung cancer
Abstract
The reduction of uridine to dihydrouridine at specific positions in tRNA is catalysed by dihydrouridine synthase (Dus) enzymes. Increased expression of human dihydrouridine synthase 2 (hDus2) has been linked to pulmonary carcinogenesis, while its knockdown decreased cancer cell line viability, suggesting that it may serve as a valuable target for therapeutic intervention. Here, the X-ray crystal structure of a construct of hDus2 encompassing the catalytic and tRNA-recognition domains (residues 1–340) determined at 1.9 Å resolution is presented. It is shown that the structure can be determined automatically by phenix.mr_rosetta starting from a bacterial Dus enzyme with only 18% sequence identity and a significantly divergent structure. The overall fold of the human Dus2 is similar to that of bacterial enzymes, but has a larger recognition domain and a unique three-stranded antiparallel β-sheet insertion into the catalytic domain that packs next to the recognition domain, contributing to domain–domain interactions. The structure may inform the development of novel therapeutic approaches in the fight against lung cancer.
1. Introduction
tRNA is extensively modified, with 105 different nucleoside modifications identified to date (Cantara et al., 2011 ▸). Modifications are catalyzed enzymatically during post-transcriptional maturation of tRNA. They occur at specific positions, in about 10% of nucleosides in total (Jühling et al., 2009 ▸), altering local chemistry and affecting tRNA conformation and flexibility (Dalluge et al., 1996 ▸; Motorin & Helm, 2010 ▸). tRNA modifications have been shown to affect human health (Nallagatla et al., 2013 ▸; Gehrig et al., 2012 ▸) and are associated with disorders including cancer (Chen et al., 2009 ▸; Rakovich et al., 2011 ▸; Zinshteyn & Gilbert, 2013 ▸; Kato et al., 2005 ▸; Kuchino & Borek, 1978 ▸; Begley et al., 2013 ▸; Spinola et al., 2005 ▸). One of the most common modified nucleosides, dihydrouridine (D) is produced by dihydrouridine synthase (Dus) by enzymatic reduction of the C5—C6 bond in uridine (U) (Fig. 1 ▸ a). The nonplanar base of dihydrouridine is unable to form stacking interactions with bases of other nucleosides, increasing flexibility (Dalluge et al., 1996 ▸). Dihydrouridine has been postulated to aid in the fidelity of translation and to define cognate interactions with partner aminoacyl-tRNA synthetases (Hendrickson, 2001 ▸ and references therein). Given that dihydrouridine increases flexibility, its incorporation has been proposed to be more important for tRNA folding in psychrophiles and mesophiles (Dalluge et al., 1997 ▸). Escherichia coli has three Dus enzymes that perform specific, nonredundant modifications (Bishop et al., 2002 ▸). In humans, there are four Dus enzymes. The hDus2 subfamily is proposed to specifically modify U20 based on sequence identity and biochemical data available for Saccharomyces cerevisiae Dus2 (Xing et al., 2004 ▸). hDus2 contains 493 residues and comprises an N-terminal catalytic domain, a central tRNA-recognition domain and a C-terminal dsRNA-binding domain (369–433) (Fig. 2 ▸ a). There is growing evidence that overexpression of hDus2 potentiates the growth of non-small cell lung carcinoma (NSCLC; Kato et al., 2005 ▸). Elevated hDus2 mRNA and protein levels were identified in a range of NSCLC cell lines, and siRNA-dependent knockdown of hDus2 decreased colony formation and cell viability, while hDus2 immunohistochemical staining correlated with patient survival and was also defined as an independent prognostic factor for the development of NSCLC (Kato et al., 2005 ▸). Additionally, increased levels of dihydrouridine incorporation have been identified in other mammalian carcinomas (Kuchino & Borek, 1978 ▸).
Figure 1.

Dihydrouridine modification. (a) Uridine and dihydrouridine. (b) U20 location shown for E. coli tRNAPhe (Byrne et al., 2010 ▸). U20 is illustrated by a van der Waals model coloured by atom type with O atoms in red, N atoms in blue and C atoms in white.
Figure 2.

The structure of hDus2 1–340 comprising the catalytic and recognition domains. (a) Domain schematic of hDus2. (b) The structure of hDus2 viewed from the putative tRNA-binding surface. The ribbon diagram is coloured from blue (N-terminus) to red (C-terminus); missing residues 118–128 are indicated by a dashed green ribbon; bound FMN is shown as sticks. (c) Secondary-structure topology diagram [colouring as in (b)] showing domain composition and connectivity. (d) FMN coordination, with FMN and active-site residues shown as sticks, hydrogen bonds shown as dashed lines, atoms coloured according to type and residues forming hydrophobic contacts designated by red semicircles.
X-ray structures are available for three Dus enzymes. The first structure to be solved was of the TM0096 protein from Thermotoga maritima (PDB entry 1vhn; Park et al., 2004 ▸), which has not yet been functionally characterized. The second was of the position 20 specific enzyme from Thermus thermophilus (TtDus; PDB entry 3bop; Yu et al., 2011 ▸). For the third enzyme, the position-16 specific E. coli DusC, two structures are available at a higher resolution (EcDusC, PDB entry 4bfa; Byrne et al., 2015 ▸) and a lower resolution (PDB entry 3w9z; Chen et al., 2013 ▸). All three enzymes have a conserved catalytic domain comprising a TIM barrel with a bound flavin mononucleotide (FMN) and a C-terminal α-helical tRNA-recognition domain that varies slightly in its relative orientation to the TIM barrel. Bound FMN functions as a cofactor during the reduction of the C5—C6 double bond of uridine (Rider et al., 2009 ▸). For TtDus and EcDusC, structural information is also available for complexes with tRNA indicating that the orientation of the tRNA substrate defines the modification specificity of the enzyme (PDB entries 3b0v, 3b0u, Yu et al.., 2011 ▸ and 4yco, 4ycp, Byrne et al., 2015 ▸). The structure of TtDus covalently bound to target tRNA was determined at 3.5 Å resolution and a higher 1.95 Å resolution structure was obtained for the enzyme cross-linked to a smaller tRNA fragment (PDB entries 3b0v and 3b0u, respectively; Yu et al., 2011 ▸). To date, no structural information on archaeal or eukaryotic Dus has been reported. Here, we present the X-ray structure of a human Dus2 protein construct comprising the catalytic and tRNA-recognition domains.
The rapid growth of the PDB has led to an increasingly large pool of search models for use in molecular replacement. In many cases search models with low identity produce solutions that are correctly placed, but the noisy electron-density maps calculated using phases from the model contain insufficient information to guide improvement of the model. In such cases, recent developments using molecular-modelling tools to rebuild the placed model have produced spectacular results (DiMaio et al., 2011 ▸; DiMaio, 2013 ▸; Terwilliger et al., 2012 ▸). As there are three possible template structures available (TmDus, TtDus and EcDusC), we investigated whether any of these could be used to solve the hDus2 structure by automated molecular replacement and rebuilding using phenix.mr_rosetta. In order to compare the phasing methods, the phases were also determined experimentally using SAD techniques.
2. Methods
2.1. Purification, crystallization and data collection
Cloning, purification, crystallization and data collection of native hDus2 1–340 was performed as described previously (Griffiths et al., 2012 ▸). Selenomethionyl hDus2 1–340 was expressed and purified in the same manner as the native protein, using immobilized metal-affinity and anion-exchange chromatography, except that E. coli B834 (DE3) cells were used for expression and were grown in minimal medium supplemented with 40 µg ml−1 l-selenomethionine (Hendrickson et al., 1990 ▸). Labelling was confirmed using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) and the protein (at 10 mg ml−1 in 20 mM Tris, 100 mM NaCl, 5 mM imidazole, 5 mM DTT pH 8) was subjected to crystallization screening using the PACT screen (Newman et al., 2005 ▸) in 0.3 µl sitting drops with 54 µl reservoir incubated at 292 K. Rhomboid-shaped crystals grew in 2 days from a condition comprising 0.1 M MES–malic acid–Tris pH 4, 25%(w/v) PEG 1500 (Newman, 2004 ▸). Cryoprotection was performed by passing crystals through reservoir solution supplemented with 200 mM NaCl, 40 mM Tris pH 8 and 32% PEG 1500 prior to flash-cooling in liquid N2. X-ray data for selenomethionyl hDus2 were collected at 100 K on the I04 beamline at Diamond Light Source (DLS), Didcot, England using an ADSC Q315r CCD detector. Indexing and integration was performed with XDS (Kabsch, 2010 ▸). POINTLESS (Evans, 2011 ▸) was used to confirm the Laue group and AIMLESS was used for scaling and merging (Evans & Murshudov, 2013 ▸). X-ray data statistics are summarized in Table 1 ▸.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Native | Selenomethionyl | |
|---|---|---|
| Data collection | ||
| X-ray source | Beamline I04, DLS | Beamline I04, DLS |
| Wavelength () | 0.9795 | 0.9795 |
| Space group | P21 | C2 |
| Unit-cell parameters (, ) | a = 46.3, b = 49.6, c = 69.6, = 95.7 | a = 73.0, b = 50.1, c = 72.1, = 90.7 |
| Resolution limits () | 46.01.9 (2.01.9) | 36.52.3 (2.42.3) |
| Unique reflections | 24243 (3515) | 11728 (1136) |
| Completeness (%) | 97.6 (97.8) | 100 (100) |
| Mutliplicity | 3.0 (2.8) | 3.8 (3.8) |
| I/(I) | 10.3 (1.7) | 9.5 (2.6) |
| R merge † (%) | 6.3 (58.5) | 8.4 (40.8) |
| R p.i.m. ‡ (%) | 4.0 (41.4) | 8.0 (38.7) |
| CC1/2 § | 0.997 (0.722) | 0.992 (0.790) |
| No. of molecules in asymmetric unit | 1 | 1 |
| V M (3Da1) | 2.06 | 1.68 |
| Solvent content (%) | 40.2 | 26.9 |
| Wilson B factor (2) | 25.5 | 26.2 |
| Structure refinement | ||
| Resolution range () | 46.01.9 | |
| No. of reflections in refinement | 22995 | |
| R factor (%) | 16.8 | |
| No. of reflections used for R free | 1236 | |
| Free R factor (%) | 20.7 | |
| No. of protein atoms | 2466 | |
| No. of water molecules | 194 | |
| B factor (2) | 36.6 | |
| R.m.s.d. | ||
| Bond lengths () | 0.012 | |
| Bond angles () | 1.5 | |
| Ramachandran statistics | ||
| Favoured (%) | 98 | |
| Allowed (%) | 2 | |
| Disallowed (%) | 0 | |
R
merge = 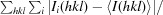
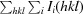 .
.
R
p.i.m. = 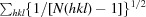
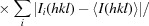 .
.
CC1/2 is the half-data-set correlation coefficient.
2.2. Initial structure determination
The structure was first determined by single-wavelength anomalous diffraction (SAD) using selenomethionyl protein. Selenium sites were identified using SHELXC/SHELXD (Sheldrick, 2008 ▸), phases were calculated and refined using Phaser (McCoy et al., 2007 ▸) and density modification was performed with Parrot (Cowtan, 2010 ▸). Buccaneer (Cowtan, 2006 ▸) was deployed for initial automated model building. Subsequent manual rebuilding was performed with Coot (Emsley & Cowtan, 2004 ▸; Emsley et al., 2010 ▸) and the model was refined using REFMAC5 (Murshudov et al., 2011 ▸). The structure of the native protein was then determined at a higher resolution by molecular replacement using MOLREP (Vagin & Teplyakov, 2010 ▸). This model was refined using isotropic B factors and four TLS groups (residues 7–109, 110–220, 221–319 and 320–339) defined by the TLS Motion Determination server (Painter & Merritt, 2006 ▸; Table 1 ▸). The coordinates and structure factors have been deposited in the Protein Data Bank and are available under accession code 4xp7.
2.3. Exploring the power of molecular replacement using distant homologues
For automated molecular replacement and rebuilding from low-identity templates, we used phenix.mr_rosetta (DiMaio et al., 2011 ▸; DiMaio, 2013 ▸; Terwilliger et al., 2012 ▸). A sequence alignment of hDus2 residues 1–340 with TtDus (PDB entry 3b0p), TmDus (PDB entry 1vhn) and EcDusC (PDB entry 4bfa) was generated using HHpred (Söding et al., 2005 ▸). Fragments were generated from the sequence of hDus2 using the Robetta fragment server (Kim et al., 2004 ▸). The alignment, fragment files from the Robetta fragment server (http://robetta.bakerlab.org/), the native data set and the sequence of hDus2 residues 1–340 were used as input for phenix.mr_rosetta executed on a single computer. 100 Rosetta models were built for each MR solution, taking between 50 and 75 CPU hours. As the best solution from the final phenix.autobuild step of the first round is (by default) rebuilt again with Rosetta, a run of phenix.mr_rosetta took around 100–150 CPU hours to generate a final model from each molecular-replacement solution. The density-modified phases and FOMs from the highest scoring cycle of phenix.autobuild in the final stage of phenix.mr_rosetta for each of the three templates were used for model building with Buccaneer (Cowtan, 2006 ▸, 2008 ▸) and refinement with REFMAC5 (Murshudov et al., 2011 ▸).
In order to compare the quality of the three models produced by these automated (i.e. no manual building) methods with that produced by experimental phasing, the Buccaneer model built into the experimentally phased map (as described previously) was used as a MR search model with the (higher resolution) native data. The placed model was used to generate density-modified phases with RESOLVE via phenix.autobuild (Terwilliger, 2000 ▸; Terwilliger et al., 2008 ▸). These density-modified phases and FOMs were combined with the native data and used for model building as described above.
For a conventional MR approach, the sequences of hDus2 1–340 and the search models TtDus (PDB entry 3b0p), TmDus (PDB entry 1vhn) and EcDusC (PDB entry 4bfa) were aligned by HHpred (Söding et al., 2005 ▸). Sequences were trimmed to the catalytic domain; search models were truncated and edited using Sculptor (Bunkóczi & Read, 2011 ▸). Phaser (McCoy et al., 2007 ▸) solutions with the highest LLG were refined by REFMAC5 using the jelly-body option and autobuilt using Buccaneer.
2.4. Sequence conservation and structure superposition
Structure-based alignment of the hDus2, S. cerevisiae Dus2, S. pombe Dus2, TtDus, EcDusC and TmDus sequences (UniProt IDs Q9NX74, P53720, O74731, Q5SMC7, P33371 and Q9WXV1, respectively) was performed using the Expresso server (Armougom et al., 2006 ▸) and presented using ESPript (Robert & Gouet, 2014 ▸). Sequences within the eukaryotic Dus2 subfamily were aligned using Clustal Omega and conservation was mapped onto the protein surface by ConSurf (Landau et al., 2005 ▸) and rendered using CCP4mg (McNicholas et al., 2011 ▸). Sequence alignment of representative members of the three eukaryotic subfamilies including Dus2, Dus1 and Dus4 was performed using Clustal Omega and presented using ESPript. The Dus3 subfamily was not included in this alignment owing to significant sequence differences (Kasprzak et al., 2012 ▸). The structures of TtDus, TmDus, EcDusC (PDB entry 4bfa) and hDus2 were aligned by secondary-structure matching using Superpose (Krissinel & Henrick, 2004 ▸) and the cartoons were rendered with CCP4mg (McNicholas et al., 2011 ▸). Electrostatic surfaces were calculated with PDB2PQR (Dolinsky et al., 2004 ▸) and APBS (Baker et al., 2001 ▸) and were rendered by PyMOL (v.1.3r1; Schrödinger). Cofactor-coordination figures were generated using LigPlot+ (Laskowski & Swindells, 2011 ▸) and the protein-topology two-dimensional cartoon was generated by Pro-Origami (Stivala et al., 2011 ▸).
3. Results and discussion
3.1. Initial structure determination by SAD
The structure of hDus2 was initially determined by the conventional SAD approach using crystals of selenomethionyl protein that diffracted to 2.3 Å resolution. Following this, we also attempted to solve this structure by MR with distant sequence homologues using phenix.mr_rosetta (DiMaio et al., 2011 ▸; Terwilliger et al., 2012 ▸) and the 1.9 Å resolution native data set. The results of the structure-determination attempts are described below.
The crystals of selenomethione-labelled hDus2, unlike the P21 crystals of the native protein, belonged to space group C2 (Table 1 ▸). There is one molecule per asymmetric unit, corresponding to a solvent content of 27% (Matthews, 1968 ▸). Density-modified SAD phases resulted in clear electron-density maps and autobuilding was able to produce an almost complete model. The FMN density was nonplanar, hence the two-electron-reduced form (PDB ligand ID FNR; Supplementary Fig. S1) was used during the refinement (Table 1 ▸).
3.2. Post hoc structure determination by molecular replacement
Previous attempts at rebuilding potential MR solutions obtained with the TmDus and EcDusC models were unsuccessful. Indeed, autobuilding with phenix.autobuild or density modification with RESOLVE followed by extensive rounds of autobuilding with either Buccaneer or ARP/wARP (Langer et al., 2008 ▸) starting from the models placed in the initial MR stage of phenix.mr_rosetta (Supplementary Fig. S2a) failed to produce solutions with an R free of less than 50%. As the sequence alignment generated with HHpred contains residues 1 to 340 of hDus2, the search models used in phenix.mr_rosetta contain parts of the recognition domains of TtDus, TmDus and EcDusC that align with the recognition domain of hDus2. The Phaser translation-function Z-score (TFZ) and LLG for the top solutions were relatively poor: TtDus, TFZ 4.7, LLG 39.7; TmDus, TFZ 5.2, LLG 26.0; EcDusC, TFZ 4.8, LLG 25.6. Correlation between the 2mF o − DF c maps calculated from these solutions and the map calculated from the final refined structure of hDus2 was ∼0.35 for TtDus and TmDus and ∼0.30 for EcDusC (Supplementary Table S1). It is therefore unsurprising that autobuilding from these MR solutions was not possible. However, despite the low sequence identity to TtDus, TmDus and EcDusC (20.3, 17.9 and 22.6%, respectively), phenix.mr_rosetta was able to generate excellent maps for hDus2 using TtDus and TmDus as search models. Autobuilding into these maps with Buccaneer produced almost complete models (Supplementary Fig. S2b, Supplementary Table S2).
Despite having the highest sequence identity to hDus2, EcDusC proved to be the poorest search model, as demonstrated by the lowest correlation between maps calculated from this model and the final refined structure, and phenix.mr_rosetta failed to generate a map suitable for autobuilding. However, during the revision stage of this manuscript we investigated whether the latest versions of PHENIX and Rosetta would be able to rebuild from the MR solution obtained for the EcDusC model, which was clearly correctly placed in the unit cell. It transpired that the current versions are able to improve this model and generate maps suitable for autobuilding and refinement using the same versions of Buccaneer/REFMAC5 as previously (Supplementary Fig. S2b, Supplementary Table S2).
Given the success of phenix.mr_rosetta, we investigated whether it was possible to solve the structure of hDus2 by more conventional approaches without remodelling by Rosetta. Superposition of TtDus, TmDus and EcDusC revealed a highly similar catalytic domain but variation in the conformation and the relative position of the C-terminal recognition domain (Supplementary Fig. S2c). Sequence alignments with hDus2 indicated homology in the catalytic domain (Supplementary Fig. S3). Hence, search models were generated using Sculptor based on the HHpred alignments used as input for phenix.mr_rosetta but encompassing only the catalytic domain. These minimal catalytic domain structures were used as search models with Phaser. A clear solution (TFZ 8.4, LLG 83.4) was found for the TtDus catalytic domain model, with a 2mF o − DF c map correlation of 0.37 to the map calculated from the final refined structure of hDus2 (Supplementary Table S3). 100 cycles of REFMAC jelly-body refinement resulted in an R and R free of 46.4 and 48.3%, respectively, and a map correlation of 0.56. Iterative rebuilding and refinement of this model with Buccaneer and REFMAC5 generated a near-complete model of hDus2 with an R free of <30%. Attempts to repeat this approach with the TmDus and EcDusC catalytic domains as search models produced reasonable map correlation after jelly-body refinement only for TmDus (Supplementary Table S3). Whilst rebuilding the TmDus solution to a complete model of hDus2 was possible, it required extensive iteration of jelly-body refinement and Buccaneer rebuilding/REFMAC refinement. In agreement, superposition of the catalytic domains shows that hDus2 is most similar to TtDus (Cα r.m.s.d. of 1.5 Å for residues 3 to 240). In contrast, TmDus (Cα r.m.s.d. of 2.0 Å for residues 5–228) and EcDusC (Cα r.m.s.d. of 1.9 Å for residues 1–240) were more divergent (Supplementary Fig. S2d, Supplementary Table S3).
3.3. Unique features in the fold and relative orientation of hDus2 domains
Like other Dus enzymes, hDus2 folds into catalytic and tRNA-recognition domains (Figs. 2 ▸ b and 2 ▸ c). The catalytic domain, containing the bound FMN cofactor (Fig. 2 ▸ d), comprises residues 7–258 of the protein, being roughly two times larger than the recognition domain (residues 259–339). Superposition of hDus2 with TtDus illustrates that the enzymes are broadly similar (Cα r.m.s.d. of 2.8 Å, 239 residues aligned; Fig. 3 ▸ a). There are two major differences between hDus2 and its bacterial enzyme homologues. Firstly, the catalytic domain contains a three-stranded antiparallel β-sheet insertion (residues 53–76; β4–β5–β6; Figs. 2 ▸ b and 2 ▸ c and Supplementary Fig. S4a). Secondly, the recognition domain is larger, comprising a five-helix bundle with a considerably longer C-terminal helix which is positioned differently with respect to the catalytic domain (Fig. 3 ▸ a and Supplementary Fig. S4b).
Figure 3.

Comparison of hDus2 with TtDus and analysis of electrostatic surface and sequence conservation. (a) Ribbon diagram showing superposition of hDus2 with TtDus (grey). The boxed area is magnified in (b), showing hDus2 interactions between the catalytic and recognition domains that are not present in bacterial Dus enzymes. (c) hDus2 superposed with the structure of the TtDus–tRNA complex (with TtDus in grey and tRNA in blue). (d) hDus2 molecular surface coloured according to electrostatic potential (±5kT/e; red, negative; blue, positive). (e) hDus2 surface coloured according to sequence conservation: high (purple) to low (yellow).
Superposition of hDus2 and TtDus (Cα r.m.s.d. of 1.5 Å for the catalytic domain) reveals differences at the interface between the catalytic and recognition domains (Fig. 3 ▸ b). The different orientation of the recognition domain in hDus2 results in helices α10 and α11 extending into the area occupied by tRNA in TtDus (Fig. 3 ▸ c). The position of the recognition domain is stabilized by the three-stranded β-sheet insertion in the catalytic domain, which packs next to the recognition domain. The β4–β5 loop (residues 58–60) of the β-sheet interacts with residues 327–334 of the recognition domain (Fig. 3 ▸ b). In addition to mediating interactions between the two domains, the β-sheet insertion extends into the tRNA-occupied area of TtDus (Fig. 3 ▸ c and Supplementary Fig. S4c), suggesting a role in protein–tRNA interactions. It is interesting to note that the three-stranded β-sheet insertion appears to be unique to the eukaryotic Dus2 subfamily (Supplementary Fig. S5).
3.4. tRNA binding and the active site
TtDus makes contacts with the tRNA T-loop through helix α6 (residues 96–101). The corresponding segment of hDus2 (residues 118–128) is not defined in the electron-density maps. TtDus also forms hydrogen bonds to the target residue U20 and to G44 in the variable-loop region via Lys175 and Arg178 of the 310-helix (residues 174–180). This helix is not present in hDus2, nor is there any significant sequence conservation in this segment (Supplementary Fig. S3), indicating there may be differences in the manner that hDus2 binds its target tRNA substrates.
hDus2 has a positively charged surface area (Fig. 3 ▸ d) which appears to be most similar to the positively charged area of TtDus, but its size is smaller than in bacterial enzymes (Supplementary Fig. S6). Proximal to the catalytic Cys116 is a loop which was not visible in the electron density (residues 118–128). Some decrease in the positively charged area may be due to two lysine residues present in this loop. Significantly, the positively charged area shows high sequence conservation (Fig. 3 ▸ e), indicating functional importance. Part of the positively charged and conserved area of hDus2 corresponds to the active site containing the bound FMN molecule. The residues coordinated to the FMN are highly conserved in TtDus, TmDus and EcDusC (Fig. 2 ▸ d, Supplementary Figs. S3 and S7), indicating a similar enzymatic mechanism. In the structures of TtDus and the lower resolution structure of EcDusC (PDB entry 3w9z; Chen et al., 2013 ▸), positive difference density in the active site was attributed to an unknown cofactor, which was modelled as a sulfate moiety in the TmDus model. Notably, no equivalent density is observed in the difference electron-density maps of hDus2.
4. Conclusions
We have determined the structure of the human tRNA-modification enzyme hDus2 that has been implicated in lung cancer (Kato et al., 2005 ▸). The structure differs in two major ways from those of bacterial Dus enzymes. Firstly, the catalytic domain contains an additional three-stranded β-sheet that is absent from the bacterial enzymes and forms an interacting surface with the recognition domain. Secondly, the recognition domain, in addition to having an additional α-helix, is positioned differently with respect to the catalytic domain. Interactions between the extended C-terminal helix of the recognition domain and the three-stranded β-sheet of the catalytic domain that were not seen in bacterial Dus enzymes appear to extend the domain interface and may stabilize the overall structure. Conservation of the catalytic residues indicates that the enzymatic mechanism is essentially the same as for the bacterial homologues. We propose that structural differences and differences in the electrostatic surface may result in altered positioning of tRNA during catalysis compared with bacterial enzymes. Whilst a complete understanding of tRNA recognition and modification awaits the determination of the X-ray structure of a complex of hDus2 with tRNA, the structural data presented here will inform the design of potent inhibitors of the enzyme.
In spite of the low sequence identity to available search models, automatic approaches using phenix.mr_rosetta were successfully used post hoc to determine the structure of hDus2, demonstrating that the structures of functional homologues, despite having sequence identities below 20%, can be used for structure determination by molecular replacement. Whilst it was in fact possible to solve the structure of hDus2 without the use of phenix.mr_rosetta. this required extensive manual intervention that included refinement and model rebuilding. Counterintuitively, the highest identity search model proved to be the worst template, complicating structure determination. In contrast, the use of phenix.mr_rosetta, while computationally expensive, required no intervention from the user to achieve a near-complete model. Retrospective analaysis suggested that it was not necessary to build as many as 100 Rosetta models for each solution. Indeed, by taking only the top MR solution for each of the three templates and building 20 Rosetta models for each solution a complete model can be produced in ∼25 h on a modest four-core desktop computer. Such an automated approach can liberate time for other ‘bottlenecks’ of biological crystallography associated with protein production and obtaining diffraction-quality crystals.
Supplementary Material
PDB reference: human dihydrouridine synthase, 4xp7
Supplementary Figures. DOI: 10.1107/S1399004715009220/rr5097sup4.pptx
Supplementary Tables.. DOI: 10.1107/S1399004715009220/rr5097sup1.pdf
Acknowledgments
We would like to thank Dr Johan Turkenburg and Sam Hart for assistance with data collection. We thank Diamond Light Source for access to beamline I04 (proposal number mx-1221) that contributed to the results presented here. This work was funded by the Wellcome Trust, grant reference 098230.
References
- Armougom, F., Moretti, S., Poirot, O., Audic, S., Dumas, P., Schaeli, B., Keduas, V. & Notredame, C. (2006). Nucleic Acids Res. 34, W604–W608. [DOI] [PMC free article] [PubMed]
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed]
- Begley, U., Sosa, M. S., Avivar-Valderas, A., Patil, A., Endres, L., Estrada, Y., Chan, C. T. Y., Su, D., Dedon, P. C., Aguirre-Ghiso, J. A. & Begley, T. (2013). EMBO Mol. Med. 5, 366–383. [DOI] [PMC free article] [PubMed]
- Bishop, A. C., Xu, J., Johnson, R. C., Schimmel, P. & de Crécy-Lagard, V. (2002). J. Biol. Chem. 277, 25090–25095. [DOI] [PubMed]
- Bunkóczi, G. & Read, R. J. (2011). Acta Cryst. D67, 303–312. [DOI] [PMC free article] [PubMed]
- Byrne, R. T., Konevega, A. L., Rodnina, M. V. & Antson, A. A. (2010). Nucleic Acids Res. 38, 4154–4162. [DOI] [PMC free article] [PubMed]
- Byrne, R. T., Jenkins, H. T., Peters, D. T., Whelan, F., Stowell, J., Aziz, N., Kasatsky, P., Rodnina, M. V., Koonin, E. V., Konevega, A. L. & Antson, A. A. (2015). Proc. Natl Acad. Sci. USA, 112, 6033–6037. [DOI] [PMC free article] [PubMed]
- Cantara, W. A., Crain, P. F., Rozenski, J., McCloskey, J. A., Harris, K. A., Zhang, X., Vendeix, F. A. P., Fabris, D. & Agris, P. F. (2011). Nucleic Acids Res. 39, D195–D201. [DOI] [PMC free article] [PubMed]
- Chen, C., Tuck, S. & Byström, A. S. (2009). PLoS Genet. 5, e1000561. [DOI] [PMC free article] [PubMed]
- Chen, M., Yu, J., Tanaka, Y., Tanaka, M., Tanaka, I. & Yao, M. (2013). Acta Cryst. F69, 834–838. [DOI] [PMC free article] [PubMed]
- Cowtan, K. (2006). Acta Cryst. D62, 1002–1011. [DOI] [PubMed]
- Cowtan, K. (2008). Acta Cryst. D64, 83–89. [DOI] [PMC free article] [PubMed]
- Cowtan, K. (2010). Acta Cryst. D66, 470–478. [DOI] [PMC free article] [PubMed]
- Dalluge, J. J., Hamamoto, T., Horikoshi, K., Morita, R. Y., Stetter, K. O. & McCloskey, J. A. (1997). J. Bacteriol. 179, 1918–1923. [DOI] [PMC free article] [PubMed]
- Dalluge, J. J., Hashizume, T., Sopchik, A. E., McCloskey, J. A. & Davis, D. R. (1996). Nucleic Acids Res. 24, 1073–1079. [DOI] [PMC free article] [PubMed]
- DiMaio, F. (2013). Acta Cryst. D69, 2202–2208. [DOI] [PMC free article] [PubMed]
- DiMaio, F., Terwilliger, T. C., Read, R. J., Wlodawer, A., Oberdorfer, G., Wagner, U., Valkov, E., Alon, A., Fass, D., Axelrod, H. L., Das, D., Vorobiev, S. M., Iwaï, H., Pokkuluri, P. R. & Baker, D. (2011). Nature (London), 473, 540–543. [DOI] [PMC free article] [PubMed]
- Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. (2004). Nucleic Acids Res. 32, W665–W667. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Gehrig, S., Eberle, M. E., Botschen, F., Rimbach, K., Eberle, F., Eigenbrod, T., Kaiser, S., Holmes, W. M., Erdmann, V. A., Sprinzl, M., Bec, G., Keith, G., Dalpke, A. H. & Helm, M. (2012). J. Exp. Med. 209, 225–233. [DOI] [PMC free article] [PubMed]
- Griffiths, S., Byrne, R. T., Antson, A. A. & Whelan, F. (2012). Acta Cryst. F68, 333–336. [DOI] [PMC free article] [PubMed]
- Hendrickson, T. L. (2001). Proc. Natl Acad. Sci. USA, 98, 13473–13475. [DOI] [PMC free article] [PubMed]
- Hendrickson, W. A., Horton, J. R. & LeMaster, D. M. (1990). EMBO J. 9, 1665–1672. [DOI] [PMC free article] [PubMed]
- Jühling, F., Mörl, M., Hartmann, R. K., Sprinzl, M., Stadler, P. F. & Pütz, J. (2009). Nucleic Acids Res. 37, D159–D162. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kasprzak, J. M., Czerwoniec, A. & Bujnicki, J. M. (2012). BMC Bioinformatics, 13, 153. [DOI] [PMC free article] [PubMed]
- Kato, T., Daigo, Y., Hayama, S., Ishikawa, N., Yamabuki, T., Ito, T., Miyamoto, M., Kondo, S. & Nakamura, Y. (2005). Cancer Res. 65, 5638–5646. [DOI] [PubMed]
- Kim, D. E., Chivian, D. & Baker, D. (2004). Nucleic Acids Res. 32, W526–W531. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Kuchino, Y. & Borek, E. (1978). Nature (London), 271, 126–129. [DOI] [PubMed]
- Landau, M., Mayrose, I., Rosenberg, Y., Glaser, F., Martz, E., Pupko, T. & Ben-Tal, N. (2005). Nucleic Acids Res. 33, W299–W302. [DOI] [PMC free article] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A. & Swindells, M. B. (2011). J. Chem. Inf. Model. 51, 2778–2786. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- Motorin, Y. & Helm, M. (2010). Biochemistry, 49, 4934–4944. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nallagatla, S. R., Jones, C. N., Ghosh, S. K., Sharma, S. D., Cameron, C. E., Spremulli, L. L. & Bevilacqua, P. C. (2013). PLoS One, 8, e57905. [DOI] [PMC free article] [PubMed]
- Newman, J. (2004). Acta Cryst. D60, 610–612. [DOI] [PubMed]
- Newman, J., Egan, D., Walter, T. S., Meged, R., Berry, I., Ben Jelloul, M., Sussman, J. L., Stuart, D. I. & Perrakis, A. (2005). Acta Cryst. D61, 1426–1431. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Park, F., Gajiwala, K., Noland, B., Wu, L., He, D., Molinari, J., Loomis, K., Pagarigan, B., Kearins, P., Christopher, J., Peat, T., Badger, J., Hendle, J., Lin, J. & Buchanan, S. (2004). Proteins, 55, 772–774. [DOI] [PubMed]
- Rakovich, T., Boland, C., Bernstein, I., Chikwana, V. M., Iwata-Reuyl, D. & Kelly, V. P. (2011). J. Biol. Chem. 286, 19354–19363. [DOI] [PMC free article] [PubMed]
- Rider, L. W., Ottosen, M. B., Gattis, S. G. & Palfey, B. A. (2009). J. Biol. Chem. 284, 10324–10333. [DOI] [PMC free article] [PubMed]
- Robert, X. & Gouet, P. (2014). Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Söding, J., Biegert, A. & Lupas, A. N. (2005). Nucleic Acids Res. 33, W244–W248. [DOI] [PMC free article] [PubMed]
- Spinola, M., Galvan, A., Pignatiello, C., Conti, B., Pastorino, U., Nicander, B., Paroni, R. & Dragani, T. A. (2005). Oncogene, 24, 5502–5509. [DOI] [PubMed]
- Stivala, A., Wybrow, M., Wirth, A., Whisstock, J. C. & Stuckey, P. J. (2011). Bioinformatics, 27, 3315–3316. [DOI] [PubMed]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., DiMaio, F., Read, R. J., Baker, D., Bunkóczi, G., Adams, P. D., Grosse-Kunstleve, R. W., Afonine, P. V. & Echols, N. (2012). J. Struct. Funct. Genomics, 13, 81–90. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Moriarty, N. W., Zwart, P. H., Hung, L.-W., Read, R. J. & Adams, P. D. (2008). Acta Cryst. D64, 61–69. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Xing, F., Hiley, S. L., Hughes, T. R. & Phizicky, E. M. (2004). J. Biol. Chem. 279, 17850–17860. [DOI] [PubMed]
- Yu, F., Tanaka, Y., Yamashita, K., Suzuki, T., Nakamura, A., Hirano, N., Suzuki, T., Yao, M. & Tanaka, I. (2011). Proc. Natl Acad. Sci. USA, 108, 19593–19598. [DOI] [PMC free article] [PubMed]
- Zinshteyn, B. & Gilbert, W. V. (2013). PLoS Genet. 9, e1003675. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human dihydrouridine synthase, 4xp7
Supplementary Figures. DOI: 10.1107/S1399004715009220/rr5097sup4.pptx
Supplementary Tables.. DOI: 10.1107/S1399004715009220/rr5097sup1.pdf