

The expression, purification and crystal structure of a recombinant fluorobody to TLH are reported.
Keywords: fluorobody, GFP scaffold, CDR3, TLH
Abstract
A fluorobody is a manmade hybrid molecule that is composed of green fluorescent protein (GFP) and a fragment of antibody, which combines the affinity and specificity of an antibody with the visibility of a GFP. It is able to provide a real-time indication of binding while avoiding the use of tags and secondary binding reagents. Here, the expression, purification and crystal structure of a recombinant fluorobody for TLH (thermolabile haemolysin), a toxin from the lethal food-borne disease bacterium Vibrio parahaemolyticus, are presented. This is the first structure of a fluorobody to be reported. Crystals belonging to space group P43212, with unit-cell parameters a = b = 63.35, c = 125.90 Å, were obtained by vapour diffusion in hanging drops and the structure was refined to an R free of 16.7% at 1.5 Å resolution. The structure shows a CDR loop of the antibody on the GFP scaffold.
1. Introduction
Antibodies are some of the most important biomolecules and are widely used in experimental biology, biomedical research, diagnostics and therapy (Peterson, 1996 ▸; Ayyar et al., 2012 ▸; Brennan et al., 2010 ▸; Gebauer & Skerra, 2009 ▸; van Dongen et al., 2007 ▸; Ross et al., 2003 ▸). Conventional antibodies are Y-shaped molecules that consist of two identical heavy chains and two identical light chains held together by interchain disulfide bonds. Each of the chains contains three of the six loops that form the binding regions. These regions are also called complementary-determining regions (CDRs) and are located in the variable fragment (Fv) of the antibody. The hypervariable regions of the antibody are surrounded by less variable β-sheet stretches (Wilson & Stanfield, 1994 ▸). The engineered scFv (single-chain variable fragment) only contains the Fv domain and a linker connecting the light chain and heavy chain together, and has decreased size and antigenicity (Ahmad et al., 2012 ▸). Recently, heavy-chain antibodies (HcAbs) have been characterized from camels and sharks. The antigen-binding site of these antibodies consists of only one single domain, in which the CDR loops, in particular the long, protruding CDR3, play a major role in antigen recognition (Wesolowski et al., 2009 ▸). Based on HcAb, a single-domain antibody (sdAb, also called a nanobody) was developed with improved stability and solubility. It also more easily penetrates tissues and has a high production yield in Escherichia coli (Djender et al., 2014 ▸). Although the CDR1, the CDR2, the framework regions between the CDRs and even the most distant Fc region can affect the binding properties of the antibody (Sela-Culang et al., 2013 ▸; Dam et al., 2008 ▸; Rodríguez-Rodríguez et al., 2012 ▸), CDR3 is considered to be the most important region for antigen binding (Xu & Davis, 2000 ▸). In some favourable cases, only the CDR3 can maintain the binding specificity (Deroo et al., 2008 ▸). Thus, it is possible to transfer the CDR3 to other scaffolds while retaining its binding properties.
Green fluorescent protein (GFP) is an intrinsic fluorescent protein with a typical β-barrel structure that mainly consists of β-sheets. It has high thermodynamic and chemical stability and has been used as a scaffold for peptide display and antibody engineering (Abedi et al., 1998 ▸). A hybrid molecule that combines the affinity and specificity of an antibody with the visibility of a GFP is called a fluorobody. With intrinsic fluorescence, it is able to provide a real-time indication of binding and to avoid the use of tags and secondary binding reagents (Espey et al., 2002 ▸). In addition, the fluorescence of the GFP also indicates that the hybrid protein is properly folded. It was found that insertions at three exposed loop regions of the superfolder GFP do not affect the stability and fluorescence of GFP, although insertion at many other sites substantially reduces GFP fluorescence (Pavoor et al., 2009 ▸). It is therefore possible to make a fluorobody by inserting a CDR loop into one of the exposed loop regions of GFP. No structure of a fluorobody has been reported yet despite its potential wide application.
For pathogen detection, a fluorobody was developed by inserting the L-CDR3 (light-chain complementary-determining region 3) of a high-affinity scFv into loop 9 of GFP (residues 171–176) (Wang et al., 2014 ▸). This fluorobody retains its fluorescence properties and binding specificity for thermolabile haemolysin (TLH), a toxin from Vibrio parahaemolyticus, a food-borne Gram-negative halophilic bacterium that causes lethal food-borne diseases and poses a serious threat to human and animal health all over the world (Burdette et al., 2008 ▸; Bresee et al., 2002 ▸). In this study, we crystallized this fluorobody and determined its structure at 1.5 Å resolution. This model will provide a molecular basis for fluorobody development and antibody research.
2. Materials and methods
2.1. Macromolecule production
The fluorobody expression vector pGEPi-sfGFP-9-LCDR3a was made by inserting L-CDR3a (228-H1QYHRSPR2T-236) into the pGEPi-sfGFP vector (donated by Dr Mengfei Ho, University of Illinois at Urbana-Champaign, USA) at positions 175S/176V of GFP loop 9. The anti-TLH scFv (Wang et al., 2012 ▸) was used as the template for amplifying L-CDR3a. PCR amplification was performed using the following primers: forward, 5′-TTAGGATCCCACCAGTATCATCGTTCCCCACGGACGCTGCAGGGATCAGTTCAACTAGCAGACCATTATCAAC-3′; reverse, 5′-GCGGCACATCGTACGGATAACCAGAAC-3′. The PCR products were digested with BamHI and NotI and inserted into pGEPi-sfGFP vector. The expressed proteins contained a 6×His tag at the C-terminus. The residue Arg2 in the L-CDR3 loop was mutated to Lys during a lysine-scanning experiment and the resulted plasmid was designated pGEPi-sfGFP-9-LCDR3a-K.
The fluorobody was expressed by transforming Escherichia coli BL21(DE3) cells (Novagen) with plasmid pGEPi-sfGFP-9-LCDR3a-K; expression was induced with 0.1 mM isopropyl β-d-1-thiogalactopyranoside for 12 h on attaining an OD of 0.6. Cultures were grown in Luria–Bertani (LB) medium containing 100 µg ml−1 kanamycin at 16°C and were harvested by centrifugation at 3000g for 20 min at 4°C. The cell pellet from 1 l of culture was resuspended in lysis buffer (20 ml 50 mM Tris–HCl pH 8.0, 300 mM NaCl) and lysed by sonication. The supernatant was separated from the cell debris by centrifugation at 18 000g for 30 min and loaded onto an Ni-affinity column (Qiagen). Protein with a 6×His tag was eluted with buffer containing 100 mM imidazole after extensive washing with lysis buffer to remove nonspecifically bound proteins. The obtained protein was dialyzed against 20 mM Tris–HCl pH 8.0 and then loaded on a RESOURCE Q column (GE Healthcare). Bound proteins were eluted in a gradient of 0–1 M NaCl and the peak fractions at around 185 mM NaCl were pooled and further purified by size-exclusion chromatography on a Superdex 75 column (GE Healthcare) pre-equilibrated with 20 mM Tris–HCl pH 8.0. The peak fractions at an elution volume of around 11 ml were collected and concentrated to 40 mg ml−1 for crystallization using a 10 kDa cutoff centrifugal filter (Millipore). Macromolecule-production information is summarized in Table 1 ▸.
Table 1. Macromolecule-production information.
| Complete amino-acid sequence of the construct produced | MKATKLVPSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLGVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNFNSHNVYITADKQKNGIKANFKIRHNIEDGSHQYHRSPKTLQGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHGMDELYKGGPGIKAAAGSGYPYDVPDYAGSGEQKLISEEDLNGAAGSGHHHHHH |
2.2. Crystallization
Crystallization trials were performed using the hanging-drop vapour-diffusion method at 18°C. Drops were prepared by mixing 0.5 µl protein solution with 0.5 µl reservoir solution and were equilibrated against 50 µl reservoir solution in the wells. Initial hits were obtained using the JCSG kit (Qiagen, USA). Diffraction-quality fluorobody crystals were grown under optimized conditions (40% PEG 400, 0.1 M HEPES pH 7.0) after 2 d. Crystallization information is summarized in Table 2 ▸.
Table 2. Data collection and processing.
| Diffraction source | Beamline 17U, SSRF |
| Wavelength () | 0.97915 |
| Temperature (K) | 100 |
| Detector | ADSC Q315 CCD |
| Crystal-to-detector distance (mm) | 200 |
| Rotation range per image () | 1 |
| Total rotation range () | 180 |
| Exposure time per image (s) | 0.5 |
| Space group | P43212 |
| a, b, c () | 63.35, 63.35, 125.90 |
| , , () | 90, 90, 90 |
| Mosaicity () | 0.305 |
| Resolution range () | 31.681.494 (1.5471.494) |
| Total No. of reflections | 581228 (57709) |
| No. of unique reflections | 42094 (4168) |
| Completeness (%) | 99.33 (100.00) |
| Multiplicity | 13.8 (13.8) |
| I/(I) | 23.41 (10.19†) |
| R r.i.m. | 0.07757 (0.2352) |
| Overall B factor from Wilson plot (2) | 14.12 |
The data set was collected at a suboptimal distance and data beyond 1.49 resolution were not collected.
2.3. Data collection and processing
Crystals were cooled directly in liquid nitrogen. Data were collected at the BL17U station of the Shanghai Synchrotron Radiation Facility (SSRF) at −173°C. A complete data set was collected at a wavelength of 0.97915 Å using 1° oscillations and an exposure time of 0.5 s per frame. The data were processed using the HKL-2000 and xia2 software (Otwinowski & Minor, 1997 ▸; Winter, 2010 ▸).
2.4. Structure solution and refinement
The structure was determined by molecular replacement with Phaser (McCoy et al., 2007 ▸; Read, 2001 ▸) using the superfolder GFP structure (PDB entry 2b3p; Pedelacq et al., 2006 ▸) as a search model. Model building and iterative refinement were performed with Coot and PHENIX (Adams et al., 2010 ▸; Emsley et al., 2010 ▸). The orientations of the amino-acid side chains and bound water molecules were modelled on the basis of 2mF obs − DF calc and mF obs − DF calc difference Fourier maps. The model figures were generated with PyMOL (v.1.3r1; Schrödinger) and Chimera (Pettersen et al., 2004 ▸). MolProbity (Chen et al., 2010 ▸) was used for Ramachandran analysis. The interaction of the loop was analyzed using LigPlot+ (Laskowski & Swindells, 2011 ▸).
3. Results and discussion
To discover the structural features of the fluorobody, it was expressed in E. coli and purified by affinity, anion-exchange and gel-filtration chromatography. The purified fluorobody showed a single band on SDS–PAGE which is consistent with the calculated molecular mass (Figs. 1 ▸ a and 1 ▸ c). The resulting protein was subjected to crystallization trials as described in §2. Several conditions yielded crystals, and diffraction-quality crystals were obtained using a condition containing PEG 400 (Fig. 1 ▸ b). The structure was determined and refined to 1.5 Å resolution with final R work and R free values of 14.5 and 16.7%, respectively (Table 3 ▸). The space group was P43212 (unit-cell parameters a = b = 63.35, c = 125.9 Å). The majority of the GFP fold was built automatically using Buccaneer (Cowtan, 2006 ▸). The L-CDR3 loop was manually rebuilt using Coot (Emsley et al., 2010 ▸), followed by several rounds of refinement using PHENIX (Adams et al., 2010 ▸). The final model contains one monomer per asymmetric unit. As predicted, the structure of the fluorobody forms a typical GFP fold with a modified L-CDR3 loop. The GFP fold is an 11-stranded β-barrel wrapped around a single central helix which spontaneously cyclizes the tripeptide Ser65-Tyr66-Gly67 to form a chromophore. The tightly packed nature of the barrel excludes solvent molecules, protecting the chromophore fluorescence from quenching by water. The fluorobody and sfGFP have a root-mean-square deviation (r.m.s.d.) of 0.354 Å over 237 GFP Cα atoms (Fig. 2 ▸). 13 of the 18 amino-acid residues of the L-CDR3 loop [IEDGS-H1Q1-YHRSPKTLQGS; the loop also includes four residues (LQGS) derived from a cloning artifact and five residues (IEDGS) derived from the original GFP loop 9] are clearly visible in the OMIT map, but the map does not show clear electron density for residues 173–177, including His1 and Gln1 of the CDR. The model contains these five residues in order to interpret some strong density that is most likely to arise from them. Accordingly, the high B factor and discontinuous electron density imply that these five residues are in a dynamic state (Fig. 3 ▸ b). Checking the crystal contacts, we found that part of the L-CDR3 loop is stabilized in the crystal by interaction with a neighbouring molecule. Two adjacent symmetry-related molecules form a homodimer by interacting with each other. The SDS–PAGE result also shows the existence of a trace amount of dimer (Fig. 1 ▸a). The L-CDR3 loop of one molecule interacts with the GFP barrel of another neighbouring molecule. The side chain of His in the loop is stabilized by forming a hydrogen bond to Asn164 and a hydrophobic interaction with Thr153 of a neighbouring GFP molecule. The side chains of Pro and Lys are stabilized by hydrophobic interaction with Phe165 and Lys166 of a neighbouring GFP molecule. Thus, these residues, as well as the Arg and Tyr residues in the loop, are reasonably well defined by the electron density in the OMIT map (Figs. 3 ▸ b and 3 ▸ c).
Figure 1.

Purification of the fluorobody. (a) The purified fluorobody was analyzed by 15% SDS–PAGE. Lane 1, the purified fluorobody. Lane M contains molecular-mass marker (labelled in kDa). (b) The crystal obtained under the optimized condition (40% PEG 400, 0.1 M HEPES pH 7.0). (c) The gel-filtration chromatographic profile of the fluorobody (Superdex 75 10/300 column).
Table 3. Structure refinement.
| Resolution range () | 31.681.494 (1.5471.494) |
| Completeness (%) | 99.33 (100.00) |
| Cutoff | 10.19 |
| No. of reflections, working set | 40098 |
| No. of reflections, test set | 1996 |
| Final R work | 0.145 |
| Final R free | 0.167 |
| Cruickshank DPI | 0.0796 |
| No. of non-H atoms | |
| Protein | 1985 |
| Polyethylene glycol | 32 |
| Water | 303 |
| Total | 2320 |
| R.m.s. deviations | |
| Bonds () | 0.009 |
| Angles () | 1.34 |
| Average B factors (2) | |
| Overall | 24.2 |
| Protein | 21.8 |
| Polyethylene glycol | 39.6 |
| Water | 37.4 |
| Ramachandran plot | |
| Favoured regions (%) | 98 |
| Additionally allowed (%) | 2 |
Figure 2.
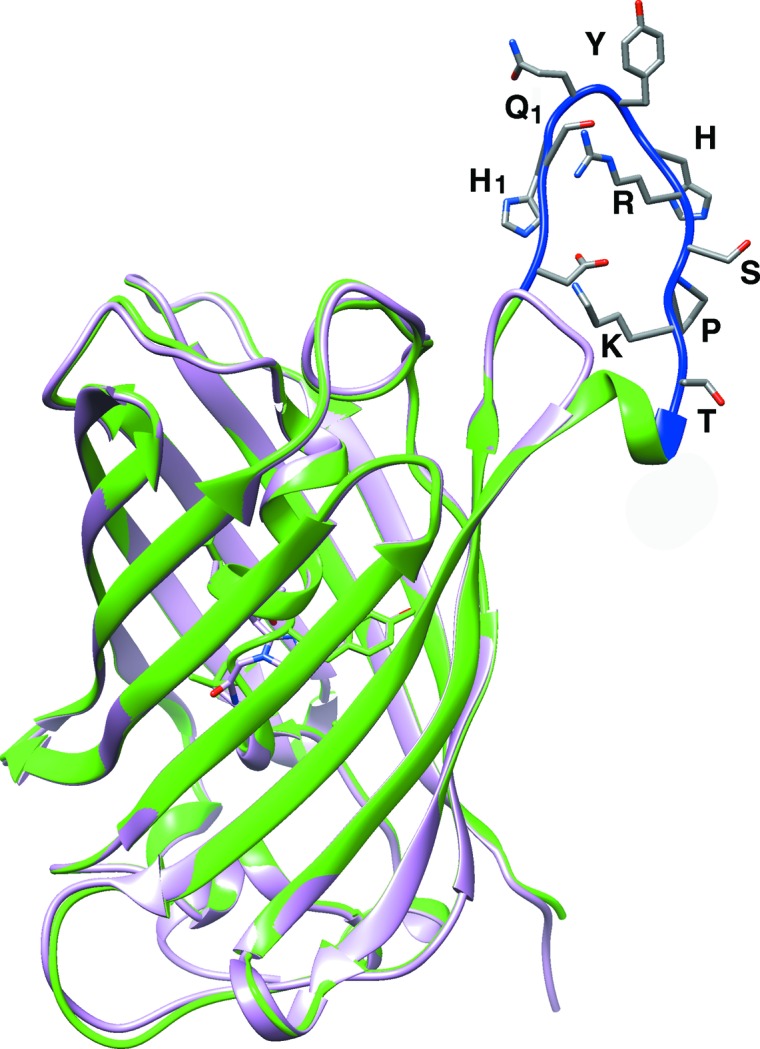
Superimposed structures of the fluorobody and sfGFP. SfGFP, the GFP part of the fluorobody and the L-CDR3 loop are shown in pink, green and blue, respectively.
Figure 3.

The L-CDR3 loop is stabilized in the crystal by interaction with the neighbouring molecule. (a) Two adjacent symmetry-related molecules, forming the homodimer by interacting with each other, are shown as green and cyan ribbons. The L-CDR3 loop is depicted in sticks. An F o − F c OMIT map (red mesh, omitting residues 171–188) shows the electron density of the L-CDR3 loop between two molecules contoured at 3σ. (b) The detailed electron density (red mesh) from the F o − F c OMIT map (2σ) shows the residues of the L-CDR3 loop (modelled as blue sticks). The residues from GFP are modelled as green sticks and cartoons. (c) The L-CDR3 loop residues Gln1, His, Pro and Lys interact with a neighbouring GFP barrel. The neighbouring GFP barrel is shown as a blue ribbon and the L-CDR3 loop is shown as a green ribbon. The residues involved in the interaction are shown in sticks. Hydrophobic interactions are shown as yellow dashed lines and two hydrogen bonds are shown as red dashed lines.
Alanine scanning showed that the Gln1, Arg and Pro residues of the CDR3 loop have the most prominent effects on antigen binding, and lysine scanning showed that the current Lys residue of the loop has the best binding affinity (Wang et al., 2014 ▸). In our structure, the Arg, Pro and Lys residues mainly help to stabilize the CDR loop structure, while residue Gln1 is in a dynamic state and may be involved in direct interactions with the antigen.
Supplementary Material
PDB reference: fluorobody, 4xgy
Acknowledgments
This work was supported by the funding from the Scientific and Technology Project of Fujian Province of China (No. 2014YZ0001), the One Hundred Person Project of the Chinese Academy of Sciences and the NSF of Fujian Province (2013J01150). The authors would like to thank the beamline scientists at the SSRF for support during data collection. We are grateful to Dr Mengfei Ho (University of Illinois at Urbana-Champaign) for donating the pGEPi-sfGFP vector.
References
- Abedi, M. R., Caponigro, G. & Kamb, A. (1998). Nucleic Acids Res. 26, 623–630. [DOI] [PMC free article] [PubMed]
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Ahmad, Z. A., Yeap, S. K., Ali, A. M., Ho, W. Y., Alitheen, N. B. M. & Hamid, M. (2012). Clin. Dev. Immunol. 2012, 980250. [DOI] [PMC free article] [PubMed]
- Ayyar, B. V., Arora, S., Murphy, C. & O’Kennedy, R. (2012). Methods, 56, 116–129. [DOI] [PubMed]
- Brennan, D. J., O’Connor, D. P., Rexhepaj, E., Ponten, F. & Gallagher, W. M. (2010). Nature Rev. Cancer, 10, 605–617. [DOI] [PubMed]
- Bresee, J. S., Widdowson, M. A., Monroe, S. S. & Glass, R. I. (2002). Clin. Infect. Dis. 35, 748–753. [DOI] [PubMed]
- Burdette, D. L., Yarbrough, M. L., Orvedahl, A., Gilpin, C. J. & Orth, K. (2008). Proc. Natl Acad. Sci. USA, 105, 12497–12502. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cowtan, K. (2006). Acta Cryst. D62, 1002–1011. [DOI] [PubMed]
- Dam, T. K., Torres, M., Brewer, C. F. & Casadevall, A. (2008). J. Biol. Chem. 283, 31366–31370. [DOI] [PMC free article] [PubMed]
- Deroo, S., Fischer, A., Beaupain, N., Counson, M., Boutonnet, N., Pletinckx, J., Loverix, S., Beirnaert, E., De Haard, H., Schmit, J. C. & Lasters, I. (2008). Mol. Immunol. 45, 1366–1373. [DOI] [PubMed]
- Djender, S., Schneider, A., Beugnet, A., Crepin, R., Desrumeaux, K. E., Romani, C., Moutel, S., Perez, F. & de Marco, A. (2014). Microb. Cell Fact. 13, 140. [DOI] [PMC free article] [PubMed]
- Dongen, G. A. M. S. van, Visser, G. W., Lub-de Hooge, M. N., de Vries, E. G. & Perk, L. R. (2007). Oncologist, 12, 1379–1389. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Espey, M. G., Xavier, S., Thomas, D. D., Miranda, K. M. & Wink, D. A. (2002). Proc. Natl Acad. Sci. USA, 99, 3481–3486. [DOI] [PMC free article] [PubMed]
- Gebauer, M. & Skerra, A. (2009). Curr. Opin. Chem. Biol. 13, 245–255. [DOI] [PubMed]
- Laskowski, R. A. & Swindells, M. B. (2011). J. Chem. Inf. Model. 51, 2778–2786. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pavoor, T. V., Cho, Y. K. & Shusta, E. V. (2009). Proc. Natl Acad. Sci. USA, 106, 11895–11900. [DOI] [PMC free article] [PubMed]
- Pedelacq, J.-D., Cabantous, S., Tran, T., Terwilliger, T. C. & Waldo, G. S. (2006). Nature Biotechnol. 24, 79–88. [DOI] [PubMed]
- Peterson, N. C. (1996). Lab. Anim. Sci. 46, 8–14. [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed]
- Read, R. J. (2001). Acta Cryst. D57, 1373–1382. [DOI] [PubMed]
- Rodríguez-Rodríguez, E. R., Ledezma-Candanoza, L. M., Contreras-Ferrat, L. G., Olamendi-Portugal, T., Possani, L. D., Becerril, B. & Riaño-Umbarila, L. (2012). J. Mol. Biol. 423, 337–350. [DOI] [PubMed]
- Ross, J., Gray, K., Schenkein, D., Greene, B., Gray, G. S., Shulok, J., Worland, P. J., Celniker, A. & Rolfe, M. (2003). Expert Rev. Anticancer Ther. 3, 107–121. [DOI] [PubMed]
- Sela-Culang, I., Kunik, V. & Ofran, Y. (2013). Front. Immunol. 4, 302. [DOI] [PMC free article] [PubMed]
- Wang, R., Fang, S., Wu, D., Lian, J., Fan, J., Zhang, Y., Wang, S. & Lin, W. (2012). Appl. Environ. Microbiol. 78, 4967–4975. [DOI] [PMC free article] [PubMed]
- Wang, R., Xiang, S., Zhang, Y., Chen, Q., Zhong, Y. & Wang, S. (2014). Appl. Environ. Microbiol. 80, 4126–4137. [DOI] [PMC free article] [PubMed]
- Wesolowski, J. et al. (2009). Med. Microbiol. Immunol. 198, 157–174. [DOI] [PMC free article] [PubMed]
- Wilson, I. A. & Stanfield, R. L. (1994). Curr. Opin. Struct. Biol. 4, 857–867. [DOI] [PubMed]
- Winter, G. (2010). J. Appl. Cryst. 43, 186–190.
- Xu, J. L. & Davis, M. M. (2000). Immunity, 13, 37–45. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: fluorobody, 4xgy