

Key Clinical Message
Diamond-Blackfan anemia (DBA) is a congenital erythroid aplasia usually diagnosed in the early infancy and associated with mutations or large deletions in 11 ribosomal protein (RP) genes. Adult patients with severe, transfusion dependence, aregenerative anemia might have a genetic-in-origin disease with an atypical presentation. Late onset nonclassical DBA should be ruled out and mutations of RP genes studied.
Keywords: Diamond-Blackfan anemia, pure red cell aplasia, RPL11 mutations
Introduction
Diamond-Blackfan anemia (DBA) is a rare genetic disease mostly diagnosed in the first year of life. Different mutations on genes codifying for large and small subunits of ribosomal proteins (RPL and RPS) have been described as disease causative. Additionally, genotype–phenotype correlations have been established. We report an atypical case of a previously healthy 35-year-old patient presenting with severe aregenerative anemia which was ultimately found to be genetic in origin. Fanconi anemia was discarded and a nonclassical DBA caused by a “de novo” and previously nondescribed mutation located in the exon 5 of the RPL11 gen was diagnosed. Transfusion dependence was avoided with a nonsteroidal medical treatment. This case report pointed out that a late onset anemia presenting in an adult patient is not incompatible with a genetic-in-origin disease with an atypical presentation.
Diamond-Blackfan anemia (DBA) is a rare disease characterized primarily by normochromic macrocytic anemia and reticulocytopenia. Classical DBA affects about seven out of every million live births and presents during the first year of life. About 40% of patients display physical malformations, mostly craniofacial (ophthalmological disorders including), and at the extremities. Recent advances in identifying the genotype that underlies DBA have shown involvement of genes encoding both large (RPL) and small (RPS) ribosomal subunit proteins. However, as mutated genes have been discovered in DBA, nonclassical cases with less distinct phenotypes are being described in adults as well as in children.
Here, we are reporting a 35-year-old patient with congenital thumb hypoplasia who presented to us with severe aregenerative anemia finally being diagnosed with a nonclassical DBA caused by a De novo and previously nondescribed mutation located in the exon 5 of the RPL11 gen.
Case Report
A Spanish 35-year-old male patient, with a surgical history of interventions in both hands because of congenital hypoplasia of the thumb of the right hand and syndactyly between first and second fingers of the left hand, attended the emergency department complaining of progressive fatigue and headache for the past 15 days. Severe normocytic normochromic reticulocytopenic anemia was disclosed (Hb 7.2 gr/dL (13–18 gr/dL), MCV 92.9 fl (80–90 fl), reticulocyte 13 × 109/L, WBC 4.3 × 109/L (4–11 × 109/L) with normal differential and platelets 342 × 109/L(150–400 × 109). Anemia studies were as follows: iron 227 μg/dL (65–175 μg/dL), ferritin 664 ng/mL (22–322 ng/mL), transferrin saturation 100% (20–50%), B12 vitamin 486 pg/mL (211–911 pg/mL), and folic acid 2.8 ng/mL (5.4–17 ng/mL). Hemoglobin electrophoresis excluded thalassemia. Renal and liver function tests were normal. Biochemical parameters of hemolysis were absent. Direct antiglobulin test (DAT) and indirect antiglobulin tests were negative. Autoimmunity screening (ANA, ENA, anti-DNA, C3–C4, ANCAp, ANCAc) was normal. Serologies for HIV, HBV, HCV, syphilis, and EBV were all negative. DNA amplification for B-19 Parvovirus by PCR on the serum was negative. Immunophenotypic study of peripheral blood was negative for clonally B and T cells. PNH clone was negative.
Bone marrow (BM) aspiration showed moderate hypoplasia of the erythroid lineage with myeloid and megakaryocytic cells of a normal aspect and without any signs of dysplasia (see Figure 1). Perls' reaction in the bone marrow smear was negative. Cariotypic analysis on BM cells revealed a normal male cariotype 46 XY.
Figure 1.
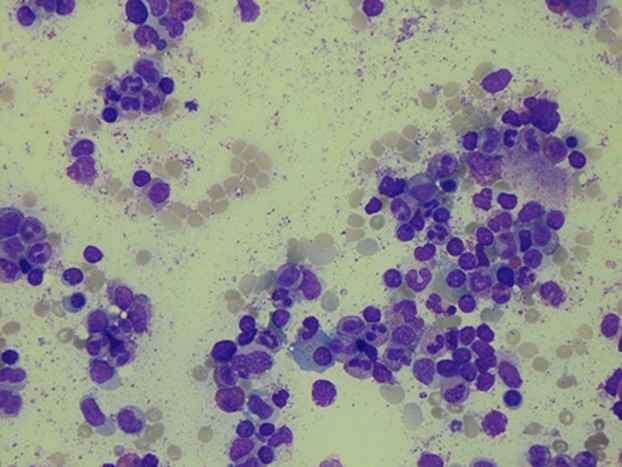
Bone marrow study. Hypoplasia of erythroid lineage.
A CT-scan of the thorax, abdomen, and pelvis and an abdominal ultrasound were both normal. Echocardiogram was normal. Skeleton plain radiographs showed fusion of the phalanges of the finger with the thumb metacarpal in the left hand and absence of the first finger in the right hand (see figure 2).
Figure 2.
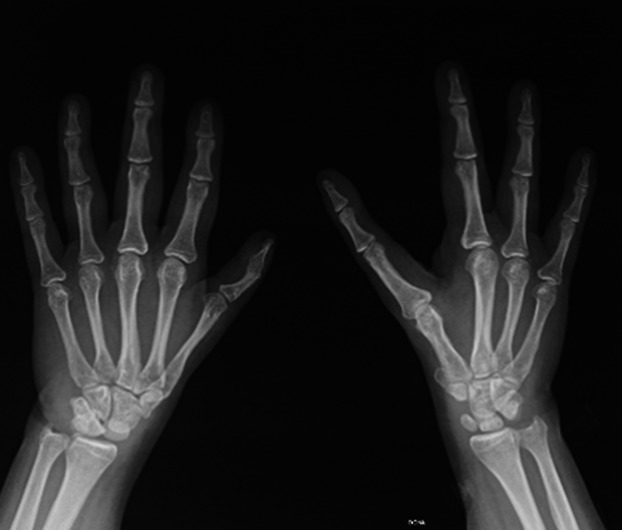
Radiologic studies. Malformations in both hands. Left hand, fusion of the phalanges of the finger with the thumb metacarpal. Right hand: Absence of the second toe.
The patient underwent a complete ophthalmologic evaluation including best corrected visual acuity, slit-lamp biomicroscopy, and dilated ophthalmoscopic examination after instillation of phenylephrine 2.5% and tropicamide 1%. No refractive defects, no epicanthus palpebralis, no strabismus, no microcornea, no microphthalmia, no posterior embryotoxon, no cataracts, and no signs of congenital glaucoma were found 8.
Fanconi Anemia was excluded by means of chromosomal fragility studies performed on the peripheral blood with C mitomycin and diepoxybutane and complementation studies for Fanconi genes in cultured skin fibroblasts that were all negative.
A molecular study for DBA genes was performed and C489_490delGCinsT heterozygous mutation in exon 5 of the RPL11 gene was detected. This mutation produces a variation in the composition of the last 16 amino acidic residues of the normal protein. This is previously nondescribed mutation for DBA development. Molecular studies of the RPL11 gene were done in both parents and her sister and disclosed normal results. According to the description of the majority of DBA patients, our patient had a “de novo” mutation. Our patient also exhibited other variations of the RPS19 (IVS4+14G>A) and RPS26 (5′UTR-22C>G) genes, that were considered as polymorphism and not pathological variations.
According to the diagnostic criteria previously established at an International Consensus Conference 1, our patient should be classified as sporadic nonclassical form of DBA (insufficient diagnosis criteria and gene mutation described in “classical” DBA) 6.
The patient was initially treated 3 with prednisone (1 mg/Kg/24 h), folic acid, and vitamin B12. He needed regular red blood cells transfusions and iron chelation with deferasirox was started. A short course of high doses of intravenous methylprednisolone (500 mg per day for 5 consecutive days) was administered and later on steroids were tapered until complete discontinuation, because of its inefficacy and adverse effects. We decided to ask for an off-label use of cyclosporine to reduce the number of transfusions. Cyclosporine was approved by ethic committee of University Hospital Principe de Asturias and the patient's written informed consent was requested. Six months after initial presentation, cyclosporine at a dose of 5 mg/kg/day, twice a day was started and subsequently eight months after presentation danazol at a dose of 400 mg per day was added. With this combination, we observed a progressive erythroid response and transfusions were avoided (last transfusion on 31st July 2012). Since January 2013 a slow reduction in cyclosporine dose was initiated until its complete discontinuation on November 2013. He was treated with acyclovir, sulfametoxazol and trimetroprim because he has been treated with cyclosporine for eighteen months. Currently, our patient is exclusively on treatment with danazol.
Two years after diagnosis the ophthalmologic evaluation did not reveal any change despite corticosteroids treatment.
A Familiar HLA typing was performed and his only sister was found to be HLA genetically identical. A molecular study of the RPL11 gene was performed on both parents and his only sister and were all normal. Until now the possibility of performing an allogeneic hematopoietic stem cell transplantation has been postponed.
Discussion
We described a real clinical case of Diamond-Blackfan anemia in adulthood 4 (debut at 35 years old). Classical DBA affects about seven per million live births and presents during the first year of life. Our case illustrates that late onset is not incompatible with the diagnosis of DBA. In the last 20 years, different mutations and deletions in nine ribosomal protein genes (RPL and PRS) have been discovered and being associated with a haploinsufficiency development of DBA. Defects in the RPS19 5,7gene, encoding the ribosomal protein S19, are the main known cause of Diamond-Blackfan anemia and account for more than 25% of cases. Two new genes 2 (RPL5, RPL11), encoding for ribosomal proteins of the large subunit, have been reported to be involved in a considerable percentage of patients. Gazda et al. and Cnejla et al. found mutations in RPL11 in about 5% and 7%, respectively, of DBA patients. Our patient has a previously undescribed mutation in exon 5 of RPL11, according to the description of the majority of DBA patients. Gazda et al. reported that RPL5 and RPL11 mutations are more frequently associated with physical malformations than are RPS19 mutations. Mutations in RPL11 were predominantly associated with thumb abnormalities (like our patient) and craniofacial malformation.
It was a de novo mutation as it was not found either in his parents or in his sister. The RPL11 gene mutations have previously been associated with congenital bone malformations as those presented at the birth in our patient. Although we may assume that our patient was born with this RPL-11 mutation, he did not develop anemia until the age of 35 years. We decided to start on cyclosporine to avoid the secondary effects of steroids but it was the addition of danazol what produced a real improvement of the hemoglobin levels. A continued and stable erythroid response has been observed under danazol therapy after cyclosporine withdrawal. Since June 2012, our patient has not received any red-blood-cell transfusion and has maintained the haemoglobin up to 10 gr/dl. The last control was in July 2014 and he has Haemoglobin 13.4 gr/dl.
In conclusion, Diamond-Blackfan anemia is a rare genetic disease mostly diagnosed in the first year of life, nevertheless late onset is not incompatible with the diagnosis of DBA. Different mutations on genes codifying for large and small subunits of ribosomal proteins have been described, as disease causative and also genotype–phenotype correlations have been established. The identification of the affected gene in a congenital disorder, such as DBA, is often the key that suddenly enables understanding of the molecular pathogenesis and development of novel therapies.
Recently, several aspects of DBA pathophysiology have been uncovered, including: genome-wide studies to find structural aberrations, and mutation analysis of all ribosomal proteins and ribogenesis factors, ribogenesis factors. More studies are required that will hopefully result in better diagnosis and treatment of this disease that profoundly impairs the quality of life of young patients and their families.
Conflict of Interest
None declared.
References
- Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br. J. Haematol. 2008;142:859–876. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarello P, Garelli E, Carando A, et al. Diamond- Blackfan anemia: genotype-phenotype correlations in Italian patients with RPL5 and RPL11 mutations. Haematologica. 2010;95:206–213. doi: 10.3324/haematol.2009.011783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos A. Muir E. How I treat Diamond-Blackfan anemia. Blood. 2010;116:3715–3723. doi: 10.1182/blood-2010-02-251090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban EP, Buchanan GR, Graham M. Frenkel EP. Diamond-Blackfan syndrome in adult patients. Am. J. Med. 1985;78:533–538. doi: 10.1016/0002-9343(85)90352-3. [DOI] [PubMed] [Google Scholar]
- Horos R. von Lindern M. Molecular mechanisms of pathology and treatment in Diamond Blackfan Anaemia. Br. J. Haematol. 2012;159:514–527. doi: 10.1111/bjh.12058. [DOI] [PubMed] [Google Scholar]
- Farruggia P, Quarello P, Garelli E, Paolicchi O, Ruffo GB, Cuccia L, et al. The spectrum of non-classical Diamond-Blackfan anemia: a case of late beginning transfusion dependency associated to a new RPL5 mutation. Pediatr. Rep. 2012;4:e25. doi: 10.4081/pr.2012.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniz H, Gastou M, Leblanc T, Hurtaud C, Crétien A, Lécluse Y, et al. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012;3:e356. doi: 10.1038/cddis.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsilou ET, Giri N, Weinstein S, Mueller C, Savage SA. Alter BP. Ocular and orbital manifestations of the inherited bone marrow failure syndromes: fanconi anemia and dyskeratosis congenita. Ophthalmology. 2010;117:615–622. doi: 10.1016/j.ophtha.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]