

Key Clinical Message
Low fraction fetal DNA in noninvasive prenatal testing in the context of fetal growth restriction and multiple congenital anomalies should alert medical professionals to the possibility of digynic triploidy. Single-nucleotide polymorphism microarray can detect the parental origin of triploidy and explain its mechanism.
Keywords: cffDNA, digyny, NIPT, SNP array, triploidy
Introduction
Triploidy is a chromosomal anomaly that is seen in 1–2% of all conceptions and up to 10% of spontaneous abortions 1. It is classified as diandry when the extra set of chromosomes is from the father and digyny when the extra set of chromosomes is of maternal origin. Most cases of diandry occur as a result of dispermy and less commonly are caused by nondisjunction during the first (MI) or the second meiotic division (MII) of spermatogenesis. On the other hand, most cases of digyny are caused by errors in MI or MII of oogenesis 2.
Although most triploid conceptions end in first trimester spontaneous abortions, there are few reports of live born infants with triploidy, with the longest reported survival of digynic triploidy at 10 ½ months 3. Based on personal communication of the authors of same report, about half of previously reported 38 triploidy cases were live-born and lived for a few minutes to 5 days 3. Niemann-Seyde et al. 4 reported a girl with diandric triploidy who survived for 10 ½ weeks and reviewed the data on 7 previously reported triploid patients who survived beyond 2 months of age. Hasegawa et al. 5 described a girl with digynic triploidy who survived for 46 days and discussed parental of origin and clinical findings of four additional triploid (three digynic and one diandric) infants who survived 4 weeks or more. Parental origin studies of triploidy showed variable diandric to digynic ratios 2,6–8, reflecting ascertainment bias and different selection criteria 8. Digyny is associated with asymmetric severe fetal growth restriction (FGR) and a very small placenta 8,9. Diandric triploid fetuses are relatively well grown or exhibit symmetric FGR accompanied by a large cystic, often molar, placenta 9.
Since the introduction of noninvasive prenatal testing (NIPT) to clinical practice, it has evolved to be a common screening test for chromosomal aberrations in pregnancies complicated by abnormal ultrasound or maternal serum screens or for advanced maternal age 10. However, there is limited data in the utility of NIPT in the detection of triploidy and the matter has never been systematically investigated. Here, we present a digynic triploidy case that escaped diagnosis despite using two methods of NIPT. Our data suggest that low fraction fetal DNA in NIPT along with multiple congenital anomalies and FGR should alert medical professionals to the possibility of digynic triploidy and demonstrate that SNP microarray can detect the parental origin of triploidy and its mechanism.
Case Presentation
The proband was born to a 24-year-old G1P0 after a pregnancy complicated by multiple fetal anomalies. There was no family history of congenital anomalies, early infant deaths, or consanguinity. The mother declined first trimester screening for fetal anomalies and chromosomal abnormalities. At 18 weeks of gestation, the fetus had been diagnosed with early-onset FGR, oligohydramnios, and bilateral brain ventriculomegaly. Severe oligohydramnios would have made the amniocentesis technically difficult and the parents declined the procedure. NIPT on cell-free fetal DNA (cffDNA) testing at 18 weeks of gestation could not be interpreted secondary to an insufficient fraction (1.3%). Repeat ultrasound assessment in our center at ∽22 weeks was notable for asymmetric bilateral ventriculomegaly, severe FGR (<5th percentile), oligohydramnios (amniotic fluid index was 5th centile), small chest, a possible right pelvic kidney, and the fetal gender could not be determined. The mother declined amniocentesis and opted to repeat cffDNA testing at 22 weeks of gestation, which demonstrated the expected representation of chromosome 21, 18, and 13 materials, absence of Y chromosome material, with cffDNA fraction of 5.2%. At ∽33 weeks, the estimated fetal weight was 601 g. The mother was counseled on the poor prognosis for her fetus but opted for full obstetric intervention, including potential cesarean section with full neonatal management. After one dose of antenatal steroids on the day prior to delivery, the proband was born via cesarean section for nonreassuring fetal status with a 0/8 biophysical profile and fetal bradycardia. A partial placental abruption, very small placenta and short, thin cord were noted at the time of delivery.
At birth, the neonate had no spontaneous respirations and a heart rate below 80 beats per minute, for which he was intubated and placed on a respirator, but soon required high frequency ventilation and nitric oxide. Despite the NIPT results, the infant was found to have male genitalia with hypospadias. On physical examination, all growth parameters were below 3rd centile for proband's gestational age. His weight was 590 g (50th centile for 23 weeks' gestation), length was 30 cm (50th centile for 23 weeks' gestation), and occipitofrontal circumference was 25 cm (50th centile for 26 weeks' gestation). Facies showed frontal bossing, hypertelorism, a bulbous nasal tip, a depressed nasal bridge, micrognathia, and low set and posteriorly rotated ears. There was cutaneous syndactyly of 2–4th toes bilaterally, as well as 3–4th fingers of the left hand, webbing of all digits of the right hand, and adducted thumbs bilaterally. His skin was thin and translucent. A head ultrasound demonstrated enlarged, dysplastic lateral ventricles with a dysplastic corpus callosum and possible partial thalamic fusion. A skeletal survey revealed thin clavicles, gracile ribs and humeri, and butterfly vertebral bodies in the upper thoracic spine. An echocardiogram showed a moderate perimembranous ventricular septal defect (VSD) and atrial-level shunt, a large patent ductus arteriosus (PDA), and dilation of the aortic root (Z score +2.3) and ascending aorta (Z score +3.2). Renal ultrasound showed small kidneys with loss of normal corticomedullary differentiation with few scattered microcysts. Ultrasound of the bladder showed an irregular contour suggestive of trabeculations. Despite maximum ventilatory support, the neonate's condition continued to decline. The family chose to redirect plans to supportive care and the neonate succumbed to respiratory failure at 16 h of life.
At autopsy, the aforementioned dysmorphic features were noted (Fig.1). There was dilatation of the lateral ventricles and fusion of the thalami. The lungs were small and hypoplastic and the testes were found intra-abdominally. The bladder was dilated with thickened wall and trabeculated mucosa associated with urethral stenosis. The placenta weighed 51.7 g and had microscopic features of chronic placental insufficiency including distal villous hypoplasia and increased syncytial knotting. The umbilical cord had three vessels with focal obliteration of the umbilical artery, consistent with flow change due to focal vascular thrombosis. Testes had immature seminiferous tubules.
Figure 1.
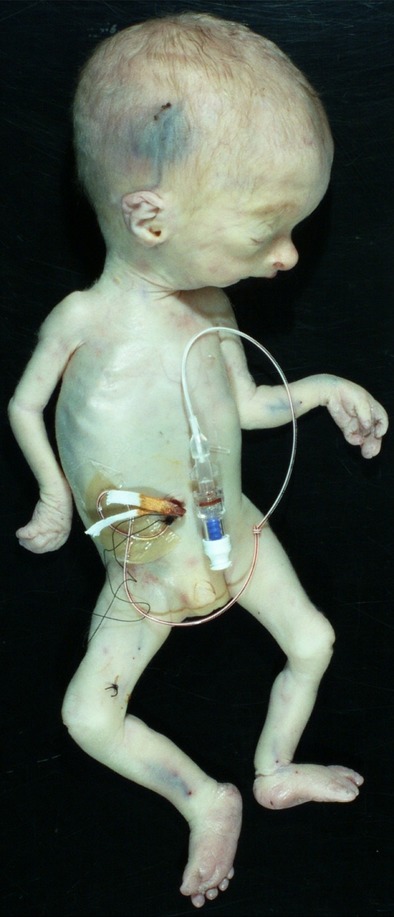
Autopsy findings in the proband. External examination demonstrates relative macrocephaly, dysmorphic facies, hypertelorism, micrognathia, and syndactyly of the third and fourth fingers on left hand.
Molecular and Cytogenetic Results
Due to multiple congenital anomalies in the neonate and discrepancy between the male phenotype and the NIPT results, FISH analysis was performed on a direct preparation from a sample of peripheral blood and revealed two hybridization signals for CEP X and one hybridization signal for SRY in all 500 nuclei analyzed. Subsequent rapid FISH analysis for select loci revealed three copies of chromosomes 13, 18, and 21 using LSI 13, CEP 18, and LSI 21, two copies of the X chromosome (CEP X), and one copy of the Y chromosome (CEP Y). A chromosomal microarray analysis (CMA), using the Affymetrix CytoScan HD array was performed with reflex to karyotype; the latter confirmed 69,XXY pattern. The CMA of the proband revealed triploidy as indicated by the presence of four-allele peak tracks on all autosomes with a normal Log2 ratio. There were two copies of chromosome X (Fig.2) and one copy of chromosome Y as interpreted from the allele peak tracks, Log2 ratio and smooth signal. Notably, an apparent copy number neutral region demonstrating a long contiguous stretch of homozygosity (LCSH) on chromosome X from bands p22.2 to q24 and spanning ∽106.28 Mb was observed and suggests that the majority of the length of chromosome X is identical across all loci. This pattern is consistent with a triploidy of digynic origin in that the two copies of chromosome X are uniparental and as by nature the Y chromosome is paternally derived, the X chromosomes are therefore maternally attributed. Regions distal to the LCSH include the pseudoautosomal regions demonstrated a Log2 ratio and allele peak tracks similar to the pattern seen in all autosomal chromosomes (Fig.2). Additionally, regions of meiotic recombination (cross-over) were observed on chromosome X encompassing bands p22.33 to p22.2 on the short arm and bands q24 to q28 on the long arm as determined by genomic stretches demonstrating heterozygosity. These findings, along with similar changes on the autosomes, indicate error in nondisjunction during MII as the most likely mechanism. The CMA also revealed a maternally inherited 762 kb gain of chromosomal material on 9p21.2 (26,456,904–27,218,960;hg19), likely of no clinical significance. Of note, because of the normalization algorithm employed during analysis, the detection of triploidy is not typically possible in array platforms containing solely copy number probes.
Figure 2.
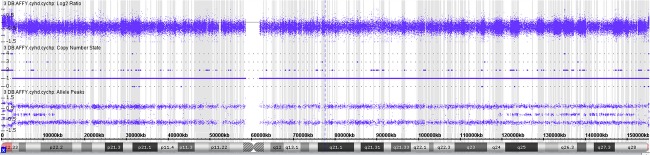
Molecular findings in the proband. Chromosomal Microarray Analysis (CMA) data from the X chromosome with a normalized copy number state ∽2 in PAR1 and PAR2, and ∽1 across the remainder of the chromosome with a long contiguous stretch of homozygosity observed proximally, and regions of heterozygosity observed distally.
Discussion
The results of NIPT in the proband make this case particularly interesting. Currently, there are four commercially available noninvasive prenatal tests in the United States: Natera's Panorama Prenatal test (NPP), Sequenom's MaterniT21 test (SM), Ariosa Diagnostics's Harmony test (ADH), and Verinata Health's Verifi test (VHV) 11. For our patient's mother, the initial NPP test was not diagnostic due to a low fraction of cffDNA. The subsequent SM test revealed no Y chromosomal material, consistent with a normal female fetus. One possibility for this difference is the type of technology used in each of these tests. The NPP test uses multiplex PCR followed by SNP-based targeted sequencing and analysis of parental genotypes 12, as opposed to the SM test which uses massively parallel shotgun sequencing and quantification of the number of reads from each chromosome 13. While both methods allow for accurate determination of individual chromosomal trisomies, the SM test uses a comparison among the relative number of reads from each chromosome. Hence, the SM test may be at a disadvantage in detecting triploidy, which affects all of the autosomes equally. In fact, while none of the other three commercial companies make a claim regarding the detection of triploidy, the NPP test in a recently published study has been able to correctly identify all four diandric trisomy cases, while all four digynic cases tested were found to have low cffDNA fraction after adjusting for maternal weight and gestational age (<4%) and consequently escaped diagnosis 14. The small size of the placenta in digynic triploidy likely contributes to low cffDNA 14. The source of cffDNA in maternal blood is dying cytotrophoblastic cells 15 and its low fraction reflects low placental mass as seen in digynic triploidy 14. The lack of observed Y chromosomal material in our proband by Sequenom NIPT can be related to an overestimation of the cffDNA fraction because of limitation in the quantification method used, which is based on differentially methylated markers in fetal and maternal DNA 13. Furthermore, the reported accuracy of detecting sex chromosome abnormalities is lower than autosomal aberrations with overall sensitivity of 96.2%, false positive rate of 0.3%, and nonreportable rate of 5% 16. The small size of Y chromosome and fewer interrogated genomic regions on Y chromosome can also lead to a weaker signal-to-noise ratio 16. These limitations were exacerbated by the triploid karyotype composition since the sex chromosome representations are evaluated as ratios of normalized read counts as compared to the total autosomal read counts. The diagnosis of digynic triploidy can also be challenging because of difficulties in performing amniocentesis due to oligohydramnios.
In conclusion, digynic triploidy is difficult to identify by NIPT due to low cffDNA fraction. SNP microarray can be an effective tool in determining the mechanism and parental origin in triploidy. A low cffDNA fraction in the context of FGR and multiple congenital anomalies should alert medical professionals to the possibility of digynic triploidy. The development of highly sensitive NIPT technologies that combine massively parallel sequencing and SNP analysis has the potential to improve the detection of digynic triploidy.
Acknowledgments
We thank the parents for agreeing to participate in the study and for their consent to publish the photographs in this article.
Ethical Approval
IRB approved consent was obtained to publish the photographs and clinical data in the medical literature.
Conflict of Interest
The authors declare no conflict of interest.
References
- Forrester MB. Merz RD. Epidemiology of triploidy in a population-based birth defects registry, Hawaii, 1986–1999. Am. J. Med. Genet. A. 2003;119A:319–323. doi: 10.1002/ajmg.a.20152. [DOI] [PubMed] [Google Scholar]
- Zaragoza MV, Surti U, Redline RW, Millie E, Chakravarti A. Hassold TJ. Parental origin and phenotype of triploidy in spontaneous abortions: predominance of diandry and association with the partial hydatidiform mole. Am. J. Hum. Genet. 2000;66:1807–1820. doi: 10.1086/302951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherard J, Bean C, Bove B, DelDuca V, Jr, Esterly KL, Karcsh HJ, et al. Long survival in a 69, XXY triploid male. Am. J. Med. Genet. 1986;25:307–312. doi: 10.1002/ajmg.1320250216. [DOI] [PubMed] [Google Scholar]
- Niemann-Seyde SC, Rehder H. Zoll B. A case of full triploidy (69, XXX) of paternal origin with unusually long survival time. Clin. Genet. 1993;43:79–82. doi: 10.1111/j.1399-0004.1993.tb04432.x. [DOI] [PubMed] [Google Scholar]
- Hasegawa T, Harada N, Ikeda K, Ishii T, Hokuto I, Kasai K, et al. Digynic triploid infant surviving for 46 days. Am. J. Med. Genet. 1999;87:306–310. doi: 10.1002/(sici)1096-8628(19991203)87:4<306::aid-ajmg5>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Miny P, Koppers B, Dworniczak B, Bogdanova N, Holzgreve W, Tercanli S, et al. Parental origin of the extra haploid chromosome set in triploidies diagnosed prenatally. Am. J. Med. Genet. 1995;57:102–106. doi: 10.1002/ajmg.1320570121. [DOI] [PubMed] [Google Scholar]
- McFadden DE, Kwong LC, Yam IY. Langlois S. Parental origin of triploidy in human fetuses: evidence for genomic imprinting. Hum. Genet. 1993;92:465–469. doi: 10.1007/BF00216452. [DOI] [PubMed] [Google Scholar]
- Daniel A, Wu Z, Bennetts B, Slater H, Osborn R, Jackson J, et al. Karyotype, phenotype and parental origin in 19 cases of triploidy. Prenat. Diagn. 2001;21:1034–1048. doi: 10.1002/pd.164. [DOI] [PubMed] [Google Scholar]
- McFadden DE. Kalousek DK. Two different phenotypes of fetuses with chromosomal triploidy: correlation with parental origin of the extra haploid set. Am. J. Med. Genet. 1991;38:535–538. doi: 10.1002/ajmg.1320380407. [DOI] [PubMed] [Google Scholar]
- Haymon L, Simi E, Moyer K, Aufox S. Ouyang DW. Clinical implementation of noninvasive prenatal testing among maternal fetal medicine specialists. Prenat. Diagn. 2014;34:416–423. doi: 10.1002/pd.4301. [DOI] [PubMed] [Google Scholar]
- Agarwal A, Sayres LC, Cho MK, Cook-Deegan R. Chandrasekharan S. Commercial landscape of noninvasive prenatal testing in the United States. Prenat. Diagn. 2013;33:521–531. doi: 10.1002/pd.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaides KH, Syngelaki A, del Mar Gil M, Atanasova V. Markova D. Validation of targeted sequencing of single-nucleotide polymorphisms for non-invasive prenatal detection of aneuploidy of chromosomes 13, 18, 21, X, and Y. Prenat. Diagn. 2013;33:575–579. doi: 10.1002/pd.4103. [DOI] [PubMed] [Google Scholar]
- Palomaki GE, Deciu C, Kloza EM, Lambert-Messerlian GM, Haddow JE, Neveux LM, et al. DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: an international collaborative study. Genet. Med. 2012;14:296–305. doi: 10.1038/gim.2011.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaides KH, Syngelaki A, del Mar Gil M, Quezada MS. Zinevich Y. Prenatal detection of fetal triploidy from cell-free DNA testing in maternal blood. Fetal Diagn. Ther. 2014;35:212–217. doi: 10.1159/000355655. [DOI] [PubMed] [Google Scholar]
- Faas BH, de Ligt J, Janssen I, Eggink AJ, Wijnberger LD, van Vugt JM, et al. Noninvasive prenatal diagnosis of fetal aneuploidies using massively parallel sequencing-by-ligation and evidence that cell-free fetal DNA in the maternal plasma originates from cytotrophoblastic cells. Expert Opin. Biol. Ther. 2012;12(Suppl 1):S19–S26. doi: 10.1517/14712598.2012.670632. [DOI] [PubMed] [Google Scholar]
- Mazloom AR, Džakula Ž, Oeth P, Wang H, Jensen T, Tynan J, et al. Noninvasive prenatal detection of sex chromosomal aneuploidies by sequencing circulating cell-free DNA from maternal plasma. Prenat. Diagn. 2013;33:591–597. doi: 10.1002/pd.4127. [DOI] [PubMed] [Google Scholar]