

Abstract
The cause(s) of sporadic Alzheimer’s disease (sAD) are complex and currently poorly understood. They likely result from a combination of genetic, environmental, proteomic and lipidomic factors that crucially occur only in the aged brain. Age-related changes in calcium levels and dynamics have the potential to increase the production and accumulation of both amyloid-β peptide (Aβ) and τ pathologies in the AD brain, although these two pathologies themselves can induce calcium dyshomeostasis, particularly at synaptic membranes. This review discuses the evidence for a role for calcium dyshomeostasis in the initiation of pathology, as well as the evidence for these pathologies themselves disrupting normal calcium homeostasis, which lead to synaptic and neuronal dysfunction, synaptotoxicity and neuronal loss, underlying the dementia associated with the disease.
Keywords: calcium, Alzheimer’s disease, amyloid-beta peptide, tau, NMDA, synapse, autophagy, aging
-
Introduction
-
Familial and sporadic AD
APP processing and the amyloid cascade hypothesis
-
-
Calcium in the initiation of pathology
-
Effects of calcium on APP processing and Aβ deposition/aggregation
Genetic linkage in calcium related genes and AD
Apoε4 and calcium
Calcium and APP processing
Synaptic regulation of Aβ production
-
Normal age-related changes in calcium homeostasis and AD pathology
Age-related reductions in autophagy
-
-
Calcium in disease progression
Linking Aβ and τ
Calcium, calpains and τ pathology
-
Calcium as a disease effector
Synaptic calcium dyshomeostasis and AD – a focus on NMDA receptors
Aβ elevates cytosolic intracellular calcium levels in vivo
Aβ and mitochondrial calcium dyshomeostasis
Current therapeutics
Conclusions
Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder, which is characterized by progressive memory loss and behavioural changes that become more apparent and more severe as the disease progresses, and accounts for up to 70% of all dementias. Underlying these phenotypic changes are the appearance of several hallmark pathologies. The first is the accumulation of the amyloid-β peptide (Aβ) into extracellular plaques, whereas the second is the formation of neurofibrillary tangles (NFTs) composed of hyperphosphorylated τ protein within neurons [1]. The consequence of the development of these two pathologies is widespread synaptic and neuronal loss, such that a brain with advanced AD can weigh 33% less than a brain from an aged-matched control. AD research has focused on two main areas – the first is in understanding what causes the accumulation of these two pathologies in the aged brain, and the second is understanding how the presence of these pathologies affects the local brain environment to cause the widespread synaptic and neuronal loss, and cognitive decline. Through understanding, it is possible that (1) the accumulation of pathologies in the aged brain can be delayed, prevented, or even reversed, and that (2) the neurodegenerative properties of the pathologies can be dampened. Over the past 20 years aberrant calcium dysregulation has been consistently implicated in AD [2, 3], either in the initiation of the disease, the progression of AD-related pathologies, or in mediating the neuronal and synaptic loss that results from the presence of the pathologies.
Calcium is an integral signalling molecule whose local and global levels are tightly regulated at a temporal and spatial level as elevations in calcium activate a number of cellular signalling pathways including those crucial for learning and memory such as CamKII and CREB, as well as cell death pathways. In most cell types’, cytosolic calcium levels are kept low (∼100 nM) relative to the extracellular space and the intracellular stores by calcium-binding buffering proteins (e.g. calbindin), and via the extrusion of cytosolic calcium across the plasma membrane through calcium ATPase pumps and exchangers, and also due to sequestration into intracellular stores such as the endoplasmic reticulum (ER) and mitochondria. Calcium influx into the cytosol occurs across the plasma membrane via store-operated calcium channels, voltage-gated calcium channels or from internal stores. The ER is the largest intracellular store, maintaining a high calcium concentration (100–500 μM) via the unidirectional pumping of cytosolic calcium into the ER lumen by SERCA. Calcium release from the ER into the cytosol occurs via two types of calcium channels: IP3Rs and RyRs. IP3R-mediated release is regulated at the plasma membrane by ligand binding to specific G-protein coupled receptors that induce phospholipase C to cleave phosphatidylinositol-4, 5-bisphosphate into diacylglycerol and IP3, which then binds to the IP3R in the ER membrane. Calcium influx across the plasma membrane occurs due to depolarization, through VGCCs, and glutamate receptors such as the NMDA receptor and the mGluR.
Familial and sporadic AD
The overwhelming majority of AD cases occurs with no obvious environmental or genetic cause and is known as sporadic AD (sAD). It typically manifests at a late age of onset of more than 65 years. Around 5% of all AD cases, however, have a Mendelian pattern of inheritance and these are referred to as familial AD (fAD). The age of onset of dementia in fAD can vary dramatically, but typically fAD cases occur at much younger ages (from 30 years or more), and their dementia is usually more severe and progresses more rapidly. Clinically, and neuropathologically, both sAD and fAD are very similar, with all patients exhibiting widespread plaque and tangle pathologies with extensive neuronal loss.
APP processing and the amyloid cascade hypothesis
Studies have shown that cases of fAD are accounted for by mutations in only three genes – the amyloid precursor protein (APP) gene [4, 5], presenilin 1 (PS1) gene [6] and presenilin 2 (PS2) gene [7]. APP is a large membrane spanning protein that contains the Aβ peptide sequence [4]. Aβ is sequentially cleaved from APP. An enzyme identified as BACE first cleaves APP [8] to produce a 99 amino acid stub within the membrane known as C99. C99 is then cleaved by a complex known as γ-secretase to release Aβ[9, 10]. Presenilins comprise the catalytically active subunit of the γ-secretase complex (which also includes nicastrin, APH-1 and PEN-2 [11]), and thus are essential for release of the Aβ peptide. Although the BACE cleavage site of APP occurs at a specific sequence, the γ-secretase cleavage has loose sequence specificity and can cleave C99 between 38 and 43 amino acids from the N-terminal, to release Aβ peptides of variable lengths. The most common lengths are 1–40 and 1–42. Aβ1–40 is 10–20 times more abundant than Aβ1–42. Mutations in presenilins associated with fAD increase the production of Aβ1–42 at the expense of shorter Aβ peptides [12], whereas mutations in APP are associated with increased BACE cleavage [13], Aβ structural misfolding [14], or an increased prevalence of Aβ1–42 at the expense of shorter Aβ peptides [15].
APP cleavage by BACE and γ-secretase to generate Aβ is not the predominant APP processing pathway. More commonly APP is cleaved by an enzyme with α-secretase activity, at a site juxtaposed to the membrane, to release the large ectodomain known as sAPPα, as well as an 83 amino acid stub within the membrane known as C83. α-secretase cleavage occurs within the Aβ sequence to preclude Aβ production. Hence, stimulation of α-secretase cleavage leads to reduced Aβ production [16], and is a possible therapeutic target for the disease.
The production of Aβ alone is not sufficient to be toxic, and many studies have highlighted the aggregation state of Aβ as being crucial to its toxicity in vitro[17, 18], its ability to impair learning and memory [19] as well as long-term potentiation (LTP) [20, 21], in vivo[22]. Aβ1–42 aggregates more readily than Aβ1–40, and forms the majority of Aβ species present within AD plaques, explaining why mutations that affect the Aβ 40/42 ratio cause fAD without affecting total Aβ levels. More recently, soluble oligomers have been identified as the disease active state of Aβ, and include dimers and trimers [23, 24] to dodecamers [19] and beyond.
The sum of these findings led to the amyloid cascade hypothesis, which states that the accumulation of Aβ causes both sAD and fAD, and the downstream consequences of Aβ accumulation lead to τ pathology and ultimately synaptic and neuronal loss [25].
Although the causes of fAD are for the most part understood, the cause(s) of sAD are poorly understood. It should be noted that both sAD and fAD are age-related – despite fAD mutations in APP and PS1/2 being present from birth, dementia onset usually requires many decades to manifest suggesting that either pathology accumulation is very slow, or that the younger brain is better able to cope with the increased Aβ pathology that these mutations lead to, either via its degradation or by protecting against the downstream effects of Aβ accumulation. With sAD, the cause of pathology development and consequent dementia is not well understood. It likely results from combinations of genetic, environmental, proteaomic and lipidomic interactions resulting in the accumulation of AD-related pathologies. Understanding both the causes for the appearance of AD-related pathologies and the effects of these pathologies on the local brain environment is crucial in order to understand, prevent or treat the disease.
This review will discuss the possibilities that calcium plays in both disease initiation, progression, and as a disease effector molecule, as summarized in Fig. 1. Notably, the presenilins, in addition to being the catalytic subunit of the γ-secretase complex, are also key regulators of ER calcium homeostasis and have been reported to regulate SERCA [26], IP3 receptors [27] and ryanodine receptors [28] and also reported to form calcium conducting ER leak channels themselves [29]. fAD linked PS mutations have profound effects on calcium signalling [30], but the significance of this to fAD disease progression is unclear as mutations that affect Aβ accumulation/increased production are sufficient to cause fAD without these additional effects on calcium caused by PS mutations. Thus it seems that, at least with respect to fAD, fAD linked PS mutations alter Aβ metabolism to give rise to fAD, and additionally affect calcium homeostasis which may or may not affect disease progression. The effects and implications of presenilin regulation of both calcium and γ-secretase activity will not be discussed in this review as it was recently covered elsewhere [31].
Figure 1.
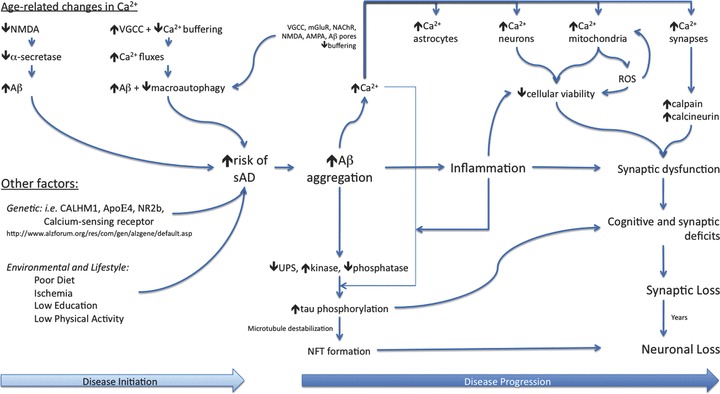
Involvement of calcium in AD progression: A number of factors can increase the likelihood of an individual developing sporadic AD. These could include changes in age-related calcium handling which result in increased calcium fluxes. Such increases are associated with increased production of Aβ, and also with reductions in macroautophagy – a clearance pathway for intracellular aggregates. Many genetic polymorphisms have been identified which can increase the risk of developing AD, including several calcium related genes. Notably, environmental and lifestyle appear to offer both great protection and also risk to developing AD. Accumulation of Aβ is considered one of the primary steps in developing AD. Aβ can aggregate into oligomers and fibrils, which can themselves alter cellular calcium homeostasis leading to increased calcium influx into neurons, astrocytes, but also into specific compartments such as mitochondria and synapses. Elevation of calcium can lead to synaptic dysfunction, and subsequent cognitive deficits. Over time these synaptic dysfunctions could lead to synaptic loss, and eventually the neuronal loss that plagues the late stage AD brain. Aβ aggregates can also lead to τ phosphorylation, via reductions in the ubiquitin-proteasome system, increases in τ kinases and decreases in τ phosphatases. Increases in calcium influx have been associated with activation of τ kinases and may also contribute to the development of NFTs.
Calcium in the initiation of pathology
Effects of calcium on APP processing and Aβ deposition/aggregation
APP processing lies at the heart of AD, and a majority of therapeutics are designed to alter APP processing to prevent the production of Aβ, to prevent the disease and to halt further progression [32]. Ultimately Aβ accumulation is the tenet of the disease, and accumulation occurs due to an increase in production during aging, a decrease in degradation or a change in predisposition to aggregate. For example, changes in APP metabolism can produce more aggregate prone Aβ1–42 at the expense of shorter Aβ peptides.
Genetic linkage in calcium related genes and AD
Calcium has been implicated both in the development of sAD through genetic susceptibility, as well as altered Aβ production and metabolism. A number of genes are associated with increased risk of developing sAD, most notably the apoɛ4 allele [33]. Recently, a polymorphism in a gene encoding a novel calcium conducting channel was found to have linkage to sAD [34], although this same polymorphism was shown not to have linkage in three separate studies of Caucasian populations [35–37]. This channel is called calcium homeostasis modulator 1 (CALHM1) and is a conserved three-transmembrane domain containing glycoprotein. It localizes to both the ER and the plasma membrane. Overexpression of this channel induces a cytosolic calcium influx pathway, which is unaffected by conventional calcium channel blockers, but prevented by the removal of extracellular calcium and non-specific ion channel pore blockers. The induction of this particular calcium influx route into the cytosol results in an increase in sAPPα production, with a concomitant reduction in Aβ, suggesting an effect on one of the α-secretase enzymes. Notably, knockdown of this channel, or the presence of the identified polymorphism for sAD (rs2986017 encoding P86L substitution) decreases calcium permeability and increases Aβ production. These data provide strong evidence that calcium signalling and influx can contribute to the initiation of AD pathology in the aged brain, and that specific calcium pathways can affect APP metabolism. Although further studies are required to replicate the linkage of the CALHM1 rs2986017 polymorphism to sAD, this important finding shows how changes in calcium influx pathways can alter APP processing and Aβ production.
Since a polymorphism in the CALHM1 gene has linkage to increased risk of sAD, it begs the question whether other known risk factors could also directly involve altered calcium homeostasis. One study has shown an association between the NMDA receptor NR2B subunit gene promoter polymorphism and development of sAD in a North China population [38]. The NMDA receptor is an important focal point in AD, both in modulating the appearance of Aβ and τ pathology, and also in mediating Aβ-induced synaptic and behavioural deficits in the disease process. Offering a direct correlation between calcium and cognitive decline, serum calcium levels correlate well with cognitive decline during aging, with elevated serum calcium levels being associated with worse cognitive function [39], and also a faster rate of decline. A regulator of serum calcium levels, the calcium-sensing receptor – is associated with development of AD, particularly with a dinucleotide repeat polymorphism within the promoter sequence [40].
Apoɛ4 and calcium
Of note, the presence of the Apoɛ4 allele is a strong risk factor for the development of sAD [33]– with ∼50% of sAD patients having at least one copy of the Apoɛ4 allele. Despite the significant impact of this lipid transporter on AD, we do not fully understand its role in disease progression. The Apoɛ genotype modulates elevated serum calcium and cognitive decline [41]. High serum calcium is associated with worse cognitive function especially in Apoɛ4 carriers, but this interaction is lost in Apoɛ2 carriers [41]. Furthermore, the Apoɛ4 allele is associated with increased calcium rises in neuronal cultures, which can mediate cell death [42], and also in SHSY5Y cultures where increased calcium influxes increase GSK3β activity leading to reduced cell viability [43]. Apoɛ4 appears to affect calcium influx into neurons via plasma membrane calcium channels [42], which, as discussed below, can alter APP processing leading to increased production of Aβ.
Calcium and APP processing
In addition to CALHM1, a number of other studies have reported altered calcium homeostasis affecting the α-secretase dependent processing of APP. Increasing calcium influx into the cytosol with the ionophore ionomycin leads to increased sAPPα production [44], whereas calcium influx through the NMDA receptor increases sAPPα production and decreases Aβ generation [45]. However, despite calcium influx through certain channels stimulating α-secretase processing of APP, the vast majority of studies have shown that increased calcium influx, and elevated cytosolic calcium levels, leads to a net increase in Aβ production [46–48].
More recently, data have highlighted the ER calcium stores as playing an important role in the regulation of Aβ production. We have shown that inhibition of SERCA, either with thapsigargin or by siRNA knockdown, leads to a robust reduction in Aβ generation whereas overexpression of SERCA increases Aβ generation [26]. In agreement with ER calcium stores playing a regulatory role in Aβ generation, knockdown of the IP3 receptor also reduces Aβ generation [27]. In concert, calcium release from the ER via the ryanodine receptor increases Aβ production [49], whereas capacitative calcium entry (CCE), a calcium influx pathway across the plasma membrane, affects APP metabolism with inhibition of CCE leading to increased Aβ 1–42 production [50]. Taken together these studies point to ER calcium as a key regulator of Aβ generation, as well as calcium influx across the plasma membrane through channels such as CALHM1 and CCE channels, and demonstrates how age- or environment-dependent changes in neuronal calcium homeostasis could alter APP processing to drive the increased production of Aβ, or altered metabolism to preferentially generate Aβ1–42.
Synaptic regulation of Aβ production
Adding a new dimension to these in vitro experiments, in vivo studies have allowed a new insight into how the workings of neuronal networks can regulate Aβ production, in both mouse models of AD and also in the human brain [51]. Synaptic activity is dependent upon calcium, both at the pre- and post-synaptic terminals, for exocytosis of neurotransmitters across the synaptic cleft, as well as initiation of action potentials at the post-synaptic membrane. In slice cultures from APP overexpressing mice, increased synaptic activity led to increased production of Aβ, whereas inhibition of synaptic activity suppressed Aβ generation [52]. Synaptic activity affects APP processing at either the α- or β-secretase levels, as levels of C99 were modulated. Using microdialysis, and subsequent analysis of interstitial Aβ, it was shown that synaptic activity also regulated Aβ production in vivo[53]. Decreased synaptic activity, elicited by tetrodotoxin (TTX) administration, reduced Aβ and partially corresponded to increased C83 levels, suggesting an increase in α-secretase or a decrease in BACE processing of APP with decreased synaptic activity. TTX inhibits action potentials by blocking Na+ channels, to prevent calcium influx at the pre-synaptic terminal and subsequent exocytosis of neurotransmitters and concomitant endocytosis of synaptic vesicles. Hence, TTX application is likely associated with a decrease in synaptic calcium and a decrease in Aβ generation/secretion. Furthermore, Aβ production and secretion are dependent upon synaptic vesicle membrane recycling; increasing synaptic activity leads to synaptic APP being recycled into endosomes where Aβ generation occurs which is then secreted from the neuron [54]. Though synaptic activity modulates both endocytosis of APP into Aβ-generating endosomes and also exocytosis of neurotransmitter containing vesicles, it is unclear if Aβ is secreted from synapses alongside neurotransmission or in a more passive manner. Furthermore, synaptic activity increases Aβ oligomer formation in a synaptic cleft zinc dependent manner [55], suggesting that synaptic activity can not only increase Aβ generation/secretion but it can also drive it to aggregate into toxic oligomeric species. Of interest, it seems that secreted Aβ at synapses is able to regulate synaptic function through depression of excitatory transmission by an NMDA receptor-mediated pathway [52], suggesting that Aβ is purposely secreted from the pre-synaptic terminal in order to play a role in synaptic plasticity. How this role differs between healthy controls and AD patients is unknown, but may suggest a calcium dependent physiological role for Aβ at the synapse.
Normal age-related changes in calcium homeostasis and AD pathology
Normal brain aging involves subtle changes in cognition and brain functioning, attributed in part to age-related changes in how neurons shuttle calcium from their various internal stores, and altered calcium influx across the plasma membrane. These changes are likely due, partially, to an increased presence of reactive oxygen species, as well as transcriptional and post-translational changes in the calcium channels and receptors themselves. Age-related changes in neuronal calcium handling have been elegantly reviewed elsewhere [3, 56, 57], but the consensus is that aged neurons tend to have increased resting cytosolic calcium levels [58–60] as well as increased cytosolic calcium fluxes due to increased calcium entry through voltage gated calcium channels [61, 62]. This increased influx, in turn, stimulates calcium release from the ER via the ryanodine receptor [63, 64] resulting in further elevated cytosolic calcium fluxes. Voltage gated calcium channels, such as the L-type, are known to be modulated by reactive oxygen species [65], as well as by Aβ[66], both of which are increased through the aging process. Furthermore, aged neurons appear to have reduced calcium buffering capabilities due to reductions in the levels of cytosolic calcium binding proteins such as calbindin D28K [67] and calreticulin [68], reduced mitochondrial buffering capabilities [69], and a decrease in SERCA function [70], at least in peripheral nerves, resulting in prolonged calcium fluxes. Interestingly, IP3 evoked calcium fluxes appear to decrease in aged neurons [71], perhaps due to a diminished chemical gradient due to elevated cytosolic calcium. Hence, age related increases in cytosolic calcium levels could conceivably alter APP processing as discussed above, and shift APP metabolism to produce increased amounts of Aβ.
Along these lines, aging is associated with a decrease in calcium currents through NMDA receptors in neurons [56]. Since current through the NMDA receptor is associated with increased α-secretase processing of APP [45], it suggests that age-related reductions in NMDA receptor calcium would no longer stimulate the α-secretase enzymes to the same degree, resulting in a shift to Aβ generation. Due to the presence of age-related changes in calcium, coupled with the observations that calcium regulates synaptic plasticity and increases in calcium leads to neuronal cell death, the calcium homeostasis disruption theory of AD was first postulated over 20 years ago [2], and remains a viable theory for the appearance of AD in the aging brain.
Age-related reductions in autophagy
Alterations in neuronal calcium may also affect AD-related pathology through indirect means. Of interest, AD and many other neurodegenerative diseases are age dependent even when mutations in key genes force the aggregation of proteins into toxic conformations. What is the protection that the youthful brain provides against these proteinaceous aggregates seen in neurodegenerative diseases that the aged brain does not afford? Protein turnover is crucial both in neurodegenerative diseases as well as normal and successful aging. Cytosolic proteins are degraded by either the ubiquitin proteasome system, or by the lysosome system through macroautophagy or chaperone mediated autophagy. The UPS system is impaired in AD patients [72], and proteasome inhibition increases levels of both τ and Aβin vivo[73]. Notably, autophagy is crucial for the removal of cytosolic aggregates especially those implicated in neurodegenerative diseases [74]. Age-related reductions in autophagy are implicated in allowing the aged brain to develop neurodegenerative diseases, whereas stimulators of macroautophagy have proven successful in mouse models of Huntington’s disease, by removing cytosolic aggregates of expanded huntingtin protein from neurons [75]. Cytosolic calcium levels are potent regulators of macroautophagy. Inhibition or depletion of the IP3 receptor, and L-type voltage gated calcium channel blockers [76] are associated with increased autophagy. Age-related increases in L-type calcium currents and overall increases in cytosolic calcium levels may negatively influence macroautophagy, allowing the buildup of cytosolic proteins in aged neurons such as hyperphosphorylated τ, α-synuclein and huntingtin.
Calcium in disease progression
Linking Aβ and τ
Although no mutations or polymorphisms in the τ gene (MAPT) are associated with sAD or fAD, τ protein is the component of the second hallmark pathology of AD – the NFTs [77]. NFTs are composed of aggregated hyperphosphorylated τ and are found in the soma and dendrites of neurons. τ is a microtubule-associated protein that acts to stabilize microtubules enabling efficient axonal transport. τ can be phosphorylated on a number of threonine and serine residues by an expanding list of kinases, most notably cyclin dependent kinase 5 (Cdk5) [78] and glycogen synthase kinase 3β[79]. Abnormal hyperphosphorylation occurs in AD and leads to the disruption of axonal transport and the subsequent relocalization and aggregation of the modified τ proteins into structures known as paired helical filaments, which comprise the NFTs. Of note, NFT formation is sufficient to cause extensive neurodegeneration on its own. Mutations in the τ gene are associated with a group of dementias known as tauopathies, including frontotemporal dementia with parkinsonism linked to chromosome 17 [80]. Due to the genetic data that gives rise to fAD, τ hyperphosphorylation is generally considered to be a downstream event of Aβ accumulation, but is thought to be a crucial effector molecule that causes synaptic and neuronal loss [81, 82]. This hypothesis is supported by data showing that Aβ accumulating transgenic mice develop cognitive impairments, but when the τ gene is knocked out the Aβ pathology remains stable but the mice no longer develop cognitive impairments [83]. We have also demonstrated recently that treatment of 3×Tg-AD mice with the sirtuin inhibitor nicotinamide prevents cognitive deficits by removing τ phosphorylated at threonine 231, with no effects on Aβ[84]. Any factor that impacts τ phosphorylation has the potential to play an important role in disease progression in AD. Given that many kinase and phosphatase pathways are activated by calcium, there has been a great deal of interest in studying the pathways that lead to τ hyperphosphorylation through both Aβ dependent and independent pathways. In the AD brain, post-mortem studies have highlighted changes in calcium channels and pumps that correlate with severity of τ pathology, including a reduction in ryanodine receptor binding [85] and IP3 receptor binding [86], in cases with high pathology, suggesting that either NFT formation down-regulates these calcium pumps which may contribute to neurodegeneration, or that a down-regulation of these pumps (perhaps by Aβ pathologies) encourages NFT formation.
Calcium, calpains and τ pathology
Aβ induced increases in calcium lead to τ hyperphosphorylation via both GSK3β and Cdk5 [87]. Cdk5 activity is regulated through its interactions with cyclin-related activator molecules such as p25, p35, p29 and p39. Aβ increases the activity of Cdk5 by promoting the Cdk5-p25 and p35 complexes [88–90]. p39/p35 is cleaved to p29/25 by calpains [91], releasing it from a membrane tether where it forms unregulated functional complexes with Cdk5 leading to τ hyperphosphorylation [92]. Activation of calpain is calcium dependent, and aberrant activation of calpains 1 and 2 has been linked to AD [93, 94]. Calpain 2 has increased activation in NFT bearing neurons and neurites [95], whereas a calpain substrate, calcineurin, is truncated in the AD brain [96]. Hence, Aβ-mediated and age related increases in cytosolic calcium can lead to the activation of calpains, which then lead to hyperphosphorylation of τvia Cdk5 [97]. Of note, a recent study highlighted how calpain activation in transgenic mouse models of AD leads to synaptic dysfunction and behavioural impairments [98]. Calpain 1 is expressed in dendritic spines [99], which have abnormally elevated basal calcium levels in close proximity to Aβ plaques [100] which would have the potential to activate these synaptic calpains. The application of a calpain inhibitor restores synaptic deficits and spatial memory to wild-type levels, demonstrating the role activated calpain plays in mediating Aβ-induced synaptic dysfunction and cognitive decline [98]. Calpain inhibitors also prevent Aβ-mediated cleavage of τ protein into toxic fragments, in particular a 17 kD fragment [101], suggesting that Aβ-induced calcium influx leads to calpain dependent neurotoxicity via cleavage of τ protein, and others including dynamin 1 [101, 102]. These effects could be prevented by blocking calcium influx through the NMDA receptor [102] implicating Aβ, NMDA receptor mediated calcium influx, calpains, τ and neurotoxicity. Along these lines, overexpression of τ itself can lead to increased NMDA receptor activation, leading to increased calcium influx and subsequent activation of calpain, cleavage of τ to produce the aforementioned toxic 17 kD fragment, and subsequent neuronal loss [103]. This suggests that Aβ-induced increases in synaptic calcium could lead to τ pathologies via calpain activation, but that τ pathologies themselves could then influence further increases in synaptic calcium levels through the NMDA receptor.
Calcium as a disease effector
The discovery of a polymorphism in the CALHM1 gene, linking calcium influx to Aβ generation and an increased risk of developing AD provided concrete evidence for a role for calcium in the initiation of sAD [34], but so far three further linkage studies have failed to replicate the association to sAD [35–37]. Despite this, the evidence that age-related changes in neuronal calcium homeostasis could affect APP processing and Aβ metabolism is compelling and offers potential therapeutic targets to try and reverse these changes in calcium, and hence APP processing. However, the appearance of Aβ in old age is only the first step in neurodegeneration and dementia, and there is overwhelming evidence that (1) Aβ induces disturbances in neuronal and synaptic calcium homeostasis and (2) that such changes in neuronal and synaptic calcium would lead to synaptic and neuronal toxicity as sustained increases in cytosolic calcium render neurons susceptible to additional metabolic insults, as well as being able to induce cell death pathways itself. Additionally, given the evidence that altered neuronal calcium fluxes and synaptic activity can alter Aβ generation and secretion, Aβ-induced calcium fluxes could further augment Aβ generation in a positive feedback pathway, accelerating disease progression.
The effects of Aβ on calcium can be categorized into several overlapping groups. The first is a large body of literature demonstrating how Aβ can influence existing calcium conducting channels directly, including L-type voltage-gated calcium channels [65, 66, 104–107], P/Q type voltage-gated calcium channels [108], glutamate receptors including the NMDA receptor [109–112], the AMPA receptor [113] and the mGluR [114], and also nicotinic receptors [15–118]. The second group consists of numerous reports that Aβ can itself form unregulated calcium conducting pores within lipid membranes [119, 120]. Thirdly, there are reports that the presence of Aβ affects calcium buffering, either by affecting intraneuronal calcium sinks/stores or by affecting steady state levels of cytosolic calcium buffering proteins, such as calbindin [121]. Finally, there are also reports that Aβ can affect synaptic transmission, which influences calcium influx at both the pre- and post-synaptic membranes, and is essential for neuronal functioning and cognitive function. The effects on synaptic transmission are likely a consequence of Aβ inducing calcium dyshomeostasis, as well as a cause of further calcium dyshomeostasis.
Synaptic calcium dyshomeostasis and AD – a focus on NMDA receptors
Synaptic loss is the best correlate to dementia in the AD brain, although Aβ and τ loads associate poorly with dementia. In addition to this, work with transgenic mouse models, which overexpress Aβ, has shown that Aβ induces memory and LTP impairments without overt neuronal loss, and can do so in an acute fashion. As such the site of neuronal plasticity, the synapse, is highlighted as a crucial target for the Aβ peptide as disturbances here would interfere with both LTP and learning and memory, but would only lead to neuronal loss over extended periods of time, possibly beyond the lifespan of rodents. Neuronal synapses contain dense concentrations of calcium conducting channels, as well as cytosolic calcium buffering proteins, and a variety of calcium dependent signalling pathways also implicated in LTP and learning and memory such as calcineurin, CaMKII and CREB, all of which have been reported to be changed in AD brains and APP overexpressing transgenic mice. Hence, in order to satisfy the observation that Aβ acutely impairs LTP and learning and memory, but chronically leads to widespread synaptic and neuronal loss, the effects of Aβ on calcium could initially be at the synaptic level, but with the ability to lead to widespread neuronal loss over time.
One of the most significant advances in recent years in AD research has been the discovery that it is the aggregation state of Aβ that is crucial to its pathophysioloigcal effects, particularly when describing calcium dependent events. Soluble Aβ oligomers appear to mediate calcium dysfunction, as well as inhibit LTP [122], a form of synaptic plasticity, which relies upon calcium fluxes and is thought to underlie learning and memory [123]. The NMDA receptor appears to be a focal point for Aβ oligomer induced calcium dysfunction and LTP inhibition [124], as well as NMDA receptor mediated synaptic loss [125], and Aβ oligomers associate and co-localize with dendritic arbours [126, 127]– post-synaptic membranes enriched in NMDA receptors. Synaptic activity can increase generation/secretion of Aβ at synapses, but can also stimulate the aggregation of monomeric Aβ into oligomeric species. This aggregation of synaptic Aβ appears to be modulated by synaptic zinc, a known facilitator of Aβ aggregation, and by the NMDA receptor, as inhibition of NMDA calcium current influx with antagonists or with zinc chelators prevents synaptic Aβ oligomerization [55]. Intriguingly, synaptic zinc is a known competitive antagonist at the NMDA receptor [128], and is released into the synaptic cleft alongside glutamate, serving to depress synaptic transmission, yet here zinc promotes Aβ oligomerization suggesting that the inhibitory effect of endogenous synaptic zinc on the NMDA receptor is not sufficient to overcome Aβ oligomerization. What is the consequence of Aβ, or Aβ oligomers, associating with NMDA receptors at synapses? It appears that Aβ may have a physiological role in regulating synaptic plasticity via the NMDA receptor, as Aβ secreted from synapses causes NMDA receptor dependent synaptic depression [52], which could arise due to increased endocytosis of the NMDA receptor in the presence of Aβ[109, 111]. This function may go awry in the AD brain where Aβ oligomer formation is promoted, as Aβ oligomers interact with NMDA receptors to promote calcium influx into post-synaptic membranes [129], or this NMDA receptor mediated synaptic depression may be enhanced as it has been reported that Aβ oligomers decrease calcium currents through the NMDA receptor resulting in synaptic loss [125].
In addition to Aβ oligomers interacting with post-synaptic NMDA receptors, they have also been shown to affect pre-synaptic calcium conducting channels including the P/Q type voltage gated calcium channels [108] at very low nanomolar concentrations. Calcium currents through the P/Q type channels were found to be suppressed in the presence of aggregated, but not monomeric, Aβ resulting in decreased synaptic transmission, as exocytosis at the pre-synaptic membrane is dependent upon calcium influx. The authors suggested that this reduced synaptic activity could lead to synaptic loss, as long-term withdrawal of synaptic activity is associated with synaptic retraction. This effect would be enhanced by a concomitant decrease in NMDA receptor calcium influxes in the post-synaptic membrane, in the presence of Aβ oligomers.
Thus it seems that there are at least two competing theories for the mode of action of Aβ oligomers at the synapse. It is well defined that Aβ oligomers inhibit LTP, and this appears to be through an NMDA receptor dependent mechanism [124]. One theory is that LTP is impaired due to an inhibition of calcium current at the post-synaptic membrane, due to inhibition of the NMDA receptor, and perhaps a reduction in pre-synaptic activity. A lack of synaptic activity would lead to synaptic retraction over time, in accordance with reduced synapses and synaptic proteins in AD and APP transgenic mice brains. The other would suggest that Aβ oligomers induce increased calcium influx at the synapse, leading to excitotoxicty, and synaptic dysfunction, and eventual synaptic and neuronal loss. This is in accordance with the evidence that dendritic spine calcium is elevated in close proximity to Aβ plaques
[100], and the evidence of activated calcium dependent proteases and calcium dependent signalling pathways at synaptic sites. Both would impair LTP, and lead to synaptic loss, but the discrepancies could be due to spatial and temporal reasons – either different populations of synapses are affected differentially, or the acute effects of Aβ oligomers are different from chronic exposure.
Aβ elevates cytosolic intracellular calcium levels in vivo
Supporting a role that chronic Aβ oligomer exposure leads to increased calcium influx, live imaging of neurites in APP overexpressing mice, revealed elevated dendritic spine basal calcium levels correlating well with close proximity to Aβ plaques [100], indicating once again that the aggregation state of Aβ is crucial but also that the net result of Aβ interactions with synaptic channels/proteins, is elevated calcium. Although the source of increased basal cytosolic calcium is not yet elucidated, it is likely a key event in synaptic dysfunction, as spines with elevated calcium displayed altered morphology tending towards a beaded or dysmorphic appearance. These morphological changes could be rescued by application of calcineurin inhibitors; calcineurin is a calcium dependent phosphatase important for learning and memory as well as LTP, indicating that increased resting calcium levels were atypically activating this phosphatase, whereas calcineurin inhibitors partially restore learning and memory deficits in APP transgenic mice [130]. In addition to calcineurin, other spine located calcium dependent enzymes have been implicated in synaptic dysfunction due to the presence of Aβ– in particular calpain inhibitors, as discussed, also prevent Aβ induced LTP and learning and memory deficits caused by the presence of the Aβ peptide [98].
In addition to neurons, astrocytic networks display elevated cytosolic calcium levels, and demonstrate an increased frequency of calcium transients both intracellulalry and intercellularly, in APP overexpressing mice [131]. The mechanism of action is distinct from that which causes increased dendritic spine elevations in calcium as astrocytic calcium is unaffected by proximity to Aβ plaques. This suggests that Aβ aggregation state is less important to the effects on astrocytes, and is likely an easily diffusible widespread Aβ species, such as a monomer or dimer. Furthermore, astrocytic calcium dyshomeostasis is not derived, nor related, to neuronal activity, as blocking action potentials does not ameliorate elevated calcium levels or transients in astrocytes. These observations lead to speculation about the implications of widespread astrocytic calcium dyshomeostasis on AD progression. For example, it shows that astrocytes could be severely impacted as a result of pathology in AD, yet this does not seem to lead to astrocyte loss but could be indirectly contributing to the synaptic and neuronal loss seen in the disease, as astrocytes play a large supporting role for neuronal function and survival. As APP is also expressed in astrocytes could these Aβ induced changes in calcium lead to changes in APP processing in astrocytes in human beings – a positive feedback on Aβ production? Further experiments will be needed to answer these questions, as well the source of elevated dendritic spine calcium in close proximity to Aβ plaques.
The relationship between neuronal calcium dyshomeostasis and proximity to Aβ plaques was confirmed by looking at cortical neuronal calcium transients in APP transgenic mice [132]. 21% of cortical neurons demonstrate hyperactivity, with increased frequency of spontaneous calcium transients, but only within 60 μM proximity to an Aβ plaque. A similar distance was reported for elevated basal dendritic spine calcium levels [100]. The source of calcium hyperactivity is dependent upon neuronal action potentials, as TTX prevents all spontaneous calcium transients. Intriguingly, calcium transient amplitudes do not differ between APP and wild-type mice, suggesting only the frequency of transients is affected. Spontaneous transients are presumed to be a consequence of reduced inhibition as GABA, the main inhibitory neurotransmitter, as activators decreased the frequency of spontaneous transients, whereas GABA inhibitors in wild-type mice increased spontaneous calcium transients. The consequences of these spontaneous calcium transients could be the increase in dendritic spine calcium reported in APP mice which lead to altered synaptic morphologies, but also, due to the ordered nature of the transients, could also trigger epileptic like seizures. Such seizures have been reported in other APP mouse models [133] in which the removal of τ was protective against aberrant neuronal activity, as well as in AD patients themselves [134, 135].
Aβ and mitochondrial calcium dyshomeostasis
Mitochondrial dyshomeostasis has been observed in AD brains, as well as in transgenic mouse models of AD, and cell culture models [136]. Mitochondria hold the key to cellular life or death, firstly through the production of ATP, but also through the release of pro-apoptotic signals such as cytochrome C. Mitochondria buffer calcium and are involved in neuronal calcium homeostasis. Aβ, and Aβ oligomers, associate with mitochondrial membranes and have recently been shown to interact with cyclophilin D – part of the mitochondrial permeability pore [69]. Notably, this interaction makes mitochondria more susceptible to swelling in response to calcium, and increases the likelihood of pore permeability transition. Pore permeability transition is associated with a collapse of mitochondrial membrane potential and release of pro-apoptotic proteins into the cell’s cytoplasm, leading to neuronal loss. Furthermore, genetic ablation of cyclophilin D protected transgenic mice against Aβ-induced cognitive deficits, implicating Aβ and mitochondria interactions in neuronal dysfunction and highlighting the importance of normal mitochondrial function to proper synaptic transmission.
Current therapeutics
Given the apparent influence of calcium disturbances in the initiation, progression or effects of the disease, can calcium modulators be used as effective therapies for the disease? Currently two classes of drug are used for the treatment of AD – the first are acetylcholinesterase inhibitors, and the second consists of a single drug, memantine, which is a partial NMDA receptor antagonist. Hence, targeting NMDA receptor mediated calcium influx in AD patients is beneficial for cognition [137], as well as in mouse models of AD [138], cell culture models of Aβ toxicity via attenuation of τ phosphorylation [139], and is also associated with a reduction in Aβ plaque pathology [140].
Other calcium influx pathway blockers have also been evaluated in AD patients, although without sustained efficacy, including the L-type voltage gated calcium channel blocker nimodipine [141], and other calcium channel blockers associated with the cardiovascular system [142]. Dimebon, an antihistamine, has been shown to improve cognition in mild to moderate AD patients [143], and has reported activity as an NMDA receptor antagonist, as well being able to block voltage gated calcium channel currents, although at concentrations that may be too high to be responsible for the improvements in cognition [144]. Thus, targeting the NMDA receptor appears to be beneficial for the disease, and future drugs may refine this activity further. In the meantime, although calcium channel blockers have not proven effect in patients with the disease, targeting calcium influx may be able to delay or prevent the onset of AD if given at the right time, or targeting specific channels may be able to prevent the synaptotoxicity of Aβ.
Conclusions
Calcium may play a vital role in any number of aspects of AD – from modulating APP processing towards the amyloidogenic processing pathway, to underlying memory and neuronal loss. In vivo evidence has shown that Aβ increases dendritic spine calcium levels, impairs LTP and induces learning and memory deficits, highlighting the synapse as a prime Aβ target, although age related changes in calcium homeostasis may allow the buildup of pathologies associated with AD, through both direct and indirect mechanisms. Synaptic transmission is highly dependent upon tightly regulated calcium at both the pre- and post-synaptic membranes and it is at these membranes all the ion channels Aβ has been reported to disrupt are clustered, likely leading to synaptic dysfunction and more, while targeting NMDA receptors with memantine is beneficial in AD patients, as well as numerous models of the disease, placing calcium dyshomeostasis firmly at the centre of the disease.
References
- 1.Alzheimer A, Stelzmann RA, Schnitzlein HN, et al. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8:429–31. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 2.Khachaturian ZS. Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol Aging. 1987;8:345–6. doi: 10.1016/0197-4580(87)90073-x. [DOI] [PubMed] [Google Scholar]
- 3.Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging Cell. 2007;6:307–17. doi: 10.1111/j.1474-9726.2007.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–6. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 5.Levy E, Carman MD, Fernandez-Madrid IJ, et al. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–6. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 6.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 7.Sherrington R, Froelich S, Sorbi S, et al. Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet. 1996;5:985–8. doi: 10.1093/hmg/5.7.985. [DOI] [PubMed] [Google Scholar]
- 8.Vassar R, Bennett BD, Babu-Khan S, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–41. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 9.Wolfe MS, Xia W, Ostaszewski BL, et al. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–7. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 10.Li YM, Xu M, Lai MT, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–94. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 11.Francis R, McGrath G, Zhang J, et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell. 2002;3:85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 12.Xia W, Zhang J, Kholodenko D, et al. Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977–82. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]
- 13.Citron M, Oltersdorf T, Haass C, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360:672–4. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 14.Nilsberth C, Westlind-Danielsson A, Eckman CB, et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4:887–93. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 15.Pype S, Moechars D, Dillen L, et al. Characterization of amyloid beta peptides from brain extracts of transgenic mice overexpressing the London mutant of human amyloid precursor protein. J Neurochem. 2003;84:602–9. doi: 10.1046/j.1471-4159.2003.01556.x. [DOI] [PubMed] [Google Scholar]
- 16.Fisher A, Michaelson DM, Brandeis R, et al. M1 muscarinic agonists as potential disease-modifying agents in Alzheimer’s disease. Rationale and perspectives. Ann N Y Acad Sci. 2000;920:315–20. doi: 10.1111/j.1749-6632.2000.tb06941.x. [DOI] [PubMed] [Google Scholar]
- 17.Pike CJ, Burdick D, Walencewicz AJ, et al. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–87. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pike CJ, Walencewicz AJ, Glabe CG, et al. In vitro aging of beta-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–4. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 19.Lesne S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 20.Cleary JP, Walsh DM, Hofmeister JJ, et al. Natural oligomers of the amyloid beta protein specifically disrupt cognitive function. Nature Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 21.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 22.Walsh DM, Klyubin I, Shankar GM, et al. The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33:1087–90. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- 23.Klyubin I, Betts V, Welzel AT, et al. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–7. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Townsend M, Shankar GM, Mehta T, et al. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–92. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 26.Green KN, Demuro A, Akbari Y, et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107–16. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung KH, Shineman D, Muller M, et al. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP(3) receptor channel gating. Neuron. 2008;58:871–83. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rybalchenko V, Hwang SY, Rybalchenko N, et al. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol. 2008;40:84–97. doi: 10.1016/j.biocel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 29.Tu H, Nelson O, Bezprozvanny A, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–93. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nature Rev. 2002;3:862–72. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 31.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454–63. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfe MS. Selective amyloid-beta lowering agents. BMC Neurosci. 2008;9:S2–4. doi: 10.1186/1471-2202-9-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roses AD. On the discovery of the genetic association of Apolipoprotein E genotypes and common late-onset Alzheimer disease. J Alzheimers Dis. 2006;9:361–6. doi: 10.3233/jad-2006-9s340. [DOI] [PubMed] [Google Scholar]
- 34.Dreses-Werringloer U, Lambert JC, Vingtdeux V, et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell. 2008;133:1149–61. doi: 10.1016/j.cell.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minster RL, Demirci FY, DeKosky ST, et al. No association between CALHM1 variation and risk of Alzheimer disease. Hum Mutat. 2009;30:E566–9. doi: 10.1002/humu.20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertram L, Schjeide BM, Hooli B, et al. No association between CALHM1 and Alzheimer’s disease risk. Cell. 2008;135:993–4. doi: 10.1016/j.cell.2008.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sleegers K, Brouwers N, Bettens K, et al. No association between CALHM1 and risk for Alzheimer dementia in a Belgian population. Hum Mutat. 2009;30:E570–4. doi: 10.1002/humu.20990. [DOI] [PubMed] [Google Scholar]
- 38.Jiang H, Jia J. Association between NR2B subunit gene (GRIN2B) promoter polymorphisms and sporadic Alzheimer’s disease in the North Chinese population. Neurosci Lett. 2009;450:356–60. doi: 10.1016/j.neulet.2008.10.075. [DOI] [PubMed] [Google Scholar]
- 39.Schram MT, Trompet S, Kamper AM, et al. Serum calcium and cognitive function in old age. J Am Geriatr Soc. 2007;55:1786–92. doi: 10.1111/j.1532-5415.2007.01418.x. [DOI] [PubMed] [Google Scholar]
- 40.Conley YP, Mukherjee A, Kammerer C, et al. Evidence supporting a role for the calcium-sensing receptor in Alzheimer disease. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:703–9. doi: 10.1002/ajmg.b.30896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Vliet P, Oleksik AM, Mooijaart SP, et al. APOE genotype modulates the effect of serum calcium levels on cognitive function in old age. Neurology. 2009;72:821–8. doi: 10.1212/01.wnl.0000343852.10018.24. [DOI] [PubMed] [Google Scholar]
- 42.Veinbergs I, Everson A, Sagara Y, et al. Neurotoxic effects of apolipoprotein E4 are mediated via dysregulation of calcium homeostasis. J Neurosci Res. 2002;67:379–87. doi: 10.1002/jnr.10138. [DOI] [PubMed] [Google Scholar]
- 43.Cedazo-Minguez A, Popescu BO, Blanco-Millan JM, et al. Apolipoprotein E and beta-amyloid (1–42) regulation of glycogen synthase kinase-3beta. J Neurochem. 2003;87:1152–64. doi: 10.1046/j.1471-4159.2003.02088.x. [DOI] [PubMed] [Google Scholar]
- 44.Petryniak MA, Wurtman RJ, Slack BE. Elevated intracellular calcium concentration increases secretory processing of the amyloid precursor protein by a tyrosine phosphorylation-dependent mechanism. Biochem J. 1996;320:957–63. doi: 10.1042/bj3200957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoey SE, Williams RJ, Perkinton MS. Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J Neurosci. 2009;29:4442–60. doi: 10.1523/JNEUROSCI.6017-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Querfurth HW, Selkoe DJ. Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry. 1994;33:4550–61. doi: 10.1021/bi00181a016. [DOI] [PubMed] [Google Scholar]
- 47.Pierrot N, Ghisdal P, Caumont AS, et al. Intraneuronal amyloid-beta1–42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem. 2004;88:1140–50. doi: 10.1046/j.1471-4159.2003.02227.x. [DOI] [PubMed] [Google Scholar]
- 48.Green KN, Smith IF, Laferla FM. Role of calcium in the pathogenesis of Alzheimer’s disease and transgenic models. Subcell Biochem. 2007;45:507–21. doi: 10.1007/978-1-4020-6191-2_19. [DOI] [PubMed] [Google Scholar]
- 49.Querfurth HW, Jiang J, Geiger JD, et al. Caffeine stimulates amyloid beta-peptide release from beta-amyloid precursor protein-transfected HEK293 cells. J Neurochem. 1997;69:1580–91. doi: 10.1046/j.1471-4159.1997.69041580.x. [DOI] [PubMed] [Google Scholar]
- 50.Yoo AS, Cheng I, Chung S, et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–72. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 51.Brody DL, Magnoni S, Schwetye KE, et al. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–4. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron. 2003;37:925–37. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 53.Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–22. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 54.Cirrito JR, Kang JE, Lee J, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deshpande A, Kawai H, Metherate R, et al. A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J Neurosci. 2009;29:4004–15. doi: 10.1523/JNEUROSCI.5980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Foster TC. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell. 2007;6:319–25. doi: 10.1111/j.1474-9726.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 57.Toescu EC, Verkhratsky A. The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell. 2007;6:267–73. doi: 10.1111/j.1474-9726.2007.00296.x. [DOI] [PubMed] [Google Scholar]
- 58.Kirischuk S, Pronchuk N, Verkhratsky A. Measurements of intracellular calcium in sensory neurons of adult and old rats. Neuroscience. 1992;50:947–51. doi: 10.1016/0306-4522(92)90217-p. [DOI] [PubMed] [Google Scholar]
- 59.Kirischuk S, Voitenko N, Kostyuk P, et al. Age-associated changes of cytoplasmic calcium homeostasis in cerebellar granule neurons in situ: investigation on thin cerebellar slices. Exp Gerontol. 1996;31:475–87. doi: 10.1016/0531-5565(95)02070-5. [DOI] [PubMed] [Google Scholar]
- 60.Villalba M, Pereira R, Martinez-Serrano A, et al. Altered cell calcium regulation in synaptosomes and brain cells of the 30-month-old rat: prominent effects in hippocampus. Neurobiol Aging. 1995;16:809–16. doi: 10.1016/0197-4580(95)00087-u. [DOI] [PubMed] [Google Scholar]
- 61.Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–56. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Landfield PW. Aging-related increase in hippocampal calcium channels. Life Sci. 1996;59:399–404. doi: 10.1016/0024-3205(96)00318-9. [DOI] [PubMed] [Google Scholar]
- 63.Gant JC, Sama MM, Landfield PW, et al. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci. 2006;26:3482–90. doi: 10.1523/JNEUROSCI.4171-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kumar A, Foster TC. Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol. 2004;91:2437–44. doi: 10.1152/jn.01148.2003. [DOI] [PubMed] [Google Scholar]
- 65.Green KN, Peers C. Divergent pathways account for two distinct effects of amyloid beta peptides on exocytosis and Ca(2+) currents: involvement of ROS and NF-kappaB. J Neurochem. 2002;81:1043–51. doi: 10.1046/j.1471-4159.2002.00907.x. [DOI] [PubMed] [Google Scholar]
- 66.Ueda K, Shinohara S, Yagami T, et al. Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem. 1997;68:265–71. doi: 10.1046/j.1471-4159.1997.68010265.x. [DOI] [PubMed] [Google Scholar]
- 67.Iacopino AM, Christakos S. Specific reduction of calcium-binding protein (28-kilodalton calbindin-D) gene expression in aging and neurodegenerative diseases. Proc Natl Acad Sci USA. 1990;87:4078–82. doi: 10.1073/pnas.87.11.4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Villa A, Podini P, Panzeri MC, et al. Cytosolic Ca2+ binding proteins during rat brain ageing: loss of calbindin and calretinin in the hippocampus, with no change in the cerebellum. Eur J Neurosci. 1994;6:1491–9. doi: 10.1111/j.1460-9568.1994.tb01010.x. [DOI] [PubMed] [Google Scholar]
- 69.Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pottorf WJ, Duckles SP, Buchholz JN. SERCA function declines with age in adrenergic nerves from the superior cervical ganglion. J Auton Pharmacol. 2000;20:281–90. doi: 10.1046/j.1365-2680.2000.00194.x. [DOI] [PubMed] [Google Scholar]
- 71.Stutzmann GE, Smith I, Caccamo A, et al. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26:5180–9. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oddo S. The ubiquitin-proteasome system in Alzheimer’s disease. J Cell Mol Med. 2008;12:363–73. doi: 10.1111/j.1582-4934.2008.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tseng BP, Green KN, Chan JL, et al. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29:1607–18. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 75.Sarkar S, Rubinsztein DC. Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS J. 2008;275:4263–70. doi: 10.1111/j.1742-4658.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- 76.Williams A, Sarkar S, Cuddon P, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nukina N, Ihara Y. One of the antigenic determinants of paired helical filaments is related to tau protein. J Biochem. 1986;99:1541–4. doi: 10.1093/oxfordjournals.jbchem.a135625. [DOI] [PubMed] [Google Scholar]
- 78.Kobayashi S, Ishiguro K, Omori A, et al. A cdc2-related kinase PSSALRE/cdk5 is homologous with the 30 kDa subunit of tau protein kinase II, a proline-directed protein kinase associated with microtubule. FEBS Lett. 1993;335:171–5. doi: 10.1016/0014-5793(93)80723-8. [DOI] [PubMed] [Google Scholar]
- 79.Flaherty DB, Soria JP, Tomasiewicz HG, et al. Phosphorylation of human tau protein by microtubule-associated kinases: GSK3beta and cdk5 are key participants. J Neurosci Res. 2000;62:463–72. doi: 10.1002/1097-4547(20001101)62:3<463::AID-JNR16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 80.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 81.Iqbal K, Alonso Adel C, Chen S, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 82.Alonso AC, Li B, Grundke-Iqbal I, et al. Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2008;5:375–84. doi: 10.2174/156720508785132307. [DOI] [PubMed] [Google Scholar]
- 83.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–4. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 84.Green KN, Steffan JS, Martinez-Coria H, et al. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci. 2008;28:11500–10. doi: 10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kelliher M, Fastbom J, Cowburn RF, et al. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and beta-amyloid pathologies. Neuroscience. 1999;92:499–513. doi: 10.1016/s0306-4522(99)00042-1. [DOI] [PubMed] [Google Scholar]
- 86.Kurumatani T, Fastbom J, Bonkale WL, et al. Loss of inositol 1,4,5-trisphosphate receptor sites and decreased PKC levels correlate with staging of Alzheimer’s disease neurofibrillary pathology. Brain Res. 1998;796:209–21. doi: 10.1016/s0006-8993(98)00347-3. [DOI] [PubMed] [Google Scholar]
- 87.Pierrot N, Santos SF, Feyt C, et al. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J Biol Chem. 2006;281:39907–14. doi: 10.1074/jbc.M606015200. [DOI] [PubMed] [Google Scholar]
- 88.Patrick GN, Zhou P, Kwon YT, et al. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem. 1998;273:24057–64. doi: 10.1074/jbc.273.37.24057. [DOI] [PubMed] [Google Scholar]
- 89.Lee KY, Clark AW, Rosales JL, et al. Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res. 1999;34:21–9. doi: 10.1016/s0168-0102(99)00026-7. [DOI] [PubMed] [Google Scholar]
- 90.Alvarez A, Munoz JP, Maccioni RB. A Cdk5-p35 stable complex is involved in the beta-amyloid-induced deregulation of Cdk5 activity in hippocampal neurons. Exp Cell Res. 2001;264:266–74. doi: 10.1006/excr.2001.5152. [DOI] [PubMed] [Google Scholar]
- 91.Kusakawa G, Saito T, Onuki R, et al. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–72. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- 92.Lee MS, Kwon YT, Li M, et al. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–4. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 93.Shimohama S, Suenaga T, Araki W, et al. Presence of calpain II immunoreactivity in senile plaques in Alzheimer’s disease. Brain Res. 1991;558:105–8. doi: 10.1016/0006-8993(91)90722-8. [DOI] [PubMed] [Google Scholar]
- 94.Saito K, Elce JS, Hamos JE, et al. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci USA. 1993;90:2628–32. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Iwamoto N, Thangnipon W, Crawford C, et al. Localization of calpain immunoreactivity in senile plaques and in neurones undergoing neurofibrillary degeneration in Alzheimer’s disease. Brain Res. 1991;561:177–80. doi: 10.1016/0006-8993(91)90766-o. [DOI] [PubMed] [Google Scholar]
- 96.Liu F, Grundke-Iqbal I, Iqbal K, et al. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–62. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- 97.Chen X, Huang T, Zhang J, et al. Involvement of calpain and p25 of CDK5 pathway in ginsenoside Rb1’s attenuation of beta-amyloid peptide25–35-induced tau hyperphosphorylation in cortical neurons. Brain Res. 2008;1200:99–106. doi: 10.1016/j.brainres.2007.12.029. [DOI] [PubMed] [Google Scholar]
- 98.Trinchese F, Fa M, Liu S, et al. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118:2796–807. doi: 10.1172/JCI34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Perlmutter LS, Siman R, Gall C, et al. The ultrastructural localization of calcium-activated protease “calpain” in rat brain. Synapse. 1988;2:79–88. doi: 10.1002/syn.890020111. [DOI] [PubMed] [Google Scholar]
- 100.Kuchibhotla KV, Goldman ST, Lattarulo CR, et al. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–25. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sinjoanu RC, Kleinschmidt S, Bitner RS, et al. The novel calpain inhibitor A-705253 potently inhibits oligomeric beta-amyloid-induced dynamin 1 and tau cleavage in hippocampal neurons. Neurochem Int. 2008;53:79–88. doi: 10.1016/j.neuint.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kelly BL, Ferreira A. Beta-amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem. 2006;281:28079–89. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- 103.Amadoro G, Ciotti MT, Costanzi M, et al. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci USA. 2006;103:2892–7. doi: 10.1073/pnas.0511065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ekinci FJ, Malik KU, Shea TB. Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to beta-amyloid. MAP kinase mediates beta-amyloid-induced neurodegeneration. J Biol Chem. 1999;274:30322–7. doi: 10.1074/jbc.274.42.30322. [DOI] [PubMed] [Google Scholar]
- 105.Green KN, Peers C. Amyloid beta peptides mediate hypoxic augmentation of Ca(2+) channels. J Neurochem. 2001;77:953–6. doi: 10.1046/j.1471-4159.2001.00338.x. [DOI] [PubMed] [Google Scholar]
- 106.Brown ST, Scragg JL, Boyle JP, et al. Hypoxic augmentation of Ca2+ channel currents requires a functional electron transport chain. J Biol Chem. 2005;280:21706–12. doi: 10.1074/jbc.M503144200. [DOI] [PubMed] [Google Scholar]
- 107.Lopez JR, Lyckman A, Oddo S, et al. Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J Neurochem. 2008;105:262–71. doi: 10.1111/j.1471-4159.2007.05135.x. [DOI] [PubMed] [Google Scholar]
- 108.Nimmrich V, Grimm C, Draguhn A, et al. Amyloid beta oligomers (A beta(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci. 2008;28:788–97. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Goto Y, Niidome T, Akaike A, et al. Amyloid beta-peptide preconditioning reduces glutamate-induced neurotoxicity by promoting endocytosis of NMDA receptor. Biochem Biophys Res Commun. 2006;351:259–65. doi: 10.1016/j.bbrc.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 110.Nomura I, Kato N, Kita T, et al. Mechanism of impairment of long-term potentiation by amyloid beta is independent of NMDA receptors or voltage-dependent calcium channels in hippocampal CA1 pyramidal neurons. Neurosci Lett. 2005;391:1–6. doi: 10.1016/j.neulet.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 111.Snyder EM, Nong Y, Almeida CG, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–8. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 112.Ye C, Walsh DM, Selkoe DJ, et al. Amyloid beta-protein induced electrophysiological changes are dependent on aggregation state: N-methyl-D-aspartate (NMDA) versus non-NMDA receptor/channel activation. Neurosci Lett. 2004;366:320–5. doi: 10.1016/j.neulet.2004.05.060. [DOI] [PubMed] [Google Scholar]
- 113.Parameshwaran K, Sims C, Kanju P, et al. Amyloid beta-peptide Abeta(1–42) but not Abeta(1–40) attenuates synaptic AMPA receptor function. Synapse. 2007;61:367–74. doi: 10.1002/syn.20386. [DOI] [PubMed] [Google Scholar]
- 114.Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu Q, Kawai H, Berg DK. beta -Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci USA. 2001;98:4734–9. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pettit DL, Shao Z, Yakel JL. beta-Amyloid(1–42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 2001;21(1–5) doi: 10.1523/JNEUROSCI.21-01-j0003.2001. : RC120 ( ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Takenouchi T, Munekata E. Inhibitory effects of beta-amyloid peptides on nicotine-induced Ca2+ influx in PC12h cells in culture. Neurosci Lett. 1994;173:147–50. doi: 10.1016/0304-3940(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 118.Wu J, Kuo YP, George AA, et al. beta-Amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J Biol Chem. 2004;279:37842–51. doi: 10.1074/jbc.M400335200. [DOI] [PubMed] [Google Scholar]
- 119.Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci USA. 1993;90:567–71. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kawahara M, Kuroda Y. Molecular mechanism of neuronal death in Alzheimer’s disease: Ca(2+)- channel formation of beta amyloid protein molecules. Tanpakushitsu Kakusan Koso. 1997;42:2002–10. [PubMed] [Google Scholar]
- 121.Popovic M, Caballero-Bleda M, Kadish I, et al. Subfield and layer-specific depletion in calbindin-D28K, calretinin and parvalbumin immunoreactivity in the dentate gyrus of amyloid precursor protein/presenilin 1 transgenic mice. Neuroscience. 2008;155:182–91. doi: 10.1016/j.neuroscience.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Walsh DM, Townsend M, Podlisny MB, et al. Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–62. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–9. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 124.Yamin G. NMDA receptor-dependent signaling pathways that underlie amyloid beta-protein disruption of LTP in the hippocampus. J Neurosci Res. 2009;87:1729–36. doi: 10.1002/jnr.21998. [DOI] [PubMed] [Google Scholar]
- 125.Shankar GM, Bloodgood BL, Townsend M, et al. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lacor PN, Buniel MC, Chang L, et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24:10191–200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Deshpande A, Mina E, Glabe C, et al. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26:6011–8. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang EP. Metal ions and synaptic transmission: think zinc. Proc Natl Acad Sci USA. 1997;94:13386–7. doi: 10.1073/pnas.94.25.13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.De Felice FG, Velasco PT, Lambert MP, et al. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 130.Dineley KT, Hogan D, Zhang WR, et al. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007;88:217–24. doi: 10.1016/j.nlm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kuchibhotla KV, Lattarulo CR, Hyman BT, et al. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323:1211–5. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Busche MA, Eichhoff G, Adelsberger H, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–9. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 133.Palop JJ, Chin J, Roberson ED, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Amatniek JC, Hauser WA, DelCastillo-Castaneda C, et al. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia. 2006;47:867–72. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- 135.Lozsadi DA, Larner AJ. Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;22:121–4. doi: 10.1159/000093664. [DOI] [PubMed] [Google Scholar]
- 136.Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hsiung GY, Feldman HH. Pharmacological treatment in moderate-to-severe Alzheimer’s disease. Expert Opin Pharmacother. 2008;9:2575–82. doi: 10.1517/14656566.9.15.2575. [DOI] [PubMed] [Google Scholar]
- 138.Minkeviciene R, Banerjee P, Tanila H. Memantine improves spatial learning in a transgenic mouse model of Alzheimer’s disease. J Pharmacol Exp Ther. 2004;311:677–82. doi: 10.1124/jpet.104.071027. [DOI] [PubMed] [Google Scholar]
- 139.Song MS, Rauw G, Baker GB, et al. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur J Neurosci. 2008;28:1989–2002. doi: 10.1111/j.1460-9568.2008.06498.x. [DOI] [PubMed] [Google Scholar]
- 140.Scholtzova H, Wadghiri YZ, Douadi M, et al. Memantine leads to behavioral improvement and amyloid reduction in Alzheimer’s-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res. 2008;86:2784–91. doi: 10.1002/jnr.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lopez-Arrieta JM, Birks J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev. 2002;3:CD000147. doi: 10.1002/14651858.CD000147. [DOI] [PubMed] [Google Scholar]
- 142.Rosenberg PB, Mielke MM, Tschanz J, et al. Effects of cardiovascular medications on rate of functional decline in Alzheimer disease. Am J Geriatr Psychiatry. 2008;16:883–92. doi: 10.1097/JGP.0b013e318181276a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Doody RS, Gavrilova SI, Sano M, et al. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372:207–15. doi: 10.1016/S0140-6736(08)61074-0. [DOI] [PubMed] [Google Scholar]
- 144.Wu J, Li Q, Bezprozvanny I. Evaluation of Dimebon in cellular model of Huntington’s disease. Mol Neurodegener. 2008;3:15. doi: 10.1186/1750-1326-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]