

Abstract
Reactive oxygen species (ROS) form as a natural by-product of the normal metabolism of oxygen and play important roles within the cell. Under normal circumstances the cell is able to maintain an adequate homeostasis between the formation of ROS and its removal through particular enzymatic pathways or via antioxidants. If however, this balance is disturbed a situation called oxidative stress occurs. Critically, oxidative stress plays important roles in the pathogenesis of many diseases, including cancer. Epigenetics is a process where gene expression is regulated by heritable mechanisms that do not cause any direct changes to the DNA sequence itself, and disruption of epigenetic mechanisms has important implications in disease. Evidence is emerging that histone deacetylases (HDACs) play decisive roles in regulating important cellular oxidative stress pathways including those involved with sensing oxidative stress and those involved with regulating the cellular response to oxidative stress. In particular aberrant regulation of these pathways by HDACs may play critical roles in cancer progression. In this review we discuss the current evidence linking epigenetics and oxidative stress and cancer, using chronic obstructive pulmonary disease and non-small cell lung cancer to illustrate the importance of epigenetics on these pathways within these disease settings.
Keywords: epigenetics, chromatin, oxidative stress, COPD, NSCLC
-
Oxidative stress, epigenetics, NSCLC and COPD
Cigarette smoke mediated cellular proliferation
Cigarette smoke mediated lung destruction
DNA methylation and lung cancer
Histone post-translational modifications and lung cancer
-
Linking epigenetics and oxidative stress to the development of COPD and NSCLC
HATs and KATs in the developing lung
HDACs and COPD
Aberrant KAT/HDAC activity in NSCLC
ATP-dependent SWI/SNF chromatin remodelling complexes and NSCLC
KATs/HDACs and oxidative stress pathways
KEAP1-NRF2-ARE signalling pathway
-
Epigenetics, COPD, NSCLC and oxidative stress pathways/genes
NRF2
Heme oxygenase 1
Hypoxia-inducible factor-1α
PGC-1α
Oxidative stress, epigenetics, glucocorticoids and COPD
Oxidative stress, acetylation and its role in the regulation of NFκB
Targeting epigenetic mechanisms in COPD and NSCLC
-
Theophylline – a novel agonist of HDACs
Current clinical trials involving epigenetic therapies in COPD and NSCLC
Dietary HDAC inhibitors as therapeutic agents to target - COPD or NSCLC?
Sulforaphane
Curcumin
Epigallocatechin 3-gallate (EGCG)
Can dietary epigenetic inhibitors work in the clinical setting?
Caveats
Conclusions
Oxidative stress, epigenetics, NSCLC and COPD
Chronic obstructive pulmonary disease (COPD), is the diagnostic term for a group of disorders that are characterized by respiratory symptoms – dyspnoea, cough and sputum production; airflow limitation and chronic inflammation of the lung [1]. It is currently the fourth leading cause of worldwide mortality, responsible for more than 2.5 million deaths per year and is expected to become the third leading cause by 2020 [2]. In addition individuals suffering from COPD also have an associated increased risk of developing lung cancer [3, 4].
Lung cancer is the leading cause of cancer death worldwide, and is associated with over 1 million deaths annually [5]. Only 13% of lung cancer patients survive more than 5 years. According to the American Cancer Society, approximately 219,440 new cases of lung cancer (both small cell and non-small cell), and 159,390 deaths will occur in the United States during 2009 (source http://www.cancer.org). Clinico-pathologically, lung cancer can be subdivided into two broad categories, non-small-cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC can then be further divided into three major types, squamous cell carcinoma (SCC), adenocarcinoma and large cell carcinoma. Lung cancer is often considered to be largely preventable as most cases can be attributed to smoking. However, significant proportions (approximately 25%) of all lung cancers worldwide are not caused by smoking. If considered in a separate category lung cancer in never smokers would rank as the seventh most common cause of cancer death worldwide, before cancers of the cervix, pancreas and prostate [5]. Because of difficulties in its early detection, lung cancer has a high morbidity, and is often associated with resistance to currently available chemotherapy and radiotherapy regimes [6].
Cigarette smoke mediated oxidative stress is well established as being causative for both cellular proliferation in lung cancer and mediating lung destruction in COPD. In the following sections we provide examples of how cigarette smoke may cause both cellular proliferation and destruction, but for extensive reviews on this subject the reader is directed to the following recent reviews [1, 7].
Cigarette smoke mediated cellular proliferation
For example, in transformed lung epithelial type-II cells cigarette smoke condensate at low concentration (0.1 μg/ml) induced cancer cell proliferation, DNA synthesis, reduced glutathione levels and intercellular adhesion molecule-1 expression without any significant change in reactive oxygen species (ROS) and superoxide radicals production [8]. Another recent study has shown that critical miRNAs involved with regulating stress response, apoptosis, proliferation, angiogenesis and expression of genes were found to be down-regulated in the lungs of rats exposed to environmental cigarette smoke for 28 days [9]. Benzo[a]pyrene (B[a]P) is one of the strongest carcinogens present in both the environment and cigarette smoke. B[a]P has now been shown to increases the proliferative potential of lung adenocarcinoma cells through the epidermal growth factor receptor (EGFR) signalling pathway by overexpression of phosphorylated EGFR protein and inducing expression of the ligands for EGFR such as amphiregulin and epiregulin [10]. Moreover, exposure of human airway epithelial cells to cigarette smoke, results in an increase in EGFR activation over time, leading to prolonged signalling by the EGFR and which may therefore contribute to uncontrolled lung cell growth [11]
Cigarette smoke mediated lung destruction
One way by which cigarette smoke extract mediates cellular destruction in the lung has been shown to be via disruption of tubulin-microtubules structure and function, and it has been suggested that this results in cellular apoptosis and subsequent tissue damage in smokers [12] In addition, p21 is overexpressed in alveolar epithelial cells in response to cigarette smoke [13], leading to cellular senescence, and if p21 is disrupted, this response is attenuated, which indicates how critical regulators designed for the protection of the host, can lead to organ damage in the presence of cigarette smoke [14].
Vascular endothelial growth factor (VEGF) is fundamental to the development and maintenance of vasculature. Expression of critical members of this signalling pathway has been shown to be reduced in the lungs of both smokers and patients with COPD compared with non-smokers [15]. Cigarette smoke down-regulated vascular endothelial growth factor receptor (VEGFR)-2 expression, endothelial nitric oxide synthase (eNOS) levels and VEGF-induced VEGFR-2 phosphorylation in human lung microvascular endothelial cells, which results in impaired VEGF-induced endothelial cell migration and angiogenesis [16].
In COPD, defective efferocytosis (apoptosis and alveolar macrophage phagocytic function) may lead to secondary necrosis and tissue damage. Cigarette smoke has been shown to significantly decrease alveolar macrophage phagocytic function in both COPD patients and healthy smokers compared with control subjects indicating that smoking-related reduction in alveolar macrophage (AM) phagocytic ability may play important roles in the destruction of lung [17]. These differences (i.e. proliferative versus destructive effects) may be due to either cigarette smoke targeting different cell types, or may be cell specific effects. Alternatively, these differential effects may be caused by differential regulation of critical pathways via aberrant epigenetic regulation.
Molecular genetic studies of lung cancer frequently reveal multiple genetic and epigenetic changes including DNA sequence alterations, copy number changes and aberrant promoter methylation [18, 19]. The role of epigenetics in cancer is increasingly recognized as a major focus for translational research. NSCLC is no exception and DNA CpG methylation is recognized as having both predictive and prognostic significance in NSCLC [20, 21]. The three main, inter-related forms of epigenetic regulation are: DNA methylation, genomic imprinting and histone modification, all of which have important roles in disease [22].
DNA methylation and lung cancer
The importance of DNA methylation in the development of lung cancer was recently demonstrated when it was shown that the transformation efficiency for immortalization of normal bronchial epithelial cells could be enhanced by low dose exposure to carcinogens. The mechanism underpinning this involved hypermethylation of 5–10 genes due to elevated expression of DNA methyltransferase 1 (DNMT1). Ablation of DNMT1 was shown to reverse this process. Moreover, stable ‘knockdown’ of DNMT1 prior to carcinogen exposure was sufficient to prevent cellular transformation [23].
Histone post-translational modifications and lung cancer
Aberrant histone post-translational modifications have also been shown to have both predictive and prognostic significance in NSCLC [24, 25], and deregulation of some of these enzymes in a bronchial epithelial cell transformation model suggest that they play important roles in the transformation process [26]. In addition, strong evidence links aberrant expression of epigenetic regulators, in particular histone deacetylases (HDACs) to COPD [27]. There is therefore an urgent need to improve our understanding of the molecular basis of epigenetic mechanisms in NSCLC and COPD.
Oxidative stress has been clearly linked to COPD and NSCLC [28] (Figs 1 and 2), and there are many pathways/genes associated with the cellular response to oxidative stress. Among these pathways include genes such as superoxide dismutases (SODs), glutathione peroxidases (GPXs), glucocorticoid receptors (GRs), heme oxygenases (HOs) and hypoxia inducible factor-1α (HIF-1α).
Figure 1.
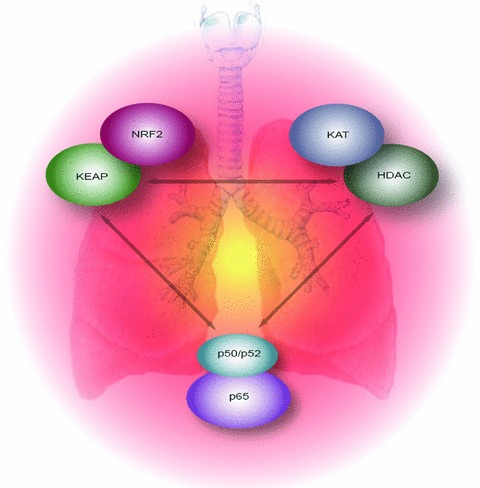
The concept. Overview of the balance between oxidative stress response mechanisms, critical regulators such as lysine acetyltransferases and HDACs and the NFκB pathway in the lung. Perturbations to any of these could lead to aberrant epigenetic regulation of pathways critical to the cells response to oxidative stress, and may lead to COPD or NSCLC.
Figure 2.
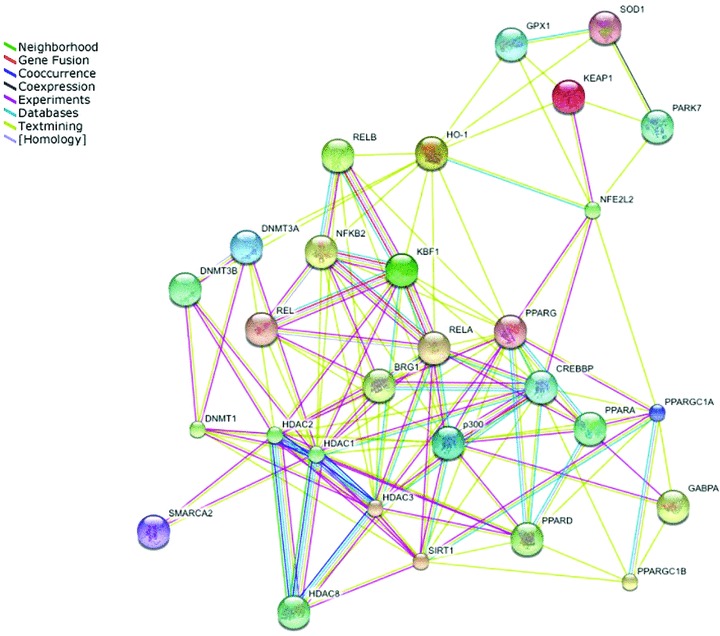
Graphical demonstration linking epigenetic regulators, the NFκB and the oxidative stress response pathways. This figure was generated using STRING 8.0 http://string.embl.de/.
In the following sections we shall discuss the role that epigenetics plays in the regulation of these genes/pathways using COPD and NSCLC to illustrate the epigenetic dysregulation associated with these diseases and discuss the potential for epigenetic targeting strategies as a therapeutic intervention for the treatment of both.
Linking epigenetics and oxidative stress to the development of COPD and NSCLC
HATs and KATs in the developing lung
Evidence is also emerging that HDACs and lysine acetyltransferases play important roles in the developing lung (Table 1). The importance of lysine acetyltransferase (KAT) activity in lung development has been shown in mouse studies, where KATs were shown to be highly expressed in the developing lung [29–34], and confirmation of their importance in lung development came from KAT3B knockout studies [35]. Recently a homeodomain protein (HOP), which in the cardiac system associates with HDAC2 to repress cardiac specific genes, was observed to be present in airway epithelium along with HDAC2. HOP was shown to represses lung-specific gene expression in an HDAC-dependent manner, and loss of HOP expression in vivo resulted in defective type 2 pneumocyte development [36].
Table 1.
KATs, KMTs and HDACs discussed in this review
| Gene | Activity | Comments | Reference |
|---|---|---|---|
| KAT2A formerly (GCN5) | Acetyltransferase | Critical for lung development | [30, 31, 34] |
| KAT2B (formerly PCAF) | Acetyltransferase | Critical for lung development | [30] |
| KAT3A | Acetyltransferase | Critical for lung development | [29, 32, 33] |
| (formerly CBP) | Mutated in lung cancer | [72] | |
| KAT3B (formerly p300) | Acetyltransferase | Critical for lung development | [31–33, 35] |
| KAT13A (formerly SRC1) | Acetyltransferase | Critical for lung development | [32] |
| HDAC1 | Deacetylase | Elevated mRNA in lung cancer | [50] |
| Protein detected in lung cancer | [51] | ||
| HDAC2 | Deacetylase | Association with HOP involved with pneumocyte development | [36] |
| Reduced levels of HDAC2 protein in lung tissue of patients with COPD | [37] | ||
| Protein detected in lung cancer | [51] | ||
| HDAC3 | Deacetylase | Elevated protein detected in NSCLC SCCs | [52] |
| HDAC5 | Deacetylase | Reduced mRNA in lung tissue of patients with COPD | [37] |
| HDAC8 | Deacetylase | Reduced mRNA in lung tissue of patients with COPD | [37] |
| Sirt1 | Deacetylase | Protein levels are reduced in lung tissue of COPD patients | [43] |
| Sirt2 | Deacetylase | Decreases ROS | [77] |
| Sirt3 | Deacetylase | Prevents oxidative stress induced apoptosis | [78] |
| KMT1A (formerly SUV39H1) | Methyltransferase | Up-regulated in cancer cell lines and following immortalization and transformation of human bronchoepithelial cells | [26] |
| KMT1B (formerly SUV39H2) | Methyltransferase | Polymorphisms associated with an increased risk for lung cancer | [73] |
| KMT1C (formerly G9a) | Methyltransferase | Up-regulated in cancer cell lines and following immortalization and transformation of human bronchoepithelial cells | [26] |
| KMT1E (formerly ESET or SETDB1) | Methyltransferase | Up-regulated in cancer cell lines and following immortalization and transformation of human bronchoepithelial cells | [26] |
| KMT4 (formerly DOT1L) | Methyltransferase | Up-regulated in cancer cell lines and following immortalization and transformation of human bronchoepithelial cells | [26] |
| KMT6 (formerly EZH2) | Methyltransferase | Up-regulated in cancer cell lines and following immortalization and transformation of human bronchoepithelial cells | [26] |
| KMT8 (formerly RIZ1) | Methyltransferase | Polymorphisms associated with an increased risk for lung cancer | [74] |
HDACs and COPD
A link between HDAC activities and COPD was identified when studies revealed that in lung tissue of patients with increasing clinical stages of COPD, graded reductions in HDAC activity, reduced levels of HDAC2 protein and lower mRNA levels for HDACs 2, 5 and 8 were observed [37]. The decreased levels of HDAC2 have also been observed in separate studies of the effects of cigarette smoking in patients with COPD [38–41], and oxidative stress has also been shown to decrease HDAC2 levels in a bronchial epithelial cell line [42]. In addition levels of the NAD+-dependent HDAC sirtuin 1 (SIRT1) have also been shown to be reduced in patients with COPD [43].
Surfactant protein A (SP-A) is a member of the collectin family of innate host defence molecules and is expressed primarily in the respiratory epithelial cells of the lung (Table 2). SP-A has been shown to be a biomarker with elevated expression in patients with COPD and in patients who smoke [44–47]. SP-A is negatively regulated by the transcription factor GATA-6 [48], in a complex containing HDAC2 and Nkx2.1 [36]. As levels of HDAC2 have been shown to be decreased in the lungs of patients with COPD, this could explain why SP-A is elevated in patients with COPD. Within macrophages in the lung environment SP-A has also been shown to decrease the phosphorylation of I-Kappa-B-Alpha (IKBA), a key regulator of NFκB activity, and nuclear translocation of p65 while additionally decreasing the phosphorylation of Akt, a major cell regulator of NFκB [49].
Table 2.
major ROS pathways discussed, epigenetic modifications or enzymes affected and cell function
| Pathway | Epigenetic modifications or enzymes affected | Cell function |
|---|---|---|
| KEAP1-NRF2-ARE | DNA methylation | Oxidative stress sensor |
| SP-A | Histone acetylation | Innate host defence |
| NFκB | Histone acetylation | Cellular stress responses |
| Lysine acetyltransferases | Induction of pro-inflammatory genes | |
| HDACs | ||
| HO-1 | Histone acetylation | Cytoprotective cellular stress responses |
| Histone methylation | ||
| SWI/SNF -like chromatin-remodelling complexes | ||
| Lysine methyltransferases | ||
| HIF-1α | Histone methylation | Regulates responses to oxidative stress |
| Histone acetylation | ||
| Lysine acetyltransferases | ||
| HDACs | ||
| PGC-1α | Histone acetylation | Regulator of antioxidant genes |
| HDACs | ||
| Histone acetyltransferases | Protects cells from oxidative stress | |
| Glucocorticoids | HDACs | Regulate expression of protective factors in oxidative stress |
| Lysine acetyltransferases |
The role of epigenetics in regulating NFκB mediated transcription is discussed in greater detail in a following section.
Aberrant KAT/HDAC activity in NSCLC
In contrast to COPD, elevated levels of HDAC1 mRNA have been observed in higher stage (stage III or IV) NSCLC [50], while a more recent study has also observed expression of all class I HDACs in lung cancer specimens [51]. Using antibody arrays, HDAC3 protein was found to be elevated in 92% of SCCs of the lung (Table 1) [52].
It is well established that HDACs form large multi-protein complexes to regulate gene expression [53]. Several members of these complexes have also been shown to be altered in NSCLC. For example, mSin3A a member of a multiple component co-repressor complex that contains HDACs has also been observed to have decreased expression in NSCLC [54].
ATP-dependent SWI/SNF chromatin remodelling complexes and NSCLC
Members of the ATP-dependent SWItch/Sucrose (SWI)/NonFermentable (SNF) chromatin remodelling complexes have also been shown to be altered in the lung. In NSCLC cell lines, the SWI/SNF complex has been found to form a larger complex containing neuron-restrictive silencer factor and its co-repressors, mSin3A and CoREST [55], and although this complex has been shown to regulate neuronal specific genes, it was suggested that deregulation of neuron-restrictive silencer factor regulated genes in NSCLC could in fact contribute to enhanced tumourigenicity [55]. Additional evidence now links other members of these SWI/SNF complexes to the pathogenesis of NSCLC adding support to this notion. Expressions of the SWI/SNF ATPase subunits, BRG1 and BRM (BRG1/BRM), have been shown to be either mutated or lost in approximately 30% of human non-small lung cancer cell lines [56–60]. Critically, BRM has been shown to be regulated epigenetically [61], and HDAC inhibitors have been shown to reactivate expression of BRM [62, 63]. When examined in primary tumours, 10% of NSCLC had concomitant loss of both BRG1 and BRM, which correlated with the poorest prognosis [56]. A multiple tissue array approach examining 12 core proteins involved with chromatin remodelling machinery in 300 NSCLC samples (150 adenocarcinomas and 150 SCCs), found two clusters which either contained BRM, Ini-1, retinoblastoma, mSin3A, HDAC1 and HAT1, or which contained BRG1, BAF155, HDAC2, BAF170 and RbAP48 (Table 1) [64]. Positive nuclear BRM (N-BRM) staining correlated with a favourable prognosis in patients with a 5-year survival of 53.5% compared with 32.3% for those patients with tumours that were negative for N-BRM (P= 0.015). Co-positivity for both N-BRM and nuclear BRG1 had an increased 5-year survival of 72% compared with 33.6% (P= 0.013) for those patients whose tumours were positive for either marker, or negative for both markers. In contrast, membranous BRM (M-BRM) staining correlated with a poorer prognosis in AD patients with a 5-year survival of 16.7% compared with those without M-BRM staining (38.1%; P= 0.016) [64].
The expression of metastasis-associated protein 1 (MTA-1) has been shown to be significantly elevated in NSCLC and was found to be associated with both tumour invasiveness and metastasis [65]. As MTA-1 is known to associate with HDACs [66–71], this would suggest that HDAC activity associated with MTA-1 is involved with NSCLC invasiveness and metastasis.
Mutations within the lysine acetyltransferase KAT3A have also been identified in a small subset of NSCLC [72], and other histone modifying enzymes have been identified for which polymorphisms within these genes are associated with an increased risk for lung cancer including lysine methyltransferase KMT1B (formerly SUV39H2) [73], and KMT8 (formerly RIZ/PRDM2) [74] (Table 1). Polymorphisms in another epigenetic regulator, Methyl-CpG binding domain 1 (MBD-1) have also been shown to be associated with an increased lung cancer risk [75]. Intriguingly, the polymorphism associated with increased risk, also had significantly higher promoter activity. As MBD-1 has been shown to associate with HDACs to repress transcription [76], this polymorphism may therefore lead to elevated MBD-1/HDAC complexes formin, resulting in aberrant gene expression leading to NSCLC development, but this will require functional testing.
KATs/HDACs and oxidative stress pathways
HDACs have been linked to the regulation of genes important for oxidative stress pathways (Figs 1 and 2). For instance, oxidative stress in adipose and kidney cells increases the levels of the NAD+ dependent HDAC SIRT2. SIRT2 then binds to FOXO3a and reduces its acetylation level. This results in increased FOXO3a DNA binding which elevates the expression of p27 (Kip1), manganese SOD and Bim. As a consequence, this then decreases cellular levels of ROS in these cells [77]. Similar studies in human cardiomyocytes have shown that SIRT3 can prevent oxidative stress-mediated cell death by deacetylating the Ku70 protein, which results in enhanced binding of Ku70 to the proapototic protein Bax, and preventing its translocation to the mitochondria, thus protecting the cardiomyocytes from oxidative stress induced apoptosis [78].
The transcription factor Bach1 (BTB and CNC homology 1, basic leucine zipper transcription factor 1) has been shown to be a repressor of the oxidative stress response in mice [79]. One mechanism by which Bach1 functions involves its role in oxidative stress-induced cellular senescence by a complex that contains p53, HDAC1 and the nuclear co-repressor N-CoR to repress target genes [80].
KEAP1-NRF2-ARE signalling pathway
One of the most important oxidative stress protective pathways is the Kelch-like ECH-Associated Protein 1 (KEAP1)- NFE2-Related
Factor 2 (NRF2)- antioxidant response element (ARE) or KEAP1-NRF2-ARE signalling pathway (Table 2, Fig. 1) which plays critical roles in protecting cells from endogenous and exogenous stresses [81], and plays important roles within the lung [82]. It has been hypothesized that low expression of the oxidative stress sensor KEAP1 is thought to be involved in carcinogenesis. Evidence supporting this notion in lung cancer has emerged whereby down-regulation of KEAP1 by DNA CpG methylation has been shown to occur in both NSCLC cell lines and tumour tissues [83], and an additional study in NSCLC cell lines showed that loss of KEAP1 function activated NRF2 and provided cellular growth advantages [84]. In COPD, the KEAP1-NRF2-ARE pathway has been shown to be disrupted but in this instance, levels of KEAP1 were found to be unchanged whereas significant decreases in NRF2 and DJ-1 protein were found. (DJ-1 is a protein that stabilizes NRF2 by impairing KEAP1-dependent proteasomal degradation of NRF2). Of these, DJ-1 expression was negatively associated with severity of COPD [85]. Another study confirmed the loss of NRF2 in lung tissues of patients with emphysema but in addition also demonstrated elevated expression of KEAP1 in these patients [86]. Overall, it would appear that dysregulation of this critical oxidative stress responsive pathway occurs in both COPD and NSCLC (Table 2).
Epigenetics, COPD, NSCLC and oxidative stress pathways/genes
NRF2
Emerging evidence is beginning to link direct chromatin remodelling to the cellular response to oxidative stress. The KEAP1-NRF2-ARE pathway is strongly implicated as an antioxidative stress pathway in the lung [82]. Under normal conditions NRF2 is found associated with the cytoplasmic inhibitor, KEAP1. Upon oxidative stress, NRF2 becomes phosphorylated and dissociates from KEAP1, whereupon it translocates to the nucleus and binds to an ARE, inducing the expression of genes that encode antioxidant-detoxifying proteins, to rescue cells against oxidative injury [87].
An early indication that NRF2 may utilize chromatin remodelling to regulate the expression of its cognate target genes came from studies of the structural motifs contained within it (Table 2). Two transcription activation domains, Neh4 and Neh5, were found to associate with KAT3B and acted synergistically to obtain maximal activation of gene expression by NRF2 [88]. Other proteins with histone modifying ability which have been shown to associate and regulate NRF2 transactivation domain activity include the lysine acetyltransferases KAT3A/KAT3B/KAT2B (formerly CBP/p300/ pCAF), and arginine methyltransferases CARM1 and PRMT1 [89] (Fig. 2).
Under conditions of oxidative stress the SWI/SNF complex component BRG1 interacts directly with NRF2 to selectively mediate induction of responsive genes [90]. More recently, the NFκB p65 subunit was shown to repress the NRF2-antioxidant response element (ARE) pathway at the transcriptional level (Fig. 2). In this regard, in cells where NFκB and NRF2 were simultaneously activated, p65 unidirectionally antagonized the transcriptional activity of NRF2, and ARE-dependent expression of haemoxygenase-1 (HO-1) was strongly suppressed. The mechanism found to cause this effect involved the selective prevention of the lysine acetyltransferase KAT3A association with p65 resulting in the inactivation of p65. This was followed by the recruitment by p65 of HDAC3 to ARE-dependent promoters to repress transcription [91].
Heme oxygenase 1
HO-1 is a cellular stress response gene which has been shown to protect the lung from oxidative stress based insults such as cigarette smoke and COPD via degradation of heme to biliverdin-IXα, carbon monoxide and iron, each with candidate roles in cytoprotection. At low concentrations, carbon monoxide can confer similar cyto- and tissue-protective effects as endogenous HO-1 expression, involving antioxidative, anti-inflammatory, anti-proliferative and anti-apoptotic effects. [92]. Regulation of HO-1 has been shown to involve chromatin remodelling (Fig. 2), and exposure of cells to heme results in de novo hyperacetylation and hypermethylation of histone H3 at the HO-1 enhancers resulting in its enhanced transcription [93]. The BRG1 catalytic subunit of SWI2/SNF2-like chromatin-remodelling complexes has been shown to utilize NRF2 to regulate expression of HO-1 in a colon cell line in response to oxidative stress [90]. Deletion of a particular region in NRF2 (the Neh5 domain) resulted in a reduction in expression of HO-1. Loss of this domain reduced the ability of KAT3B and BRG1 (Brahma-related gene 1) to associating with NRF2, thereby reducing their co-operative enhancement of HO-1 promoter activity [94].
Hypoxia-inducible factor-1α
Hypoxia is a common event in solid tumours [95], but is also strongly associated with NSCLC [96] and COPD [97]. Similarly, levels of HIF-1α or other markers of hypoxia such as Carbonic Anhydrase IX have also been linked to NSCLC [98–101]. Knockdown siRNA experiments targeting HIF-1α in a Lewis mouse model of lung cancer have been shown to prolong survival by reducing cancer cell proliferation, angiogenesis and apoptosis, and other cellular responses in the lung tumour tissue [102].
In a study examining the progressive structural changes in the bronchial epithelium with subepithelial fibrous remodelling, it was shown that HIF-1α was expressed in epithelial cells associated with increased progression of structural changes, increased reticular basement membrane (RBM) thickness, and with a reduction in the number of blood vessels in the subepithelium of COPD patients [103], while in a clinical study of healthy human volunteers, when placed under conditions of hypoxia, it was found that hypoxia induced oxidative stress via an overgeneration of ROS, which was positively correlated with an up-regulation of HIF-1α, VEGF and DNA oxidation [104].
HIF-1α has recently been shown to regulate epigenetic regulators including lysine demethylases [105–107].
Epigenetic mechanisms have been shown to affect HIF-1α mainly through stabilization of the protein itself, by HDACs [108–111]. In addition, the MTA-1 has also been shown to both stabilize HIF-1α and enhance tumour metastasis via angiogenesis through direct associations with HDACs [112, 113]. Indeed, in many cell culture models inhibition of HDACs has been shown to both destabilize HIF-1α and reduce angiogenesis [108, 114–118].
HIF-1α functionally associates with lysine acetyltransferases (KATs and HDACs) to regulate gene expression in response to hypoxia [111, 119–122]. These associations are known to be essential to drive hypoxia-responsive transcription, from studies in mice which were engineered to carry deletions in the domains which associate with these proteins. The domains necessary for KAT3A and KAT3B interactions with HIF-1α were genetically non-redundant and indispensable for HIF-1α transactivation function, while a similar yet larger effect was observed for the domain required for HIF-1α association with HDACs (35–50% compared to 70%) [123]. Indeed for some target HIF-1α responsive genes both lysine acetyltransferases and HDACs were required for greater than 90% of the genes response, suggesting that a functional interaction between lysine acetylases and HDACs are essential for nearly all HIF-1α responsive transcription [123].
PGC-1α
Peroxisome proliferator-activated receptor-γ co-activator 1α (PGC-1α) was originally isolated from a brown fat cDNA library linked to adaptive thermogenesis [124]. Since then it has been associated with various other important pathways including mitochondrial oxidative metabolism, adipocyte cell fate decision, glucose homeostasis and hepatic gluconeogenesis [125]. It is well established that PGC-1α acts as a transcriptional co-activator associating with both HDACs [126] and lysine acetyltransferases [127] to regulate expression of genes (Table 2). Under various conditions of stress PGC-1α has been shown to increase the expression of major antioxidant enzymes including copper/zinc SOD (SOD1), manganese SOD (SOD2), catalase and GPx1 [128–131]. PGC-1α can regulate the expression of uncoupling protein 2 and uncoupling protein 3, both of which are now understood to be able to mitigate ROS formation [132, 133]. Nitric oxide, a critical regulator of oxidative stress has also been shown to regulate the protection of mitochondria by modulating PGC-1α expression, resulting in changes to the expression of mitochondrial ROS detoxification system members [134].
The PGC-1 transcriptional co-activators are major regulators of several crucial aspects of energy metabolism, and NRF1 and NRF2 are key targets of the PGC-1s in mitochondrial biogenesis [82]. In a neural cell model studying the metabolism of ROS, levels of PGC-1α were induced in cells given an oxidative stressor, H2O2, resulting in greatly enhance protection from oxidative-stressor-mediated death [82, 128].
Oxidative stress, epigenetics, glucocorticoids and COPD
Glucocorticoids are a standard treatment option for patients with COPD. In particular they are frequently used for the treatment of patients with acute exacerbations of their disease [135]. However, many COPD patients develop resistance or have a reduced sensitivity to glucocorticoids [136]. Glucocorticoids act by binding to the GR, normally found in the cytoplasm. This binding results in translocation of the receptor to the nucleus, where it can suppress the expression of responsive genes in particular pro-inflammatory gene expression.
One of the genes negatively regulated by glucocorticoids is SP-A which, as described, has a role in regulating NFκB in airway epithelial cells [137]. GR regulates the expression of the SP-A gene via chromatin remodelling and assembly of complexes containing either HDACs or KATs to functionally regulate transcription of this gene [138]. It has been suggested that IKBA, a key regulator of NFκB activity is a major contributor to the anti-inflammatory effects of SP-A [137].
As levels of HDAC2 have been shown to be decreased in the lungs of patients with COPD, this would indicate the reason why SP-A is elevated in patients with COPD. It has been suggested that accumulation of IKBA, a key regulator of NFκB activity is a major contributor to the anti-inflammatory effects of SP-A [137]. Indeed, within macrophages isolated from the lung environment, SP-A has in fact been shown to decrease the phosphorylation of IKBA, resulting in decreases of nuclear translocation of p65 and phosphorylation of Akt, a major cell regulator of NFκB, and resulting in a decreased inflammatory response [49].
The UPC3 gene, implicated as a protective factor in oxidative stress has also been shown to be regulated via glucocorticoid mediated chromatin remodelling. In muscle cells, glucocorticoids activate UPC3 transcription via histone acetylation through the activity of KAT3B, and repress its transcription through the activity of the HDAC SIRT1 [139].
Indeed acetylation is an important regulator of GR at many levels. Intriguingly, GR activation requires HDAC1 to act as a co-activator (Table 2), in that HDAC1 is required for the induction of some genes by the GR [140], while HDAC6 has been shown to be important in the maturation of GR via heat shock protein 90 (Hsp90). In cells deficient for HDAC6, Hsp90-dependent maturation of the GR is compromised, resulting in reduced in ligand binding, altered nuclear translocation and aberrant transcriptional activation [141].
Using a synthetic glucocorticoid (dexamethasone), investigators have demonstrated that GR mediated repression of genes regulated by the pro-inflammatory cytokine interleukin-1β (IL-1β) utilize the activities of lysine acetyltransferases and HDACs. Initial studies found that dexamethasone inhibits IL-1β-induced acetylation at histone H4 lysine 8 at a target promoter in a concentration-dependent manner. Two mechanisms for this were identified. In one, GR acts as a direct inhibitor of KAT3A lysine acetyltransferase activity. In the other mechanism, GR recruited HDAC2 to the p65-KAT3A complex [142]. Additional studies determined that the major mechanism required the recruitment of HDAC2 to this p65-KAT3A complex [143].
Oxidative stress has been implicated in affecting glucocorticoid sensitivity in patients with COPD. One potential mechanism to explain this may be that levels of specific HDACs (in particular HDAC2) are decreased in the lungs of COPD patients (Table 2). Critically as a consequence, loss of GR acetylation induces glucocorticoid insensitivity towards NFκB-mediated gene expression [144], and restoration of HDAC2 by overexpression in glucocorticoid-insensitive alveolar macrophages from patients with COPD is able to restore glucocorticoid sensitivity [144].
Oxidative stress, acetylation and its role in the regulation of NFκB
Chronic inflammation is a pervasive element strongly associated with cancer [145]. Indeed, pro-inflammatory cascades are important elements in oxidative stress mediated responses in COPD [146] and NSCLC [147]. One of the critical mediators of pro-inflammatory cascade signalling pathways involves the activation or regulation of NFκB [148]. The NFκB/Rel family consists of five subunits, but NFκB typically consists of a heterodimeric protein comprised a p50 and a p65 (RelA) subunit.
One of the first indicators that oxidative stress affects NFκB activation within the lung came from studies in cells which showed that oxidative stress and TNF-α activated NFκB/AP-1 in a lung cancer cell line model [149], which resulted in induced histone acetylation and enhanced pro-inflammatory IL-8 release from these cells [149]. The activation of NFκB itself relies heavily on the activities of lysine acetyltransferases and HDACs (Fig. 1). Initial studies found that the lysine acetyltransferases KAT3B and KAT3A acted as key co-activators in regulating NFκB driven gene expression [150–152], through associations with the RelA/p65 subunit, whereas another lysine acetyltransferase KAT13A was found to also potentiate NFκB transactivation through association with the other subunit p50 [153]. Indeed within the setting of the lung, oxidative stress induces the expression of pro-inflammatory genes such as IL-6 and IL-8 via NFκB recruitment of lysine acetyltransferase activity of KAT3A or KAT3B to stimulate the transcription of these genes (Fig. 2) [154–156]. Once lysine acetyltransferases were found to associate with NFκB, interactions between HDACs and NFκB were quickly found to be necessary to occur in order to repress expression of NFκB regulated genes or to control the levels of expression of NFκB induced genes (Fig. 2) [157–159].
One of the critical regulators of NFκB transcriptional activation is I-Kappa-B Kinase-Alpha (IKKA) [160]. Cytoplasmic sequestration of p65 by IKBA is sufficient to translocate nuclear co-repressors silencing mediator for retinoic acid and thyroid hormone receptor (SMRT) to the cytoplasm [161]. As SMRT normally acts to repress transcription through association with HDACs [162], this results in derepressed transcription of its target genes. The association between p65 and IKKA’s ability to directly bind SMRT could be inhibited in a dose-dependent manner by the KAT3A lysine acetyltransferase further implicating a role for lysine acetylation in this process [163]. It was subsequently shown that IKKA activates NFκB mediated transcription by phosphorylating (SMRT), resulting in the exchange of transcriptional co-repressor (HDAC3) for co-activator (KAT3B) complexes, allowing KAT3B to acetylate RelA/p65 (Fig. 2) [164, 165].
Inhibition of NFκB activation has been shown whereby the protein Daxx binds to the region of RelA/p65 that contains the main sites of acetylation mediated by KAT3B/KAT3A [166]. Presumably, this may prevent acetylation of RelA/p65 by direct interference, but it must also be noted that Daxx itself has been shown to directly associate with HDAC2 [167], and so may represent a mechanism by which KATs and HDACs compete for critical lysines on NFκB subunits. In this regard, small ubiquitin-like modifier modification of KAT3A has been shown to negatively modulate its transcriptional potential by recruiting a Daxx complex containing HDAC2 [168].
Finally, cigarette smoke, a critical element in airway induced oxidative stress has been linked to NFκB activation in part through post-translational modification of HDAC1, HDAC2, and HDAC3 protein by nitrotyrosine and aldehyde-adduct formation [169]. SIRT1 has also been shown to regulate NFκB transactivation by physically associating with one of its subunits RelA/p65, and by removing acetylation at a critical lysine on RelA/p65 prevents NFκB dependent transcription [170]. Cigarette smoke has also been shown to down-regulate SIRT1 in the lung, resulting in increased acetylation of RelA/p65 with concomitant activation of NFκB mediated pro-inflammatory cytokine release [171].
A large literature now exists concerning the role of lysine acetylation in the regulation of NFκB, and for a comprehensive review of the role of KATs and HDACs in the regulation of NFκB the reader is directed to following review by Calao et al., [172].
Targeting epigenetic mechanisms in COPD and NSCLC
Epigenetic targeting remains an attractive target for pharmacological intervention in disease. Two main epigenetic therapeutic avenues have been extensively explored for the treatment of NSCLC. These are aimed at targeting DNA methylation via DNMT inhibitors (DNMTi), and at targeting histone acetylation via HDAC inhibitors. As DNA methylation is a common event in lung cancer [18–21], many in vitro studies involving DNMTi have been carried out demonstrating the loss of expression of critical genes in NSCLC and showing that such genes can be reactivated in response to DNMTi treatment [173–183]. Intriguingly it has recently been shown that DNA CpG hypermethylation and down-regulation of the oxidative stress sensor KEAP1 is common in NSCLC cell lines and tumour tissues, and can be reactivated following treatments with the DNMTi 5-aza-2′-dexoycytidine (Decitabine) [83].
We have recently extensively reviewed the potential for therapeutic targeting of HDACs in NSCLC and other disease conditions involving chronic inflammation in great detail and the reader is directed to the following reviews [184, 185]. Several of these epigenetic inhibitors have received FDA approval for the treatment of various diseases including Vorinostat (suberoylanilide hydroxamic acid, a HDi) and Dacogen (5-aza-2′-deoxycytidine, a DNMTi) and Vidaza (5-Azacytidine, a DNMTi) [186–188].
Theophylline – a novel agonist of HDACs
One of the most interesting recent new developments in relation to epigenetic targeting of COPD has been the discovery that theophylline an agent developed for the treatment of COPD can act as an agonist of HDACs [40]. As decreased HDAC activity has been observed in patients suffering from COPD [37], several clinical trials are in process to assess the clinical efficacy of this drug in both COPD and NSCLC (Table 3).
Table 3.
Clinical Trials involving epigenetic therapies in COPD and NSCLC
| Company | Intervention | Phase | Clinical trial identifier* | Published* |
|---|---|---|---|---|
| Ottawa Health Research Institute | Theophylline | III | NCT00299858 | |
| Ontario Thoracic Society | ||||
| University of Glasgow | Theophylline versus | III | NCT00119496 | [265] |
| GlaxoSmithKline | Rosiglitazone | |||
| Chest, Heart & Stroke Scotland | ||||
| Chief Scientists Office | ||||
| Hospital Universitari Son Dureta | Theophylline | I? | NCT00671151 | [266, 267] |
| Fondo de Investigación Sanitaria (FIS) | ||||
| Sociedad Española de Neumologìa y Cirugìa Torácica | ||||
| National Cancer Institute of Canada | Theophylline | III | NCT00003684 | |
| Imperial College London | Theophylline | II | NCT00241631 | [37] |
| Mitsubishi Tanabe Pharma Corporation | ||||
| Medical Research Council | ||||
| Assaf-Harofeh Medical Center | Theophylline | ? | NCT00893009 | |
| National Cancer Institute of Canada | Theophylline | III | NCT00003684 | |
| Argenta Discovery Ltd | Theophylline and Budesonide | II | NCT00634413 | |
| NCI – Center for Cancer Research-Medical Oncology | Decitabine and FR901228 | I | NCT00041158 | |
| National Cancer Institute (NCI) | ||||
| Arthur G. James Cancer Hospital and Richard J. Solove Research Institute | Decitabine and valproic acid | I | NCT00084981 | |
| NCI | ||||
| NCI – Center for Cancer Research-Medical Oncology | Decitabine | I | NCT00019825 | |
| NCI | ||||
| Sidney Kimmel Comprehensive Cancer Center | Azacitidine and MS-275 | I/II | NCT00387465 | |
| NCI | ||||
| Memorial Sloan-Kettering Cancer Center | Phenylbutyrate and Azacitidine | II | NCT00006019 | |
| NCI | ||||
| H. Lee Moffitt Cancer Center and Research Institute | LBH589 and Erlotinib | I/II | NCT00738751 | |
| Genentech | ||||
| Novartis | ||||
| NCI – Center for Cancer Research-Medical Oncology | MS-275 | I | NCT00020579 | |
| NCI | ||||
| Titan Pharmaceuticals | Pivanex | II | NCT00073385 | |
| Dartmouth-Hitchcock Medical Center | Vorinostat | ? | NCT00735826 | |
| Merck | ||||
| Fred Hutchinson Cancer Research Center | Vorinostat, Paclitaxel and radiation therapy | I/II | NCT00662311 | |
| NCI | ||||
| Nereus Pharmaceuticals, Inc. | NPI-0052 and Vorinostat | I | NCT00667082 | |
| Penn State University | Carboplatin, Paclitaxel, Bevacizumab and Vorinostat | I | NCT00702572 | |
| California Cancer Consortium | Carboplatin and Paclitaxel with or without Vorinostat | II | NCT00481078 | |
| NCI |
As found on clinicaltrials.gov (http://clinicaltrials.gov).
Current clinical trials involving epigenetic therapies in COPD and NSCLC
A large number of clinical trials in patients with COPD or NSCLC involving epigenetic therapies have been either completed or are ongoing (Table 3), and as these and other trials proceed, greater understanding of the potential side effects and dose-limiting toxicities of these drugs begins to emerge, and for the most part these first generation inhibitors have shown well-tolerated safety profiles [184].
Dietary HDAC inhibitors as therapeutic agents to target COPD or NSCLC?
Dietary interventions with cruciferous vegetables (and in particular Broccoli) may prove to have a critical role in the treatment of COPD or in preventing the development of lung cancer.
Sulforaphane
A compound found in broccoli, L-sulforaphane (SFN) was originally identified as a potential chemopreventative compound, which could protect against oxidative stress by activating phase II antioxidant enzymes [189]. Recent work has however found that this compound also acts as a HDAC inhibitor [190].
Evidence is now emerging that this compound may have efficacy in treating both COPD and NSCLC. In patients with COPD, loss of the DJ-1 protein has been shown to result in a decrease of NRF2 protein levels [85], and in vitro cell culture models where DJ-1 was either silenced by siRNA, or DJ-1 deficient mouse embryonic fibroblasts were employed found that treatments with sulforaphane up-regulated expression of NRF2 mediated antioxidant enzymes [85]. In NSCLC cell line models, sulforaphane has been shown to cause apoptosis by various mechanisms including binding to tubulin and disrupting microtubule polymerization [191–193], causing cell cycle arrest [194] and by sensitizing cells to tumours necrosis factor-related apoptosis-inducing ligand (TRAIL) [195]. Sulforaphane has also been shown to have efficacy in a mouse model. In this model, the A/J mouse model, following administration of tobacco carcinogens, lung adenomas appear during weeks 16 to 19 after administration, and adenocarcinomas occur generally during weeks 28 to 36. When the effects of sulforaphane on this model were tested, no effects on adenoma formation were observed but significant inhibition in the progression to adenocarcinomas was observed in the group fed sulforaphane, (1.5 μmol/g diet), compared to the control [196].
Curcumin
Another compound that has been shown to target epigenetic mechanisms may also play a role in the treatment of COPD is curcumin (diferuloylmethane). This is a compound derived from turmeric and has been shown to have potent anti-inflammatory properties and antioxidant properties [197, 198], and has been suggested to be a dietary intervention option in the treatment of inflammatory lung disease [199]. Curcumin has received a lot of attention as an ‘anti-cancer’ agent including studies on its potential effectiveness in lung cancer [200–203], and clinical trials have shown that it is well tolerated and lacks significant toxicity [204]. One of the mechanisms by which curcumin appears to act in its capacity as an anti-cancerous agent involves its ability to inhibit both lysine acetyltransferases [205–208], and HDACs [209, 210], although other studies have found that curcumin has no effect on HDACs [208].
Curcumin inhibits NFκB mediated responses either by either preventing the phosphorylation of IKBA [211], or by preventing its degradation [210], thereby reducing nuclear translocation NFκB.
It has also been suggested that curcumin may also prevent NFκB mediated chromatin remodelling through its ability to inhibit lysine acetyltransferases. The inhibition of NFκB in turn leads to the down-regulation of pro-inflammatory cytokines such as Chemokine, CXC Motif, Ligand CXCL1 and CXCL2 [212], or up-regulation of genes that control pro-inflammatory genes such as mitogen-activated protein kinase phosphatase-5 [213].
Curcumin and similar dietary compounds have been shown to induce HO-1 and NRF2 in breast cells [214] (Fig. 1), and so may also induce related antioxidant defences in COPD and NSCLC [215, 216]. Furthermore, curcumin has been shown to restore HDAC2 levels, and restore steroid sensitivity in monocytes exposed to cigarette smoke raising the possibility that this compound may have efficacy in the treatment of COPD [217]. Intriguingly, curcumin and L-sulphoraphane have been shown to act synergistically to down-regulate pro-inflammatory markers raising the possibility that these compounds could be combined in therapies targeting chronic inflammation such as COPD or NSCLC [218].
One reason for the differences observed in the mechanism of action of curcumin may relate to either the concentrations/doses used and or the cells targeted. Obviously, this may have profound effects on the potential clinical utility of this compound, and will require further evaluation.
Epigallocatechin 3-gallate (EGCG)
Another dietary polyphenol, which may prove of use in the treatment of COPD or NSCLC, is the green tea polyphenol EGCG. This compound has been shown to inhibit DNMTs [219, 220], and as such may function to affect epigenetic regulation of genes during oxidative stress. Indeed, EGCG has been shown to have efficacy in enhancing the expression of antioxidant genes in a mouse model of pulmonary fibrosis but the molecular mechanism underpinning this has yet to be determined [221]. Suggestive evidence has also emerged from a colon cancer study in mice where EGCG was found to up-regulate the expression of NRF2 [222], and gene expression arrays on the effect of EGCG on bronchial epithelial cells has identified genes affected by this compound [223]. In addition, EGCG has been shown to attenuate cigarette smoke induced NFκB activation in normal human bronchial epithelial cells [224], and has also been shown to inhibit IKK activation and IL-8 gene expression in NSCLC cells [225]. Several studies have shown the efficacy of EGCG as a potential chemopreventative treatment for NSCLC [226–240].
Caveats to the potential usefulness of EGCG in the treatment of pulmonary disease are that it has been linked to the development of asthma [241].
Can dietary epigenetic inhibitors work in the clinical setting?
Initial epidemiological evidence suggested a link between dietary isothiocyanate (of which sulforaphane is a member) intake and a reduced risk of developing lung cancer [242–246]. Strong evidence is now emerging within the clinical setting that broccoli or cruciferous vegetable consumption has a protective benefit in smokers, specifically in former smokers. In a placebo-controlled dose escalation trial to evaluate the effects of sulforaphane, on the expression of glutathione-s-transferase M1, glutathione-s-transferase P1, nicotinamide adenine dinucleotide phosphate (NADPH) quinone oxidoreductase and HO-1 in the upper airway of human subjects, using nasal lavage and RT-PCR, significant increases in expression of all the phase II enzymes compared to baseline were observed for the Sulforaphane treated patients, whereas in the control non-sulforaphane containing alfalfa sprouts, no changes to phase II enzymes were observed [247]. In a similar hospital-based, case-controlled study with lung cancer cases and controls matched on the basis of smoking status it was determined that among smokers, the protective effect of cruciferous vegetable intake ranged from a 20% reduction in risk to a 55% reduction in risk depending on the type of vegetable consumed and the duration and intensity of smoking. In particular, it was found that the consumption of raw cruciferous vegetables was associated with a risk reduction of lung cancer for current smokers. Additionally in patients with actual cancer, the strongest reduction was found for patients with either squamous or small-cell carcinoma, the two subtypes most strongly associated with heavy smoking [248]. Finally, a small cross-over study designed to examine the protective effect of broccoli intake in smokers and non-smokers, found that while broccoli intake (200 g/day) provided significant protection against oxidative stress induced DNA damage, no alterations to HDAC activity occurred over the trial period [249].
These results clearly indicate that dietary sulforaphane intake may have an important chemopreventative role in NSCLC, and may also be important in the treatment of COPD.
Caveats
Despite all the evidence that indicates that epigenetic mechanisms may be important therapeutic targets in the treatment of oxidative stress based conditions within the lung, there are some caveats which require consideration. For instance, in the lungs of rats treated with sulforaphanes glucosinolate precursor (glucoraphanin), levels of phase I carcinogen-activating enzymes, including activators of carcinogenic polycyclic aromatic hydrocarbons have been shown to be strongly induced suggesting that regular administration of glucoraphanin could actually increase rather than decrease lung cancer risk, especially in smokers as these individuals would be exposed to tobacco smoke related mutagens and carcinogens [250].
HDi have been shown to reactivate BRM [62], but inhibit its function, and subsequent removal of the HDi restores functional activity [63]. As such, it may be important to design staggered therapeutic interventions with HDi to allow the critical cellular functions of reactivated genes to take effect.
Issues of bioavailability may also be encountered in relation to efficacy of dietary epigenetic interventions in the treatment of COPD and NSCLC. For instance it is well established that curcumin is not easily bioavailable [251]. In a recent phase II clinical trial in patients with advanced pancreatic cancer, even though patients received up to 8 g of curcumin by mouth daily until disease progression, with restaging every 2 months, the bioavailable levels of curcumin achieved in plasma were within the range of 22 to 41 ng/ml [252]. However, curcumin bioavailability has been increased through heat treatment [253], or by the synthesis of novel derivatives [254], and should lead to better compounds for use in the clinical setting. In contrast the bioavailability of SFN is increased if cruciferous vegetables are eaten raw rather than heat treated [255–257]. It is clear that further efforts are required to understand the bioavailability and pharmacokinetics of these dietary derived natural compounds. For instance, in a recent study in human volunteers it was found that repeated ingestion of broccoli did not lead to higher plasma levels of SFN [258].
Finally, while HDi may be beneficial for the treatment of disease, they may not be of great use in targeting oxidative stress induced disease. In this regard, they have been shown to induce apoptosis in cancer cells by inducing oxidative stress [259–261].
Lung cancer and COPD are leading causes of morbidity and mortality in the United States and worldwide. They share a common environmental risk factor in cigarette smoke exposure and a genetic predisposition represented by the incidence of these diseases in only a fraction of smokers. The presence of COPD increases the risk of lung cancer up to 4.5-fold. However, in addition one outstanding issue concerns the fact that a small number of patients who develop lung cancer or COPD are non-smokers raising the possibility that they may be (i) completely different diseases which will require different treatments or (ii) there are genetic/epigenetic susceptibilities or environmental exposures that could drive similar changes in ROS stress and epigenetic modifications without the need for cigarette smoke exposure. We feel that currently we are not yet in a position to distinguish between these two possibilities. At a recent workshop held by the National Heart, Lung, and Blood Institute (NHLBI) and the National Cancer Institute (NCI), it was decided that future studies should have as their main objectives: (1) to clarify common epidemiological characteristics of lung cancer and COPD; (2) to identify shared genetic and epigenetic risk factors; (3) to identify and validate biomarkers, molecular signatures and imaging-derived measurements of each disease and (4) to determine common and disparate pathogenetic mechanisms [262].
Another area of concern pertains to the different effects of cigarette smoke on cells. This may be due to different cell types, different degrees of antioxidant capacity in the different cell types or are other factors such as chemical and environmental carcinogens may also play important roles? Indeed, some chemical carcinogens such as Benzo[a]pyrene (B[a]P) are present in both the environment and cigarette smoke, and this compound has been shown to increase cellular proliferation in lung cancer cells [10]. Small airways in patients with COPD also have evidence for proliferative responses, particularly with respect to smooth muscle and to epithelial cells [263, 264]. Clearly, future studies will be required to molecular dissect the genetic epigenetic and molecular alterations underpinning these differential responses.
Conclusions
It is clearly evident that the hypoxic environment encountered in COPD and NSCLC results in the development of oxidative stress. Throughout this review we have linked epigenetics as a mechanism by which cells regulate oxidative stress responsive genes and pathways. A fine balance between the epigenetic regulatory mechanisms and their intended targets to both dampen pro-inflammatory conditions and control cellular oxidative stress pathways exists (summarized in Fig. 1), which if perturbed may lead to aberrant epigenetic control and subsequently to exacerbations to pre-existing conditions (e.g. COPD), and which may in the end result in tumourigenesis.
Throughout this review we have examined how epigenetics may be involved in the regulation of oxidative stress responses on many levels and via various pathways. It is clear that they are intimately linked as shown figuratively in Fig. 2. Finally, we have discussed the current potential for targeted epigenetic therapies for the treatment of COPD or NSCLC, either via pharmacological intervention or through the use of dietary chemopreventatives. The available data would indicate that there is compelling evidence for further studies into epigenetic intervention strategies to treat diseases associated with oxidative stress.
References
- 1.Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31:1334–56. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- 2.Kiri VA, Fabbri LM, Davis KJ, et al. Inhaled corticosteroids and risk of lung cancer among COPD patients who quit smoking. Respir Med. 2008;103:85–90. doi: 10.1016/j.rmed.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Houghton AM, Mouded M, Shapiro SD. Common origins of lung cancer and COPD. Nat Med. 2008;14:1023–4. doi: 10.1038/nm1008-1023. [DOI] [PubMed] [Google Scholar]
- 4.Turner MC, Chen Y, Krewski D, et al. Chronic obstructive pulmonary disease is associated with lung cancer mortality in a prospective study of never smokers. Am J Respir Crit Care Med. 2007;176:285–90. doi: 10.1164/rccm.200612-1792OC. [DOI] [PubMed] [Google Scholar]
- 5.Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers – a different disease. Nat Rev Cancer. 2007;7:778–90. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 6.Sato M, Shames DS, Gazdar AF, et al. A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol. 2007;2:327–43. doi: 10.1097/01.JTO.0000263718.69320.4c. [DOI] [PubMed] [Google Scholar]
- 7.Stampfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol. 2009;9:377–84. doi: 10.1038/nri2530. [DOI] [PubMed] [Google Scholar]
- 8.Kaushik G, Kaushik T, Khanduja S, et al. Cigarette smoke condensate promotes cell proliferation through disturbance in cellular redox homeostasis of transformed lung epithelial type-II cells. Cancer Lett. 2008;270:120–31. doi: 10.1016/j.canlet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 9.Izzotti A, Calin GA, Arrigo P, et al. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009;23:806–12. doi: 10.1096/fj.08-121384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kometani T, Yoshino I, Miura N, et al. Benzo[a]pyrene promotes proliferation of human lung cancer cells by accelerating the epidermal growth factor receptor signaling pathway. Cancer Lett. 2009;278:27–33. doi: 10.1016/j.canlet.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Khan EM, Lanir R, Danielson AR, et al. Epidermal growth factor receptor exposed to cigarette smoke is aberrantly activated and undergoes perinuclear trafficking. FASEB J. 2008;22:910–7. doi: 10.1096/fj.06-7729com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das A, Bhattacharya A, Chakrabarti G. Cigarette smoke extract induces disruption of structure and function of tubulin-microtubule in lung epithelium cells and in vitro. Chem Res Toxicol. 2009;22:446–59. doi: 10.1021/tx8002142. [DOI] [PubMed] [Google Scholar]
- 13.Tsuji T, Aoshiba K, Nagai A. Cigarette smoke induces senescence in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2004;31:643–9. doi: 10.1165/rcmb.2003-0290OC. [DOI] [PubMed] [Google Scholar]
- 14.Yao H, Yang SR, Edirisinghe I, et al. Disruption of p21 attenuates lung inflammation induced by cigarette smoke, LPS, and fMLP in mice. Am J Respir Cell Mol Biol. 2008;39:7–18. doi: 10.1165/rcmb.2007-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marwick JA, Stevenson CS, Giddings J, et al. Cigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling complex in rat lungs and patients with COPD: morphological impact of VEGFR-2 inhibition. Am J Physiol Lung Cell Mol Physiol. 2006;290:L897–908. doi: 10.1152/ajplung.00116.2005. [DOI] [PubMed] [Google Scholar]
- 16.Edirisinghe I, Yang SR, Yao H, et al. VEGFR-2 inhibition augments cigarette smoke-induced oxidative stress and inflammatory responses leading to endothelial dysfunction. FASEB J. 2008;22:2297–310. doi: 10.1096/fj.07-099481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodge S, Hodge G, Ahern J, et al. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2007;37:748–55. doi: 10.1165/rcmb.2007-0025OC. [DOI] [PubMed] [Google Scholar]
- 18.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–80. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Risch A, Plass C. Lung cancer epigenetics and genetics. Int J Cancer. 2008;123:1–7. doi: 10.1002/ijc.23605. [DOI] [PubMed] [Google Scholar]
- 20.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–17. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 21.Toyooka S, Matsuo K, Gazdar AF. DNA methylation in lung cancer. N Engl J Med. 2008;358:2513–4. doi: 10.1056/NEJMc080835. [DOI] [PubMed] [Google Scholar]
- 22.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 23.Damiani LA, Yingling CM, Leng S, et al. Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res. 2008;68:9005–14. doi: 10.1158/0008-5472.CAN-08-1276. [DOI] [PubMed] [Google Scholar]
- 24.Barlesi F, Giaccone G, Gallegos-Ruiz MI, et al. Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol. 2007;25:4358–64. doi: 10.1200/JCO.2007.11.2599. [DOI] [PubMed] [Google Scholar]
- 25.Van Den Broeck A, Brambilla E, Moro-Sibilot D, et al. Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clin Cancer Res. 2008;14:7237–45. doi: 10.1158/1078-0432.CCR-08-0869. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe H, Soejima K, Yasuda H, et al. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008 doi: 10.1186/1475-2867-8-15. ; doi: 10.1186/1475-2867-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnes PJ. Role of HDAC2 in the Pathophysiology of COPD. Annu Rev Physiol. 2008;71:451–64. doi: 10.1146/annurev.physiol.010908.163257. [DOI] [PubMed] [Google Scholar]
- 28.Ciencewicki J, Trivedi S, Kleeberger SR. Oxidants and the pathogenesis of lung diseases. J Allergy Clin Immunol. 2008;122:456–68. doi: 10.1016/j.jaci.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q, Xiao H, Isobe K. Histone acetyltransferase activities of cAMP-regulated enhancer-binding protein and p300 in tissues of fetal, young, and old mice. J Gerontol A Biol Sci Med Sci. 2002;57:B93–8. doi: 10.1093/gerona/57.3.b93. [DOI] [PubMed] [Google Scholar]
- 30.Yamauchi T, Yamauchi J, Kuwata T, et al. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc Natl Acad Sci USA. 2000;97:11303–6. doi: 10.1073/pnas.97.21.11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phan HM, Xu AW, Coco C, et al. GCN5 and p300 share essential functions during early embryogenesis. Dev Dyn. 2005;233:1337–47. doi: 10.1002/dvdy.20445. [DOI] [PubMed] [Google Scholar]
- 32.Naltner A, Wert S, Whitsett JA, et al. Temporal/spatial expression of nuclear receptor coactivators in the mouse lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1066–74. doi: 10.1152/ajplung.2000.279.6.L1066. [DOI] [PubMed] [Google Scholar]
- 33.Partanen A, Motoyama J, Hui CC. Developmentally regulated expression of the transcriptional cofactors/histone acetyltransferases CBP and p300 during mouse embryogenesis. Int J Dev Biol. 1999;43:487–94. [PubMed] [Google Scholar]
- 34.Xu W, Edmondson DG, Evrard YA, et al. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat Genet. 2000;26:229–32. doi: 10.1038/79973. [DOI] [PubMed] [Google Scholar]
- 35.Shikama N, Lutz W, Kretzschmar R, et al. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J. 2003;22:5175–85. doi: 10.1093/emboj/cdg502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin Z, Gonzales L, Kolla V, et al. Hop functions downstream of Nkx2.1 and GATA6 to mediate HDAC-dependent negative regulation of pulmonary gene expression. Am J Physiol Lung Cell Mol Physiol. 2006;291:L191–9. doi: 10.1152/ajplung.00385.2005. [DOI] [PubMed] [Google Scholar]
- 37.Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–76. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 38.Szulakowski P, Crowther AJ, Jimenez LA, et al. The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174:41–50. doi: 10.1164/rccm.200505-725OC. [DOI] [PubMed] [Google Scholar]
- 39.Ito K, Caramori G, Lim S, et al. Expression and activity of histone deacetylases in human asthmatic airways. Am J Respir Crit Care Med. 2002;166:392–6. doi: 10.1164/rccm.2110060. [DOI] [PubMed] [Google Scholar]
- 40.Ito K, Lim S, Caramori G, et al. A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc Natl Acad Sci USA. 2002;99:8921–6. doi: 10.1073/pnas.132556899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito K, Lim S, Caramori G, et al. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J. 2001;15:1110–2. [PubMed] [Google Scholar]
- 42.Ito K, Hanazawa T, Tomita K, et al. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun. 2004;315:240–5. doi: 10.1016/j.bbrc.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 43.Rajendrasozhan S, Yang SR, Kinnula VL, et al. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:861–70. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo X, Lin HM, Lin Z, et al. Surfactant protein gene A, B, and D marker alleles in chronic obstructive pulmonary disease of a Mexican population. Eur Respir J. 2001;18:482–90. doi: 10.1183/09031936.01.00043401. [DOI] [PubMed] [Google Scholar]
- 45.Behera D, Balamugesh T, Venkateswarlu D, et al. Serum surfactant protein-A levels in chronic bronchitis and its relation to smoking. Indian J Chest Dis Allied Sci. 2005;47:13–7. [PubMed] [Google Scholar]
- 46.Kobayashi H, Kanoh S, Motoyoshi K. Serum surfactant protein-A, but not surfactant protein-D or KL-6, can predict preclinical lung damage induced by smoking. Biomarkers. 2008;13:385–92. doi: 10.1080/13547500801903651. [DOI] [PubMed] [Google Scholar]
- 47.Ohlmeier S, Vuolanto M, Toljamo T, et al. Proteomics of human lung tissue identifies surfactant protein a as a marker of chronic obstructive pulmonary disease. J Proteome Res. 2008;7:5125–32. doi: 10.1021/pr800423x. [DOI] [PubMed] [Google Scholar]
- 48.Bruno MD, Korfhagen TR, Liu C, et al. GATA-6 activates transcription of surfactant protein A. J Biol Chem. 2000;275:1043–9. doi: 10.1074/jbc.275.2.1043. [DOI] [PubMed] [Google Scholar]
- 49.Henning LN, Azad AK, Parsa KV, et al. Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J Immunol. 2008;180:7847–58. doi: 10.4049/jimmunol.180.12.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sasaki H, Moriyama S, Nakashima Y, et al. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer. 2004;46:171–8. doi: 10.1016/j.lungcan.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 51.Nakagawa M, Oda Y, Eguchi T, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–74. [PubMed] [Google Scholar]
- 52.Bartling B, Hofmann HS, Boettger T, et al. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer. 2005;49:145–54. doi: 10.1016/j.lungcan.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 53.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–18. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki H, Ouchida M, Yamamoto H, et al. Decreased expression of the SIN3A gene, a candidate tumor suppressor located at the prevalent allelic loss region 15q23 in non-small cell lung cancer. Lung Cancer. 2008;59:24–31. doi: 10.1016/j.lungcan.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 55.Watanabe H, Mizutani T, Haraguchi T, et al. SWI/SNF complex is essential for NRSF-mediated suppression of neuronal genes in human nonsmall cell lung carcinoma cell lines. Oncogene. 2006;25:470–9. doi: 10.1038/sj.onc.1209068. [DOI] [PubMed] [Google Scholar]
- 56.Reisman DN, Sciarrotta J, Wang W, et al. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res. 2003;63:560–6. [PubMed] [Google Scholar]
- 57.Girard L, Zochbauer-Muller S, Virmani AK, et al. Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res. 2000;60:4894–906. [PubMed] [Google Scholar]
- 58.Wong AK, Shanahan F, Chen Y, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 2000;60:6171–7. [PubMed] [Google Scholar]
- 59.Medina PP, Carretero J, Fraga MF, et al. Genetic and epigenetic screening for gene alterations of the chromatin-remodeling factor, SMARCA4/BRG1, in lung tumors. Genes Chromosomes Cancer. 2004;41:170–7. doi: 10.1002/gcc.20068. [DOI] [PubMed] [Google Scholar]
- 60.Medina PP, Romero OA, Kohno T, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat. 2008;29:617–22. doi: 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- 61.Bourachot B, Yaniv M, Muchardt C. Growth inhibition by the mammalian SWI-SNF subunit Brm is regulated by acetylation. EMBO J. 2003;22:6505–15. doi: 10.1093/emboj/cdg621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamamichi N, Yamamichi-Nishina M, Mizutani T, et al. The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene. 2005;24:5471–81. doi: 10.1038/sj.onc.1208716. [DOI] [PubMed] [Google Scholar]
- 63.Glaros S, Cirrincione GM, Muchardt C, et al. The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene. 2007;26:7058–66. doi: 10.1038/sj.onc.1210514. [DOI] [PubMed] [Google Scholar]
- 64.Fukuoka J, Fujii T, Shih JH, et al. Chromatin remodeling factors and BRM/RG1 expression as prognostic indicators in non-small cell lung cancer. Clin Cancer Res. 2004;10:4314–24. doi: 10.1158/1078-0432.CCR-03-0489. [DOI] [PubMed] [Google Scholar]
- 65.Sasaki H, Moriyama S, Nakashima Y, et al. Expression of the MTA1 mRNA in advanced lung cancer. Lung Cancer. 2002;35:149–54. doi: 10.1016/s0169-5002(01)00329-4. [DOI] [PubMed] [Google Scholar]
- 66.Iguchi H, Imura G, Toh Y, et al. Expression of MTA1, a metastasis-associated gene with histone deacetylase activity in pancreatic cancer. Int J Oncol. 2000;16:1211–4. doi: 10.3892/ijo.16.6.1211. [DOI] [PubMed] [Google Scholar]
- 67.Mazumdar A, Wang RA, Mishra SK, et al. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol. 2001;3:30–7. doi: 10.1038/35050532. [DOI] [PubMed] [Google Scholar]
- 68.Xue Y, Wong J, Moreno GT, et al. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–61. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 69.Toh Y, Kuninaka S, Endo K, et al. Molecular analysis of a candidate metastasis-associated gene, MTA1: possible interaction with histone deacetylase 1. J Exp Clin Cancer Res. 2000;19:105–11. [PubMed] [Google Scholar]
- 70.Yao YL, Yang WM. The metastasis-associated proteins 1 and 2 form distinct protein complexes with histone deacetylase activity. J Biol Chem. 2003;278:42560–8. doi: 10.1074/jbc.M302955200. [DOI] [PubMed] [Google Scholar]
- 71.You A, Tong JK, Grozinger CM, et al. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc Natl Acad Sci USA. 2001;98:1454–8. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kishimoto M, Kohno T, Okudela K, et al. Mutations and deletions of the CBP gene in human lung cancer. Clin Cancer Res. 2005;11:512–9. [PubMed] [Google Scholar]
- 73.Yoon KA, Hwangbo B, Kim IJ, et al. Novel polymorphisms in the SUV39H2 histone methyltransferase and the risk of lung cancer. Carcinogenesis. 2006;27:2217–22. doi: 10.1093/carcin/bgl084. [DOI] [PubMed] [Google Scholar]
- 74.Yoon KA, Park S, Hwangbo B, et al. Genetic polymorphisms in the Rb-binding zinc finger gene RIZ and the risk of lung cancer. Carcinogenesis. 2007;28:1971–7. doi: 10.1093/carcin/bgm156. [DOI] [PubMed] [Google Scholar]
- 75.Jang JS, Lee SJ, Choi JE, et al. Methyl-CpG binding domain 1 gene polymorphisms and risk of primary lung cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:2474–80. doi: 10.1158/1055-9965.EPI-05-0423. [DOI] [PubMed] [Google Scholar]
- 76.El-Osta A, Baker EK, Wolffe AP. Profiling methyl-CpG specific determinants on transcriptionally silent chromatin. Mol Biol Rep. 2001;28:209–15. doi: 10.1023/a:1015744625049. [DOI] [PubMed] [Google Scholar]
- 77.Wang F, Nguyen M, Qin FX, et al. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–14. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 78.Sundaresan NR, Samant SA, Pillai VB, et al. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008;28:6384–401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Igarashi K, Sun J. The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal. 2006;8:107–18. doi: 10.1089/ars.2006.8.107. [DOI] [PubMed] [Google Scholar]
- 80.Dohi Y, Ikura T, Hoshikawa Y, et al. Bach1 inhibits oxidative stress-induced cellular senescence by impeding p53 function on chromatin. Nat Struct Mol Biol. 2008;15:1246–54. doi: 10.1038/nsmb.1516. [DOI] [PubMed] [Google Scholar]
- 81.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 82.Spiegelman BM. Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found Symp. 2007;287:60–3. [PubMed] [Google Scholar]
- 83.Wang R, An J, Ji F, et al. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun. 2008;373:151–4. doi: 10.1016/j.bbrc.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 84.Ohta T, Iijima K, Miyamoto M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–9. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 85.Malhotra D, Thimmulappa R, Navas-Acien A, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. 2008;178:592–604. doi: 10.1164/rccm.200803-380OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86.Goven D, Boutten A, Lecon-Malas V, et al. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax. 2008;63:916–24. doi: 10.1136/thx.2007.091181. [DOI] [PubMed] [Google Scholar]
- 87.Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8:76–87. doi: 10.1089/ars.2006.8.76. [DOI] [PubMed] [Google Scholar]
- 88.Katoh Y, Itoh K, Yoshida E, et al. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6:857–68. doi: 10.1046/j.1365-2443.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- 89.Lin W, Shen G, Yuan X, et al. Regulation of Nrf2 transactivation domain activity by p160 RAC3/SRC3 and other nuclear co-regulators. J Biochem Mol Biol. 2006;39:304–10. doi: 10.5483/bmbrep.2006.39.3.304. [DOI] [PubMed] [Google Scholar]
- 90.Zhang J, Ohta T, Maruyama A, et al. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Mol Cell Biol. 2006;26:7942–52. doi: 10.1128/MCB.00700-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta. 2008;1783:713–27. doi: 10.1016/j.bbamcr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 92.Ryter SW, Kim HP, Nakahira K, et al. Protective functions of heme oxygenase-1 and carbon monoxide in the respiratory system. Antioxid Redox Signal. 2007;9:2157–73. doi: 10.1089/ars.2007.1811. [DOI] [PubMed] [Google Scholar]
- 93.Sun J, Brand M, Zenke Y, et al. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci USA. 2004;101:1461–6. doi: 10.1073/pnas.0308083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang J, Hosoya T, Maruyama A, et al. Nrf2 Neh5 domain is differentially utilized in the transactivation of cytoprotective genes. Biochem J. 2007;404:459–66. doi: 10.1042/BJ20061611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–43. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 96.Goudar RK, Vlahovic G. Hypoxia, angiogenesis, and lung cancer. Curr Oncol Rep. 2008;10:277–82. doi: 10.1007/s11912-008-0043-6. [DOI] [PubMed] [Google Scholar]
- 97.Raguso CA, Guinot SL, Janssens JP, et al. Chronic hypoxia: common traits between chronic obstructive pulmonary disease and altitude. Curr Opin Clin Nutr Metab Care. 2004;7:411–7. doi: 10.1097/01.mco.0000134372.78438.09. [DOI] [PubMed] [Google Scholar]
- 98.Le QT, Chen E, Salim A, et al. An evaluation of tumor oxygenation and gene expression in patients with early stage non-small cell lung cancers. Clin Cancer Res. 2006;12:1507–14. doi: 10.1158/1078-0432.CCR-05-2049. [DOI] [PubMed] [Google Scholar]
- 99.Le QT, Shi G, Cao H, et al. Galectin-1: a link between tumor hypoxia and tumor immune privilege. J Clin Oncol. 2005;23:8932–41. doi: 10.1200/JCO.2005.02.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Swinson DE, Jones JL, Cox G, et al. Hypoxia-inducible factor-1 alpha in non small cell lung cancer: relation to growth factor, protease and apoptosis pathways. Int J Cancer. 2004;111:43–50. doi: 10.1002/ijc.20052. [DOI] [PubMed] [Google Scholar]
- 101.Swinson DE, Jones JL, Richardson D, et al. Carbonic anhydrase IX expression, a novel surrogate marker of tumor hypoxia, is associated with a poor prognosis in non-small-cell lung cancer. J Clin Oncol. 2003;21:473–82. doi: 10.1200/JCO.2003.11.132. [DOI] [PubMed] [Google Scholar]
- 102.Kamlah F, Eul BG, Li S, et al. Intravenous injection of siRNA directed against hypoxia-inducible factors prolongs survival in a Lewis lung carcinoma cancer model. Cancer Gene Ther. 2009;16:195–205. doi: 10.1038/cgt.2008.71. [DOI] [PubMed] [Google Scholar]
- 103.Polosukhin VV, Lawson WE, Milstone AP, et al. Association of progressive structural changes in the bronchial epithelium with subepithelial fibrous remodeling: a potential role for hypoxia. Virchows Arch. 2007;451:793–803. doi: 10.1007/s00428-007-0469-5. [DOI] [PubMed] [Google Scholar]
- 104.Pialoux V, Mounier R, Brown AD, et al. Relationship between oxidative stress and HIF-1alpha mRNA during sustained hypoxia in humans. Free Radic Biol Med. 2009;46:321–6. doi: 10.1016/j.freeradbiomed.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 105.Pollard PJ, Loenarz C, Mole DR, et al. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem J. 2008;416:387–94. doi: 10.1042/BJ20081238. [DOI] [PubMed] [Google Scholar]
- 106.Beyer S, Kristensen MM, Jensen KS, et al. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem. 2008;283:36542–52. doi: 10.1074/jbc.M804578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wellmann S, Bettkober M, Zelmer A, et al. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem Biophys Res Commun. 2008;372:892–7. doi: 10.1016/j.bbrc.2008.05.150. [DOI] [PubMed] [Google Scholar]
- 108.Kim SH, Jeong JW, Park JA, et al. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol Rep. 2007;17:647–51. [PubMed] [Google Scholar]
- 109.Kasper LH, Brindle PK. Mammalian gene expression program resiliency: the roles of multiple coactivator mechanisms in hypoxia-responsive transcription. Cell Cycle. 2006;5:142–6. doi: 10.4161/cc.5.2.2353. [DOI] [PubMed] [Google Scholar]
- 110.Qian DZ, Kachhap SK, Collis SJ, et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66:8814–21. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 111.Kato H, Tamamizu-Kato S, Shibasaki F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem. 2004;279:41966–74. doi: 10.1074/jbc.M406320200. [DOI] [PubMed] [Google Scholar]
- 112.Moon HE, Cheon H, Chun KH, et al. Metastasis-associated protein 1 enhances angiogenesis by stabilization of HIF-1alpha. Oncol Rep. 2006;16:929–35. [PubMed] [Google Scholar]
- 113.Yoo YG, Kong G, Lee MO. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J. 2006;25:1231–41. doi: 10.1038/sj.emboj.7601025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim SH, Kim KW, Jeong JW. Inhibition of hypoxia-induced angiogenesis by sodium butyrate, a histone deacetylase inhibitor, through hypoxia-inducible factor-1alpha suppression. Oncol Rep. 2007;17:793–7. [PubMed] [Google Scholar]
- 115.Liang D, Kong X, Sang N. Effects of histone deacetylase inhibitors on HIF-1. Cell Cycle. 2006;5:2430–5. doi: 10.4161/cc.5.21.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fath DM, Kong X, Liang D, et al. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J Biol Chem. 2006;281:13612–9. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kong X, Lin Z, Liang D, et al. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26:2019–28. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mie Lee Y, Kim SH, Kim HS, et al. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1alpha activity. Biochem Biophys Res Commun. 2003;300:241–6. doi: 10.1016/s0006-291x(02)02787-0. [DOI] [PubMed] [Google Scholar]
- 119.Xenaki G, Ontikatze T, Rajendran R, et al. PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene. 2008;27:5785–96. doi: 10.1038/onc.2008.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ruas JL, Poellinger L, Pereira T. Role of CBP in regulating HIF-1-mediated activation of transcription. J Cell Sci. 2005;118:301–11. doi: 10.1242/jcs.01617. [DOI] [PubMed] [Google Scholar]
- 121.Ruas JL, Poellinger L, Pereira T. Functional analysis of hypoxia-inducible factor-1 alpha-mediated transactivation. Identification of amino acid residues critical for transcriptional activation and/or interaction with CREB-binding protein. J Biol Chem. 2002;277:38723–30. doi: 10.1074/jbc.M205051200. [DOI] [PubMed] [Google Scholar]
- 122.Carrero P, Okamoto K, Coumailleau P, et al. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol. 2000;20:402–15. doi: 10.1128/mcb.20.1.402-415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kasper LH, Boussouar F, Boyd K, et al. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005;24:3846–58. doi: 10.1038/sj.emboj.7600846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Puigserver P, Wu Z, Park CW, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–39. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 125.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–22. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–60. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 127.Puigserver P, Adelmant G, Wu Z, et al. Activation of PPARgamma coactivator-1 through transcription factor docking. Science. 1999;286:1368–71. doi: 10.1126/science.286.5443.1368. [DOI] [PubMed] [Google Scholar]
- 128.St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 129.Kukidome D, Nishikawa T, Sonoda K, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55:120–7. [PubMed] [Google Scholar]
- 130.St-Pierre J, Lin J, Krauss S, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278:26597–603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 131.Valle I, Alvarez-Barrientos A, Arza E, et al. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–73. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 132.Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol. 2000;35:811–20. doi: 10.1016/s0531-5565(00)00135-2. [DOI] [PubMed] [Google Scholar]
- 133.Bezaire V, Seifert EL, Harper ME. Uncoupling protein-3: clues in an ongoing mitochondrial mystery. FASEB J. 2007;21:312–24. doi: 10.1096/fj.06-6966rev. [DOI] [PubMed] [Google Scholar]
- 134.Borniquel S, Valle I, Cadenas S, et al. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. 2006;20:1889–91. doi: 10.1096/fj.05-5189fje. [DOI] [PubMed] [Google Scholar]
- 135.Quon BS, Gan WQ, Sin DD. Contemporary management of acute exacerbations of COPD: a systematic review and metaanalysis. Chest. 2008;133:756–66. doi: 10.1378/chest.07-1207. [DOI] [PubMed] [Google Scholar]
- 136.Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. 2008;134:394–401. doi: 10.1378/chest.08-0440. [DOI] [PubMed] [Google Scholar]
- 137.Wu Y, Adam S, Hamann L, et al. Accumulation of inhibitory kappaB-alpha as a mechanism contributing to the anti-inflammatory effects of surfactant protein-A. Am J Respir Cell Mol Biol. 2004;31:587–94. doi: 10.1165/rcmb.2004-0003OC. [DOI] [PubMed] [Google Scholar]
- 138.Islam KN, Mendelson CR. Glucocorticoid/glucocorticoid receptor inhibition of surfactant protein-A (SP-A) gene expression in lung type II cells is mediated by repressive changes in histone modification at the SP-A promoter. Mol Endocrinol. 2008;22:585–96. doi: 10.1210/me.2007-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Amat R, Solanes G, Giralt M, et al. SIRT1 is involved in glucocorticoid-mediated control of uncoupling protein-3 gene transcription. J Biol Chem. 2007;282:34066–76. doi: 10.1074/jbc.M707114200. [DOI] [PubMed] [Google Scholar]
- 140.Qiu Y, Zhao Y, Becker M, et al. HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Mol Cell. 2006;22:669–79. doi: 10.1016/j.molcel.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 141.Kovacs JJ, Murphy PJ, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 142.Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ito K, Jazrawi E, Cosio B, et al. p65-activated histone acetyltransferase activity is repressed by glucocorticoids: mifepristone fails to recruit HDAC2 to the p65-HAT complex. J Biol Chem. 2001;276:30208–15. doi: 10.1074/jbc.M103604200. [DOI] [PubMed] [Google Scholar]
- 144.Ito K, Yamamura S, Essilfie-Quaye S, et al. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373–80. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- 146.De Boer WI, Yao H, Rahman I. Future therapeutic treatment of COPD: struggle between oxidants and cytokines. Int J Chron Obstruct Pulmon Dis. 2007;2:205–28. [PMC free article] [PubMed] [Google Scholar]
- 147.Azad N, Rojanasakul Y, Vallyathan V. Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev. 2008;11:1–15. doi: 10.1080/10937400701436460. [DOI] [PubMed] [Google Scholar]
- 148.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rahman I, Gilmour PS, Jimenez LA, et al. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 2002;234–235:239–48. [PubMed] [Google Scholar]
- 150.Gerritsen ME, Williams AJ, Neish AS, et al. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA. 1997;94:2927–32. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Perkins ND, Felzien LK, Betts JC, et al. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–7. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 152.Wadgaonkar R, Phelps KM, Haque Z, et al. CREB-binding protein is a nuclear integrator of nuclear factor-kappaB and p53 signaling. J Biol Chem. 1999;274:1879–82. doi: 10.1074/jbc.274.4.1879. [DOI] [PubMed] [Google Scholar]
- 153.Na SY, Lee SK, Han SJ, et al. Steroid receptor coactivator-1 interacts with the p50 subunit and coactivates nuclear factor kappaB-mediated transactivations. J Biol Chem. 1998;273:10831–4. doi: 10.1074/jbc.273.18.10831. [DOI] [PubMed] [Google Scholar]
- 154.Vanden Berghe W, De Bosscher K, Boone E, et al. The nuclear factor-kappaB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J Biol Chem. 1999;274:32091–8. doi: 10.1074/jbc.274.45.32091. [DOI] [PubMed] [Google Scholar]
- 155.Bartling TR, Drumm ML. Oxidative Stress Causes IL8 Promoter Hyperacetylation in Cystic Fibrosis Airway Cell Models. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2007-0464OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rajendrasozhan S, Yang SR, Edirisinghe I, et al. Deacetylases and NF-kappaB in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid Redox Signal. 2008;10:799–811. doi: 10.1089/ars.2007.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lee SK, Kim JH, Lee YC, et al. Silencing mediator of retinoic acid and thyroid hormone receptors, as a novel transcriptional corepressor molecule of activating protein-1, nuclear factor-kappaB, and serum response factor. J Biol Chem. 2000;275:12470–4. doi: 10.1074/jbc.275.17.12470. [DOI] [PubMed] [Google Scholar]
- 158.Chen L, Fischle W, Verdin E, et al. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–7. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 159.Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–77. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Karin M. The IkappaB kinase - a bridge between inflammation and cancer. Cell Res. 2008;18:334–42. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- 161.Espinosa L, Santos S, Ingles-Esteve J, et al. p65-NFkappaB synergizes with Notch to activate transcription by triggering cytoplasmic translocation of the nuclear receptor corepressor N-CoR. J Cell Sci. 2002;115:1295–303. doi: 10.1242/jcs.115.6.1295. [DOI] [PubMed] [Google Scholar]
- 162.Nagy L, Kao HY, Chakravarti D, et al. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–80. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 163.Espinosa L, Ingles-Esteve J, Robert-Moreno A, et al. IkappaBalpha and p65 regulate the cytoplasmic shuttling of nuclear corepressors: cross-talk between Notch and NFkappaB pathways. Mol Biol Cell. 2003;14:491–502. doi: 10.1091/mbc.E02-07-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hoberg JE, Popko AE, Ramsey CS, et al. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26:457–71. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hoberg JE, Yeung F, Mayo MW. SMRT derepression by the IkappaB kinase alpha: a prerequisite to NF-kappaB transcription and survival. Mol Cell. 2004;16:245–55. doi: 10.1016/j.molcel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 166.Park J, Lee JH, La M, et al. Inhibition of NF-kappaB acetylation and its transcriptional activity by Daxx. J Mol Biol. 2007;368:388–97. doi: 10.1016/j.jmb.2007.02.047. [DOI] [PubMed] [Google Scholar]
- 167.Hollenbach AD, McPherson CJ, Mientjes EJ, et al. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J Cell Sci. 2002;115:3319–30. doi: 10.1242/jcs.115.16.3319. [DOI] [PubMed] [Google Scholar]
- 168.Kuo HY, Chang CC, Jeng JC, et al. SUMO modification negatively modulates the transcriptional activity of CREB-binding protein via the recruitment of Daxx. Proc Natl Acad Sci USA. 2005;102:16973–8. doi: 10.1073/pnas.0504460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yang SR, Chida AS, Bauter MR, et al. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;291:L46–57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- 170.Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–80. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yang SR, Wright J, Bauter M, et al. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. 2007;292:L567–76. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- 172.Calao M, Burny A, Quivy V, et al. A pervasive role of histone acetyltransferases and deacetylases in an NF-kappaB-signaling code. Trends Biochem Sci. 2008;33:339–49. doi: 10.1016/j.tibs.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 173.Miller-Kasprzak E, Jagodzinski PP. 5-Aza-2’-deoxycytidine increases the expression of anti-angiogenic vascular endothelial growth factor 189b variant in human lung microvascular endothelial cells. Biomed Pharmacother. 2008;62:158–63. doi: 10.1016/j.biopha.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 174.Yue W, Dacic S, Sun Q, et al. Frequent inactivation of RAMP2, EFEMP1 and Dutt1 in lung cancer by promoter hypermethylation. Clin Cancer Res. 2007;13:4336–44. doi: 10.1158/1078-0432.CCR-07-0015. [DOI] [PubMed] [Google Scholar]
- 175.Chen Y, Pacyna-Gengelbach M, Ye F, et al. Insulin-like growth factor binding protein-related protein 1 (IGFBP-rP1) has potential tumour-suppressive activity in human lung cancer. J Pathol. 2007;211:431–8. doi: 10.1002/path.2132. [DOI] [PubMed] [Google Scholar]
- 176.Shames DS, Girard L, Gao B, et al. A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med. 2006;3:e486. doi: 10.1371/journal.pmed.0030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chang HC, Cho CY, Hung WC. Downregulation of RECK by promoter methylation correlates with lymph node metastasis in non-small cell lung cancer. Cancer Sci. 2007;98:169–73. doi: 10.1111/j.1349-7006.2006.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chen H, Suzuki M, Nakamura Y, et al. Aberrant methylation of RASGRF2 and RASSF1A in human non-small cell lung cancer. Oncol Rep. 2006;15:1281–5. [PubMed] [Google Scholar]
- 179.Tada Y, Brena RM, Hackanson B, et al. Epigenetic modulation of tumor suppressor CCAAT/enhancer binding protein alpha activity in lung cancer. J Natl Cancer Inst. 2006;98:396–406. doi: 10.1093/jnci/djj093. [DOI] [PubMed] [Google Scholar]
- 180.Smith LT, Lin M, Brena RM, et al. Epigenetic regulation of the tumor suppressor gene TCF21 on 6q23-q24 in lung and head and neck cancer. Proc Natl Acad Sci USA. 2006;103:982–7. doi: 10.1073/pnas.0510171102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Izumi H, Inoue J, Yokoi S, et al. Frequent silencing of DBC1 is by genetic or epigenetic mechanisms in non-small cell lung cancers. Hum Mol Genet. 2005;14:997–1007. doi: 10.1093/hmg/ddi092. [DOI] [PubMed] [Google Scholar]
- 182.Gal-Yam EN, Saito Y, Egger G, et al. Cancer epigenetics: modifications, screening, and therapy. Annu Rev Med. 2008;59:267–80. doi: 10.1146/annurev.med.59.061606.095816. [DOI] [PubMed] [Google Scholar]
- 183.Tessema M, Belinsky SA. Mining the epigenome for methylated genes in lung cancer. Proc Am Thorac Soc. 2008;5:806–10. doi: 10.1513/pats.200805-045TH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lawless MW, Norris S, O’Byrne KJ, et al. Targeting histone deacetylases for the treatment of disease. J Cell Mol Med. 2009;13:826–52. doi: 10.1111/j.1582-4934.2008.00571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lawless MW, Norris S, O’Byrne KJ, et al. Targeting Histone Deacetylases for the treatment of immune, endocrine & metabolic disorders. Endocr Metab Immune Disord – Drug Targets. 2009;9:84–107. doi: 10.2174/187153009787582441. [DOI] [PubMed] [Google Scholar]
- 186.Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 187.Kaminskas E, Farrell AT, Wang YC, et al. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist. 2005;10:176–82. doi: 10.1634/theoncologist.10-3-176. [DOI] [PubMed] [Google Scholar]
- 188.Gore SD, Jones C, Kirkpatrick P. Decitabine. Nat Rev Drug Discov. 2006;5:891–2. doi: 10.1038/nrd2180. [DOI] [PubMed] [Google Scholar]
- 189.Talalay P, Fahey JW, Holtzclaw WD, et al. Chemoprotection against cancer by phase 2 enzyme induction. Toxicol Lett. 1995;82–83:173–9. doi: 10.1016/0378-4274(95)03553-2. [DOI] [PubMed] [Google Scholar]
- 190.Myzak MC, Karplus PA, Chung FL, et al. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–74. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 191.Mi L, Wang X, Govind S, et al. The role of protein binding in induction of apoptosis by phenethyl isothiocyanate and sulforaphane in human non-small lung cancer cells. Cancer Res. 2007;67:6409–16. doi: 10.1158/0008-5472.CAN-07-0340. [DOI] [PubMed] [Google Scholar]
- 192.Mi L, Xiao Z, Hood BL, et al. Covalent binding to tubulin by isothiocyanates. A mechanism of cell growth arrest and apoptosis. J Biol Chem. 2008;283:22136–46. doi: 10.1074/jbc.M802330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mi L, Chung FL. Binding to protein by isothiocyanates: a potential mechanism for apoptosis induction in human nonsmall lung cancer cells. Nutr Cancer. 2008;60:12–20. doi: 10.1080/01635580802381287. [DOI] [PubMed] [Google Scholar]
- 194.Liang H, Lai B, Yuan Q. Sulforaphane induces cell-cycle arrest and apoptosis in cultured human lung adenocarcinoma LTEP-A2 cells and retards growth of LTEP-A2 xenografts in vivo. J Nat Prod. 2008;71:1911–4. doi: 10.1021/np800233q. [DOI] [PubMed] [Google Scholar]
- 195.Jin CY, Moon DO, Lee JD, et al. Sulforaphane sensitizes tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis through downregulation of ERK and Akt in lung adenocarcinoma A549 cells. Carcinogenesis. 2007;28:1058–66. doi: 10.1093/carcin/bgl251. [DOI] [PubMed] [Google Scholar]
- 196.Conaway CC, Wang CX, Pittman B, et al. Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res. 2005;65:8548–57. doi: 10.1158/0008-5472.CAN-05-0237. [DOI] [PubMed] [Google Scholar]
- 197.Menon VP, Sudheer AR. Antioxidant and anti-inflammatory properties of curcumin. Adv Exp Med Biol. 2007;595:105–25. doi: 10.1007/978-0-387-46401-5_3. [DOI] [PubMed] [Google Scholar]
- 198.Rahman I. Dietary polyphenols mediated regulation of oxidative stress and chromatin remodeling in inflammation. Nutr Rev. 2008;66:S42–5. doi: 10.1111/j.1753-4887.2008.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Venkatesan N, Punithavathi D, Babu M. Protection from acute and chronic lung diseases by curcumin. Adv Exp Med Biol. 2007;595:379–405. doi: 10.1007/978-0-387-46401-5_17. [DOI] [PubMed] [Google Scholar]
- 200.Anand P, Sundaram C, Jhurani S, et al. Curcumin and cancer: an “old-age” disease with an “age-old” solution. Cancer Lett. 2008;267:133–64. doi: 10.1016/j.canlet.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 201.Lee J, Jung HH, Im YH, et al. Interferon-alpha resistance can be reversed by inhibition of IFN-alpha-induced COX-2 expression potentially via STAT1 activation in A549 cells. Oncol Rep. 2006;15:1541–9. [PubMed] [Google Scholar]
- 202.Andjelkovic T, Pesic M, Bankovic J, et al. Synergistic effects of the purine analog sulfinosine and curcumin on the multidrug resistant human non-small cell lung carcinoma cell line (NCI-H460/R) Cancer Biol Ther. 2008;7:1024–32. doi: 10.4161/cbt.7.7.6036. [DOI] [PubMed] [Google Scholar]
- 203.Sen S, Sharma H, Singh N. Curcumin enhances Vinorelbine mediated apoptosis in NSCLC cells by the mitochondrial pathway. Biochem Biophys Res Commun. 2005;331:1245–52. doi: 10.1016/j.bbrc.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 204.Sharma RA, Euden SA, Platton SL, et al. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res. 2004;10:6847–54. doi: 10.1158/1078-0432.CCR-04-0744. [DOI] [PubMed] [Google Scholar]
- 205.Morimoto T, Sunagawa Y, Kawamura T, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. 2008;118:868–78. doi: 10.1172/JCI33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Marcu MG, Jung YJ, Lee S, et al. Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem. 2006;2:169–74. doi: 10.2174/157340606776056133. [DOI] [PubMed] [Google Scholar]
- 207.Balasubramanyam K, Varier RA, Altaf M, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163–71. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 208.Kang J, Chen J, Shi Y, et al. Curcumin-induced histone hypoacetylation: the role of reactive oxygen species. Biochem Pharmacol. 2005;69:1205–13. doi: 10.1016/j.bcp.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 209.Liu HL, Chen Y, Cui GH, et al. Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating B-NHL cell line Raji proliferation. Acta Pharmacol Sin. 2005;26:603–9. doi: 10.1111/j.1745-7254.2005.00081.x. [DOI] [PubMed] [Google Scholar]
- 210.Chen Y, Shu W, Chen W, et al. Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch 1 in Raji cells. Basic Clin Pharmacol Toxicol. 2007;101:427–33. doi: 10.1111/j.1742-7843.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 211.Plummer SM, Holloway KA, Manson MM, et al. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signalling complex. Oncogene. 1999;18:6013–20. doi: 10.1038/sj.onc.1202980. [DOI] [PubMed] [Google Scholar]
- 212.Bachmeier BE, Mohrenz IV, Mirisola V, et al. Curcumin downregulates the inflammatory cytokines CXCL1 and -2 in breast cancer cells via NFkappaB. Carcinogenesis. 2008;29:779–89. doi: 10.1093/carcin/bgm248. [DOI] [PubMed] [Google Scholar]
- 213.Nonn L, Duong D, Peehl DM. Chemopreventive anti-inflammatory activities of curcumin and other phytochemicals mediated by MAP kinase phosphatase-5 in prostate cells. Carcinogenesis. 2007;28:1188–96. doi: 10.1093/carcin/bgl241. [DOI] [PubMed] [Google Scholar]
- 214.Andreadi CK, Howells LM, Atherfold PA, et al. Involvement of Nrf2, p38, B-Raf, and nuclear factor-kappaB, but not phosphatidylinositol 3-kinase, in induction of hemeoxygenase-1 by dietary polyphenols. Mol Pharmacol. 2006;69:1033–40. doi: 10.1124/mol.105.018374. [DOI] [PubMed] [Google Scholar]
- 215.Dickinson DA, Iles KE, Zhang H, et al. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. FASEB J. 2003;17:473–5. doi: 10.1096/fj.02-0566fje. [DOI] [PubMed] [Google Scholar]
- 216.Iqbal M, Sharma SD, Okazaki Y, et al. Dietary supplementation of curcumin enhances antioxidant and phase II metabolizing enzymes in ddY male mice: possible role in protection against chemical carcinogenesis and toxicity. Pharmacol Toxicol. 2003;92:33–8. doi: 10.1034/j.1600-0773.2003.920106.x. [DOI] [PubMed] [Google Scholar]
- 217.Meja KK, Rajendrasozhan S, Adenuga D, et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am J Respir Cell Mol Biol. 2008;39:312–23. doi: 10.1165/rcmb.2008-0012OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Cheung KL, Khor TO, Kong AN. Synergistic effect of combination of phenethyl isothiocyanate and sulforaphane or curcumin and sulforaphane in the inhibition of inflammation. Pharm Res. 2009;26:224–31. doi: 10.1007/s11095-008-9734-9. [DOI] [PubMed] [Google Scholar]
- 219.Fang MZ, Wang Y, Ai N, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63:7563–70. [PubMed] [Google Scholar]
- 220.Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol. 2005;68:1018–30. doi: 10.1124/mol.104.008367. [DOI] [PubMed] [Google Scholar]
- 221.Sriram N, Kalayarasan S, Sudhandiran G. Enhancement of antioxidant defense system by epigallocatechin-3-gallate during bleomycin induced experimental pulmonary fibrosis. Biol Pharm Bull. 2008;31:1306–11. doi: 10.1248/bpb.31.1306. [DOI] [PubMed] [Google Scholar]
- 222.Yuan JH, Li YQ, Yang XY. Inhibition of epigallocatechin gallate on orthotopic colon cancer by upregulating the Nrf2-UGT1A signal pathway in nude mice. Pharmacology. 2007;80:269–78. doi: 10.1159/000106447. [DOI] [PubMed] [Google Scholar]
- 223.Vittal R, Selvanayagam ZE, Sun Y, et al. Gene expression changes induced by green tea polyphenol (-)-epigallocatechin-3-gallate in human bronchial epithelial 21BES cells analyzed by DNA microarray. Mol Cancer Ther. 2004;3:1091–9. [PubMed] [Google Scholar]
- 224.Syed DN, Afaq F, Kweon MH, et al. Green tea polyphenol EGCG suppresses cigarette smoke condensate-induced NF-kappaB activation in normal human bronchial epithelial cells. Oncogene. 2007;26:673–82. doi: 10.1038/sj.onc.1209829. [DOI] [PubMed] [Google Scholar]
- 225.Chen PC, Wheeler DS, Malhotra V, et al. A green tea-derived polyphenol, epigallocatechin-3-gallate, inhibits IkappaB kinase activation and IL-8 gene expression in respiratory epithelium. Inflammation. 2002;26:233–41. doi: 10.1023/A:1019718718977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Xu Y, Ho CT, Amin SG, et al. Inhibition of tobacco-specific nitrosamine-induced lung tumorigenesis in A/J mice by green tea and its major polyphenol as antioxidants. Cancer Res. 1992;52:3875–9. [PubMed] [Google Scholar]
- 227.Komori A, Yatsunami J, Okabe S, et al. Anticarcinogenic activity of green tea polyphenols. Jpn J Clin Oncol. 1993;23:186–90. [PubMed] [Google Scholar]
- 228.Mimoto J, Kiura K, Matsuo K, et al. (-)-Epigallocatechin gallate can prevent cisplatin-induced lung tumorigenesis in A/J mice. Carcinogenesis. 2000;21:915–9. doi: 10.1093/carcin/21.5.915. [DOI] [PubMed] [Google Scholar]
- 229.Yang GY, Liao J, Kim K, et al. Inhibition of growth and induction of apoptosis in human cancer cell lines by tea polyphenols. Carcinogenesis. 1998;19:611–6. doi: 10.1093/carcin/19.4.611. [DOI] [PubMed] [Google Scholar]
- 230.Okabe S, Suganuma M, Hayashi M, et al. Mechanisms of growth inhibition of human lung cancer cell line, PC-9, by tea polyphenols. Jpn J Cancer Res. 1997;88:639–43. doi: 10.1111/j.1349-7006.1997.tb00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Cao J, Xu Y, Chen J, Klaunig JE. Chemopreventive effects of green and black tea on pulmonary and hepatic carcinogenesis. Fundam Appl Toxicol. 1996;29:244–50. doi: 10.1006/faat.1996.0028. [DOI] [PubMed] [Google Scholar]
- 232.Fujimoto N, Sueoka N, Sueoka E, et al. Lung cancer prevention with (-)-epigallocatechin gallate using monitoring by heterogeneous nuclear ribonucleoprotein B1. Int J Oncol. 2002;20:1233–9. [PubMed] [Google Scholar]
- 233.Yang GY, Liao J, Li C, et al. Effect of black and green tea polyphenols on c-jun phosphorylation and H(2)O(2) production in transformed and non-transformed human bronchial cell lines: possible mechanisms of cell growth inhibition and apoptosis induction. Carcinogenesis. 2000;21:2035–9. doi: 10.1093/carcin/21.11.2035. [DOI] [PubMed] [Google Scholar]
- 234.Witschi H, Espiritu I, Ly M, et al. Chemoprevention of tobacco smoke-induced lung tumors by inhalation of an epigallocatechin gallate (EGCG) aerosol: a pilot study. Inhal Toxicol. 2004;16:763–70. doi: 10.1080/08958370490490400. [DOI] [PubMed] [Google Scholar]
- 235.Yang CS, Liao J, Yang GY, et al. Inhibition of lung tumorigenesis by tea. Exp Lung Res. 2005;31:135–44. doi: 10.1080/01902140490495525. [DOI] [PubMed] [Google Scholar]
- 236.Yang J, Wei D, Liu J. Repressions of MMP-9 expression and NF-kappa B localization are involved in inhibition of lung carcinoma 95-D cell invasion by (-)-epigallocatechin-3-gallate. Biomed Pharmacother. 2005;59:98–103. doi: 10.1016/j.biopha.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 237.Sadava D, Whitlock E, Kane SE. The green tea polyphenol, epigallocatechin-3-gallate inhibits telomerase and induces apoptosis in drug-resistant lung cancer cells. Biochem Biophys Res Commun. 2007;360:233–7. doi: 10.1016/j.bbrc.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 238.Yan Y, Cook J, McQuillan J, et al. Chemopreventive effect of aerosolized polyphenon E on lung tumorigenesis in A/J mice. Neoplasia. 2007;9:401–5. doi: 10.1593/neo.07160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Banerjee S, Manna S, Mukherjee S, et al. Black tea polyphenols restrict benzopyrene-induced mouse lung cancer progression through inhibition of Cox-2 and induction of caspase-3 expression. Asian Pac J Cancer Prev. 2006;7:661–6. [PubMed] [Google Scholar]
- 240.Ogasawara M, Matsunaga T, Suzuki H. Differential effects of antioxidants on the in vitro invasion, growth and lung metastasis of murine colon cancer cells. Biol Pharm Bull. 2007;30:200–4. doi: 10.1248/bpb.30.200. [DOI] [PubMed] [Google Scholar]
- 241.Shirai T, Reshad K, Yoshitomi A, et al. Green tea-induced asthma: relationship between immunological reactivity, specific and non-specific bronchial responsiveness. Clin Exp Allergy. 2003;33:1252–5. doi: 10.1046/j.1365-2222.2003.01744.x. [DOI] [PubMed] [Google Scholar]
- 242.Cauchi S, Han W, Kumar SV, et al. Haplotype-environment interactions that regulate the human glutathione S-transferase P1 promoter. Cancer Res. 2006;66:6439–48. doi: 10.1158/0008-5472.CAN-05-4457. [DOI] [PubMed] [Google Scholar]
- 243.Wang LI, Giovannucci EL, Hunter D, et al. Dietary intake of Cruciferous vegetables, Glutathione S-transferase (GST) polymorphisms and lung cancer risk in a Caucasian population. Cancer Causes Control. 2004;15:977–85. doi: 10.1007/s10552-004-1093-1. [DOI] [PubMed] [Google Scholar]
- 244.Zhao B, Seow A, Lee EJ, et al. Dietary isothiocyanates, glutathione S-transferase -M1, -T1 polymorphisms and lung cancer risk among Chinese women in Singapore. Cancer Epidemiol Biomarkers Prev. 2001;10:1063–7. [PubMed] [Google Scholar]
- 245.London SJ, Yuan JM, Chung FL, et al. Isothiocyanates, glutathione S-transferase M1 and T1 polymorphisms, and lung-cancer risk: a prospective study of men in Shanghai, China. Lancet. 2000;356:724–9. doi: 10.1016/S0140-6736(00)02631-3. [DOI] [PubMed] [Google Scholar]
- 246.Spitz MR, Duphorne CM, Detry MA, et al. Dietary intake of isothiocyanates: evidence of a joint effect with glutathione S-transferase polymorphisms in lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2000;9:1017–20. [PubMed] [Google Scholar]
- 247.Riedl MA, Saxon A, Diaz-Sanchez D. Oral sulforaphane increases Phase II antioxidant enzymes in the human upper airway. Clin Immunol. 2009;130:244–51. doi: 10.1016/j.clim.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Tang L, Zirpoli GR, Reid ME, et al. Cruciferous vegetable intake is inversely associated with lung cancer risk among smokers. Seventh Annual AACR International Conference on Frontiers in Cancer Prevention Research. 2008 : Poster B132. [Google Scholar]
- 249.Riso P, Martini D, Visioli F, et al. Effect of broccoli intake on markers related to oxidative stress and cancer risk in healthy smokers and nonsmokers. Nutr Cancer. 2009;61:232–7. doi: 10.1080/01635580802425688. [DOI] [PubMed] [Google Scholar]
- 250.Paolini M, Perocco P, Canistro D, et al. Induction of cytochrome P450, generation of oxidative stress and in vitro cell-transforming and DNA-damaging activities by glucoraphanin, the bioprecursor of the chemopreventive agent sulforaphane found in broccoli. Carcinogenesis. 2004;25:61–7. doi: 10.1093/carcin/bgg174. [DOI] [PubMed] [Google Scholar]
- 251.Lao CD, Ruffin MTt, Normolle D, et al. Dose escalation of a curcuminoid formulation. BMC Complement Altern Med doi: 10.1186/1472-6882-6-10. . doi: 10.1186/1472-6882-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Dhillon N, Aggarwal BB, Newman RA, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14:4491–9. doi: 10.1158/1078-0432.CCR-08-0024. [DOI] [PubMed] [Google Scholar]
- 253.Kurien BT, Singh A, Matsumoto H, et al. Improving the solubility and pharmacological efficacy of curcumin by heat treatment. Assay Drug Dev Technol. 2007;5:567–76. doi: 10.1089/adt.2007.064. [DOI] [PubMed] [Google Scholar]
- 254.Ferrari E, Lazzari S, Marverti G, et al. Synthesis, cytotoxic and combined cDDP activity of new stable curcumin derivatives. Bioorg Med Chem. 2009;17:3043–52. doi: 10.1016/j.bmc.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 255.Vermeulen M, Klopping-Ketelaars IW, Van Den Berg R, et al. Bioavailability and kinetics of sulforaphane in humans after consumption of cooked versus raw broccoli. J Agric Food Chem. 2008;56:10505–9. doi: 10.1021/jf801989e. [DOI] [PubMed] [Google Scholar]
- 256.Conaway CC, Getahun SM, Liebes LL, et al. Disposition of glucosinolates and sulforaphane in humans after ingestion of steamed and fresh broccoli. Nutr Cancer. 2000;38:168–78. doi: 10.1207/S15327914NC382_5. [DOI] [PubMed] [Google Scholar]
- 257.Hanlon N, Coldham N, Gielbert A, et al. Absolute bioavailability and dose-dependent pharmacokinetic behaviour of dietary doses of the chemopreventive isothiocyanate sulforaphane in rat. Br J Nutr. 2008;99:559–64. doi: 10.1017/S0007114507824093. [DOI] [PubMed] [Google Scholar]
- 258.Hanlon N, Coldham N, Gielbert A, et al. Repeated intake of broccoli does not lead to higher plasma levels of sulforaphane in human volunteers. Cancer Lett doi: 10.1016/j.canlet.2009.04.004. . doi: 10.1016/j.canlet.2009-04-004. [DOI] [PubMed] [Google Scholar]
- 259.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839–52. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 260.Feng R, Oton A, Mapara MY, et al. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol. 2007;139:385–97. doi: 10.1111/j.1365-2141.2007.06772.x. [DOI] [PubMed] [Google Scholar]
- 261.Portanova P, Russo T, Pellerito O, et al. The role of oxidative stress in apoptosis induced by the histone deacetylase inhibitor suberoylanilide hydroxamic acid in human colon adenocarcinoma HT-29 cells. Int J Oncol. 2008;33:325–31. [PubMed] [Google Scholar]
- 262.Punturieri A, Szabo E, Croxton TL, et al. Lung cancer and chronic obstructive pulmonary disease: needs and opportunities for integrated research. J Natl Cancer Inst. 2009;101:554–9. doi: 10.1093/jnci/djp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Aoshiba K, Nagai A. Differences in airway remodeling between asthma and chronic obstructive pulmonary disease. Clin Rev Allergy Immunol. 2004;27:35–43. doi: 10.1385/CRIAI:27:1:035. [DOI] [PubMed] [Google Scholar]
- 264.Bai TR, Knight DA. Structural changes in the airways in asthma: observations and consequences. Clin Sci. 2005;108:463–77. doi: 10.1042/CS20040342. [DOI] [PubMed] [Google Scholar]
- 265.Spears M, McSharry C, Thomson NC. Peroxisome proliferator-activated receptor-gamma agonists as potential anti-inflammatory agents in asthma and chronic obstructive pulmonary disease. Clin Exp Allergy. 2006;36:1494–504. doi: 10.1111/j.1365-2222.2006.02604.x. [DOI] [PubMed] [Google Scholar]
- 266.Cosio BG, Tsaprouni L, Ito K, et al. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med. 2004;200:689–95. doi: 10.1084/jem.20040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Cosio BG, Iglesias A, Rios A, et al. Low-dose theophylline enhances the anti-inflammatory effects of steroids during exacerbations of COPD. Thorax. 2009;64:424–9. doi: 10.1136/thx.2008.103432. [DOI] [PubMed] [Google Scholar]