

Abstract
Hypertension is associated with neuroinflammation and increased sympathetic tone. Interference with neuroinflammation by an anti-inflammatory reagent or overexpression of interleukin-10 in the brain was found to attenuate hypertension. However, the cellular mechanism of neuroinflammation, as well as its impact on neurogenic regulation of blood pressure, is unclear. Here, we found that hypertension, induced by either angiotensin II or L-NG-nitro-l-arginine methyl ester, is accompanied by microglial activation as manifested by microgliosis and pro-inflammatory cytokine up-regulation. Targeted depletion of microglia significantly attenuated neuroinflammation, glutamate receptor expression in the paraventricular nucleus, plasma vasopressin level, kidney norepinephrine concentration and blood pressure. Furthermore, when microglia were pre-activated and transferred into the brains of normotensive mice, there was a significantly prolonged pressor response to intracerebroventricular injection of angiotensin II; and inactivation of microglia eliminated these effects. These data demonstrate that microglia, the resident immune cells in the brain, are the major cellular factors in mediating neuroinflammation and modulating neuronal excitation, which contributes to the elevated blood pressure.
Keywords: Microglia, Hypertension, Neuroinflammation, sympathetic nerve activity, Angiotensin
Introduction
Hypertension, particularly resistant hypertension, is associated with enhanced sympathetic tone, and can be substantially managed by renal nerve ablation1 and baroreceptor nerve stimulation2. The sympathetic outflow is controlled by several important nuclei and their circuits in the central nervous system (CNS), especially the hypothalamic paraventricular nucleus (PVN) and the rostroventrolateral medulla and the nucleus tractus solitaries in hindbrain3, 4. Perturbations of these nuclei have been implicated in hypertension. Although neurons in those regions are the major cells in modulating sympathetic outflow, what factors mediate the elevation of neuronal activity in hypertension stay elusive. Emerging studies indicate that hypertension is accompanied with extensive neuroinflammation, and that central anti-inflammatory treatment significantly alleviated hypertension5–7. Thus, determining the cellular mechanism of neuroinflammation and neuronal modulation in hypertension is critical to fully understand central regulation of blood pressure.
The CNS has long been considered as immune privileged due to the blood-brain barrier, the brain’s lack of lymphatic drainage to lymph nodes and suboptimal capacity to present antigen8. Microglia are the primary immune cells in the CNS. They are derived from primitive macrophages emanating from the embryonic yolk sac9. In the CNS, microglia proliferate and maintain homeostasis with limited contribution from peripheral blood-borne cells10. Recent transcriptome analyses revealed that microglia have a distinct phenotype from macrophages in other tissues, suggesting unique characteristics of microglia11, 12. As surveillance cells, microglia are highly sensitive to pathologic disturbance in the brain and play major roles in the progressive pathology of neurodegenerative diseases, such as Alzheimer’s disease13. Upon stimulation, microglia promptly undergo a series of morphologic and phenotypic changes, eventually releasing mediators that can directly modulate neuronal activities13, 14. Moreover, many lines of evidence indicate that microglia are highly involved in shaping neuronal behavior via sculpting dendritic spine formation and modulating neurotransmitter receptor presentation on the synaptic terminals in physiological conditions15, 16.
In the present study, we examined microglia in hypertension and found that microglia were activated in a different pattern from peripheral monocytes. When microglia were depleted by intracerebroventricular (i.c.v.) administration of diphtheria toxin (DT) into the transgenic CD11b-diphtheria toxin receptor (DTR) mice, the neuroinflammation and blood pressure increase induced by either angiotensin (Ang) II or L-NG-nitro-l-arginine methyl ester (L-NAME) were significantly attenuated. In contrast, adoptive transfer of activated microglia prolonged pressor responses to central application of Ang II. Taken together, our findings indicate that microglia are the key players in the neurogenic regulation of hypertension.
Methods
All surgical and experimental procedures were approved by the IACUC of Cedars Sinai Medical Center. A detailed Methods section is available in the online-only Data Supplement.
Results
Microglial activation pattern in hypertension
To characterize activation states of microglia in established hypertension, C57BL/6 mice were treated with subcutaneous (s.c.) infusion of Ang II or by feeding L-NAME in drinking water for 4 weeks. Systolic blood pressure reached 130 mmHg in the 1st week and sustained in the following 3 weeks (Figure S1A). Four weeks after the induction of hypertension, mice were sacrificed and microglia were analyzed. In both models, there was a significant increase of microglia in the PVN and motor cortex of hypertensive brains compared to the normotensive brains, as manifested by an increased area of Iba1 staining (Figure S1C–D). In contrast to the ramified appearance of naïve microglia, hypertensive microglia showed soma enlargement and process retraction. Thus, hypertension is associated with microgliosis, a characteristic of microglial activation17.
To define the characteristics of microglia in hypertension, we dissociated microglia from the brains of normotensive mice or mice made hypertensive with Ang II or L-NAME. Since we previously found that there were increases of TNFα and IL-1β expression in the brain of hypertensive rats5, 16, we first evaluated microglial expression of these pro-inflammatory cytokines using intracellular staining and flow cytometry (FCM) analysis. After dissociated from the mouse brains, microglia were cultured in vitro for 6 hours in the presence of bredfeldin A which blocks the secretion of protein from cells18. There was elevation of TNFα- and IL-1β-expressing, as well as mild but significantly increased IL-6-expressing microglia in L-NAME-treated mice compared to those in normotensive animals (Figure 1A). Although the numbers of TNFα-, IL-1β- or IL-6-expressing microglia from Ang II-treated mice were not conspicuously altered in the resting state (data not shown), there were remarkably more cells expressing these cytokines after LPS treatment in Ang II hypertensive microglia than normotensive microglia, which indicates their pre-activation (Figure 1B).
Figure 1. Enhanced production of proinflammatory cytokines by microglia of hypertensive mice.
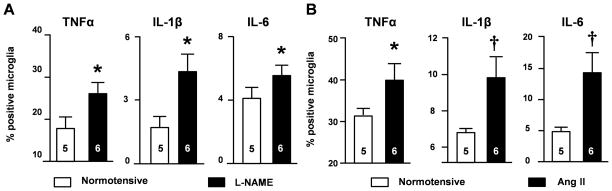
(A) The percentages of CD11b+CD45low microglia expressing proinflammatory cytokines in normotensive mice and mice after 4-weeks L-NAME treatment. (B) The percentages of microglia expressing proinflammatory cytokines from normotensive mice and Ang II-treated mice for 4 weeks are shown. Cells were stimulated with 10 ng/ml LPS. *P<0.05; †P <0.01 by unpaired t-test.
Current concepts of microglial activation arise, in part, from research into macrophage biology. In macrophages, M1 (pro-inflammatory classical activation) and M2 (alternative activation) represent extremes in a continuum of activation states19. We thus investigated the M1-associated markers (MHC class II, CCR7, IFNγR and iNOS) and the activation markers representing M2 state (CD36, mannose receptor, Tie2, CCR2 and IL-4Rα) in hypertension-associated microglia. After 4 weeks of Ang II or L-NAME treatment, microglia were dissociated and enriched by Percoll gradient centrifugation followed by FCM analysis. Intriguingly, all these molecules except iNOS were upregulated in the hypertensive microglia (Figure 2A–B). This activation profile is specific to microglia since the monocytes from Ang II-induced hypertensive mice had only increased IL-4Rα and decreased MHC class II expression compared to their normotensive counterparts (Figure 2B).
Figure 2. Surface activation markers expressed by hypertensive microglia.

Microglia were dissociated from normotensive mice or mice treated with Ang II or L-NAME for 4 weeks. Their surface expression of M1- and M2-associated activation markers were quantified by FCM. (A) Representative overlapped histograms of microglia from normotensive mice or Ang II-induced hypertensive mice. (B) Fold changes of mean florescence intensity (MFI) of activation markers between hypertensive microglia (Ang II or L-NAME) and normotensive microglia. Blood monocytes isolated from normotensive mice and Ang II-induced hypertensive mice were also analyzed. The statistics was analyzed by unpaired t-test of MFI between hypertensive cells and normotensive cells. *P<0.05; †P <0.01; ‡P <0.001 by unpaired t-test.
Loss of microglia alleviated blood pressure and neuroendocrinological factors associated with hypertension
To investigate the role of microglia in hypertension development, we first used a microglial depletion strategy. Transgenic CD11b-DTR mice express the DTR under the control of the endogenous CD11b promoter20. In the CNS, only microglia but not neuronal or other glial cells express DTR in these mice. A single i.c.v. injection of DT resulted in a dose dependent reduction of microglia in both the PVN and motor cortex (Figure 3A). At a dose of 1000 pg/g, DT caused an over 90% loss of microglia, which was confirmed by both immunohistochemistry (Figure 3A) and FCM analysis (the CD11b+CD45low population in Figure 3B). To be noted, i.c.v. DT injection (1 000 pg/g/d) did not change total blood monocytes (CD11b+Ly6G−F4/80low) or inflammatory monocytes (CD11b+Ly6G−Ly6Chigh) (Figure 3B). At the dose we used, DT was not toxic to neurons or astrocytes in the CD11b-DTR mice, as NeuN or GFAP staining of DT-treated PVN showed no change in the density or distribution of neurons or astrocytes, respectively, in comparison to the untreated CD11b-DTR mice (Figure 3C). To be noted, F4/80 and CD31 staining showed no difference in the density of perivascular macrophages between DT-treated and saline-treated CD11b-DTR mice (Figure 3C). Further, DT injection did not cause any change in body weight and general behavior of mice. Our results showed that i.c.v. infusion of DT to CD11b-DTR mice is a valid model to investigate physiological changes in microglial loss without causing change in the brain or circulating monocytes. Due to the efficiency of microglial depletion with the dose of 1000 pg/g/d, we utilized this dose in all following studies.
Figure 3. Verification of microglial depletion.

(A) Three days after i.c.v. injection of DT in the indicated doses, the brain of CD11b-DTR mice were perfused and the coronal sections of PVN and motor cortex were stained for Iba1. (B) FCM analysis of microglia (CD11b+CD45low) and blood total monocytes (CD11b+F4/80low) and inflammatory monocytes (CD11b+Ly6Chigh) in saline- and DT-treated CD11b-DTR mice. Representative dot plots from 10 mice of each treatment. (C) The densities of neuron (NeuN+), astrocytes (GFAP+), and perivascular macrophages (blood vessels (CD31+) and macrophages (F4/80+)) in the PVN of saline- and DT-treated CD11b-DTR mice.
CD11b-DTR mice were treated with Ang II or L-NAME. After 2–3 weeks, when hypertension was established and neuroinflammation had developed, microglia were depleted by i.c.v. infusion of DT. In both models, microglial depletion caused a gradual reduction in blood pressure and by 2 weeks, the mice had a 20 mmHg lower blood pressure than the i.c.v. saline-infused CD11b-DTR mice (Figure 4A). Notably, i.c.v. DT did not alter resting blood pressure in naive CD11b-DTR or C57BL/6J mice, nor did it change the blood pressure responses when C57BL/6J mice were infused with Ang II (Figure S2). To examine the effects of microglial depletion on neuroinflammation induced by hypertension, brains were harvested and the PVN were dissected at the end of the protocol. The expression of TNFα and IL-1β were analyzed by ELISA. As shown in Figure 4B, hypertension resulted in significant increases of both cytokines. Remarkably, loss of microglia reduced these cytokines to normal levels. These data strongly support our hypothesis that microglia are central in neuroninflammation and blood pressure regulation.
Figure 4. Microglia are important for sustained hypertension.

(A) Systolic blood pressure was measured in CD11b-DTR mice treated with Ang II (left) by telemetry transducer or L-NAME (right) by tailcuff. After hypertension induction, mice were treated with i.c.v. DT or saline in the indicated periods. Expression of TNFα and IL-1β (B) and GluN2A (C) in the PVN of naïve, Ang II or Ang II + i.c.v. DT treated CD11b-DTR mice were analyzed by ELISA and Western Blot, respectively. (D) The levels of kidney norepinephrine (NE) and plasma vasopressin (AVP) were analyzed by ELISA. *P<0.05 by Two-way ANOAVE in (A) and by one-way ANOVA in (B–D).
To understand whether microglia affect neuronal plasticity in hypertension, we examined the expression of the N-methyl-D-aspartate (NMDA) receptor, an excitatory synaptic receptor reported to be critical for neurogenic hypertension21. In the hypertensive PVN, the expression of NMDA subunit GluN2A was up-regulated by 2 folds (Figure 4C). Importantly, microglial depletion abolished such an increase, suggesting that loss of microglia may prevent hypertension-associated neuronal excitation. Vasopressin (AVP) and norepinephrine (NE) are two CNS-regulated hormones which are associated with blood pressure increase3. To better understand the downstream events of neuronal excitation, we examined the levels of plasma AVP and kidney NE in hypertensive mice. Consistent with NMDA receptor expression, both hormones were significantly increased in Ang II-treated hypertensive mice (Figure 4D). Microglial depletion suppressed AVP and NE levels in Ang II-treated mice to the normal levels.
Transfer of activated microglia changed blood pressure response
To further confirm the central role of microglia on blood pressure regulation, we adoptively transferred N9 cells, a murine microglial cell line, to the cerebroventricle of naïve C57BL/6J mice. N9 cells are homogeneousness, and they lack the contamination of astrocytes which is a concern when using cultured microglia from newborn brain. Some cells were primed in vitro with either Ang II (100 nM/L for 12 h) or LPS (10 ng/ml for 6 h). Twenty-four hours after their i.c.v. transfer, the recipient mice were anesthetized, and their basal blood pressure and heart rate were recorded (Outline in Figure 5A). There was no difference in baseline blood pressure and heart rate across the groups (Supplemental Table S1). When we induced a transient blood pressure increase by a single i.c.v. injection of Ang II (50 ng), there was an acute pressor response with a 5–10 mmHg rise (Figure 5B) in all groups. Interestingly, there was a significantly prolonged pressor response in mice receiving either Ang II- or LPS-primed microglia compared to the mice receiving naïve microglia or saline. This was specific for activated microglia because transferring Ang II-primed astrocytes did not change the duration of the pressor response. Minocycline has been widely used as an anti-inflammatory reagent affecting microglia22. We incorporated another two cohorts of mice which were transferred with microglia primed with Ang II or LPS in the presence of minocycline. The prolonged pressor responses observed with activated microglia were completely abolished when microglia were co-incubated with minocycline before transferring (Figure 5C).
Figure 5. Adoptive transfer of activated microglia prolonged pressor responses to i.c.v. Ang II stimulation.

(A) Outline of experimental protocol. (B) Representative traces of arterial blood pressure and mean blood pressure of recipient mice in response to i.c.v. single dose of Ang II (50 ng). The recipients were previously transferred i.c.v. with saline (Sham) or 1×105 naïve microglia (Naïve MG) or pre-primed microglia. In addition, a group of mice were transferred with 1×105 Ang II-primed astrocytes. Dashed line indicates the time point when i.c.v. injection of Ang II. (C) Quantification of magnitude and duration of blood pressure responses described in B. * P<0.05 vs. naïve; † P<0.01 vs. naive microglia; ‡ P<0.0001 vs. Ang II-primed microglia by one-way ANOVA. (D) Densitometry quantification of western blot on PVN GluN2A standardized to α-tubulin. * P<0.01; † P <0.001 by one-way ANOVA.
To confirm these blood pressure changes were associated with changes in neuronal profiling, PVN tissues were dissected from the recipients 24 hours after transfer. There was a two-fold increase in GluN2A level in LPS-primed group compared to the group receiving naïve microglia (Figure 5D). Minocycline co-incubation with LPS fully abrogated this increase. These experiments suggest that activated microglia may potentiate neuronal responses to hypertensive stimulant by enhancing the expression of NMDA subunit GluN2A in the PVN.
Discussion
Hypertension is associated with neuroinflammation; however, the cellular mechanism of neuroinflammation is unknown and it is unclear whether neuroinflammation contributes to the progression of hypertension. Here, we studied murine Ang II and L-NAME models, and found that rampart microglial activation is a hallmark of hypertension-associated neuroinflammation. Since Ang II and L-NAME induce hypertension through different mechanisms, we surmise that microglial activation is a characteristic of hypertension. To approve this hypothesis, future study of human samples will be critical. In both hypertensive models, microglial depletion significantly decreased blood pressure, neuroinflammation, and the levels of peripheral hypertensive hormones NE and AVP. In contrast, adoptive transfer of activated microglia predisposed recipients to hypertensive stimulant. In conclusion, this study shows that microglia are key modulators in the development of neurogenic hypertension.
The cause-and-effect relation between hypertension and neuroinflammation is under debating. In this present study, we focused on the role of microglia in established hypertension and in pressor responses to i.c.v. Ang II. Our depletion data and adoptive transfer data clearly show that activated microglia contribute to blood pressure regulation. However, depletion of microglia did not correct blood pressure to normal level in established hypertension and transfer of microglia did not by itself gave rise to blood pressure increase. Therefore, microglia activation is secondary to hypertensive insults but not an initiator of hypertension in our models. Microglia activation and hypertension forms a feed-forward loop.
Our study unveils a very unique activation pattern of microglia associated with hypertension. Both M1 (classical activation) and M2 (alternative activation) markers are up-regulated in hypertensive microglia but not in hypertensive monocytes. M1 and M2 represent extremes of a continuum in a universe of macrophage activation states. The up-regulation of both M1 and M2 markers have not been reported in any other tissue macrophages in any other models. Such a distinctive phenotype of microglia echoes recent transcriptome analyses which revealed that microglia, although considered the macrophages in brain, are distinguishable from peripheral myeloid cells11, 12. Whether this is due to the unique brain environment or the hypertensive factors or both needs further investigation.
In this study, we devised a microglial depletion strategy by infusing DT i.c.v to CD11b-DTR mice. A previous study also investigated microglia biology through a DT depletion strategy. In that study, the authors s.c. injected DT to CD11b-DTR mice at the age of P3 when their blood-brain barrier was incomplete22. However, peripheral monocytes and macrophages were also ablated by that approach. Our approach with i.c.v. DT infusion efficiently depleted microglia but leaving other CNS cells and circulating monocytes intact, suggesting the exclusive depletion of microglia. Thus, our study may provide a unique model to investigate microglia biology to a variety of pathophysiological settings in the future.
Microglia are the major sources to produce inflammatory molecules and neurotrophic factors. Pro-inflammatory cytokines such as IL-1β and TNFα can directly or indirectly modulate neuronal activities23, 24. One mechanism is that they can enhance neuronal excitation by increasing NMDA receptor expression in the postsynapes25, 26. A recent study shows that brain-derived neurotrophic factor (BDNF) produced by microglial are of importance in the regulation of neuronal activity by increasing the expression of NMDA receptors on the postsynaptic terminals16. Given the essential role of NMDA receptors in neuronal excitation and plasticity21, signals triggering NMDA receptor presentation are the key factors on neuronal activity. NMDA receptor is a tetrametric glutamate-gated ion channel, assembled by an essential subunit, NR1, and one or more components of a second family of subunits NR2A to D and NR3A/B27. Kinase–mediated phosphorylation directly changes the activity and density of NMDA receptor subunits on the membrane27. We found that the expression of the NMDA receptor in the PVN was significantly up-regulated in the presence of microglia but was decreased when microglia were depleted, suggesting that activated microglia could exacerbate hypertension through augmenting neuronal excitation. Future studies of the molecules mediating microglia and neuronal activation in hypertension will provide potential targets for the management of essential hypertension.
Supplementary Material
Perspective.
Our work provides direct evidence that hypertension elicits a unique activation pattern of microglia. Using depletion and adoptive transfer strategies, we evidenced that microglia are the major cellular modulators of neurogenic hypertension. Hypertension is generally accompanied with elevated sympathetic tone. To understand the central regulation of hypertension is critical to develop efficient treatment to essential hypertension. Our work unveils that microglia are central to neuroinflammation and neuronal regulation of hypertension, which provides a mechanistic insight to this disease. To pinpoint the molecules which mediates microglia-neuron interaction may provide potential targets for the management of essential hypertension.
Novelty and significance.
What is new?
Hypertension elicits a unique activation pattern of microglia with the up-regulation of markers associated with both classical and alternative activation.
Using depletion and adoptive transfer strategies, we evidenced that microglia are the major cellular modulators of neurogenic hypertension.
What is relevant?
Hypertension is generally accompanied with elevated sympathetic tone. To understand the central regulation of hypertension is critical to develop efficient treatment to essential hypertension. Our study unveils that microglia are central to neuroinflammation and neuronal regulation of hypertension, which provides a mechanistic insight to this disease.
Summary
Our study shows that rampart activation of microglia is a characteristic of hypertension. We provide direct evidence that microglia are the major modulator of neuroinflammation and central control of blood pressure.
Acknowledgments
Sources of Funding
This work was supported by American Heart Association grants 11SDG6770006 (to P.S.) and 13BGIA14680069 (to X.Z.S.), NIH grants R01 HL110353-26 (to K.E.B.) and R01 NS075930 (to P.L.), and UCLA CTSI grant UT1TR000124 (to P.S.).
Footnotes
Disclosures
None.
References
- 1.Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension. N Engl J Med. 2009;361:932–934. doi: 10.1056/NEJMc0904179. [DOI] [PubMed] [Google Scholar]
- 2.Heusser K, Tank J, Engeli S, Diedrich A, Menne J, Eckert S, Peters T, Sweep FC, Haller H, Pichlmaier AM, Luft FC, Jordan J. Carotid baroreceptor stimulation, sympathetic activity, baroreflex function, and blood pressure in hypertensive patients. Hypertension. 2010;55:619–626. doi: 10.1161/HYPERTENSIONAHA.109.140665. [DOI] [PubMed] [Google Scholar]
- 3.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- 4.Toney GM, Chen QH, Cato MJ, Stocker SD. Central osmotic regulation of sympathetic nerve activity. Acta Physiol Scand. 2003;177:43–55. doi: 10.1046/j.1365-201X.2003.01046.x. [DOI] [PubMed] [Google Scholar]
- 5.Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C, Raizada MK. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56:297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zubcevic J, Jun JY, Kim S, Perez PD, Afzal A, Shan Z, Li W, Santisteban MM, Yuan W, Febo M, Mocco J, Feng Y, Scott E, Baekey DM, Raizada MK. Altered inflammatory response is associated with an impaired autonomic input to the bone marrow in the spontaneously hypertensive rat. Hypertension. 2014;63:542–550. doi: 10.1161/HYPERTENSIONAHA.113.02722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S, Yang ZM, Francis J. Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension. Cardiovasc Res. 2009;82:503–512. doi: 10.1093/cvr/cvp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol. 2007;28:5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ Immunological Genome Consortium. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16:1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14:463–477. doi: 10.1038/nri3705. [DOI] [PubMed] [Google Scholar]
- 14.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Jalabi W, Hu W, Park HJ, Gale JT, Kidd GJ, Bernatowicz R, Gossman ZC, Chen JT, Dutta R, Trapp BD. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun. 2014;5:4486. doi: 10.1038/ncomms5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 18.Nebenfuhr A, Ritzenthaler C, Robinson DG. Brefeldin A: Deciphering an enigmatic inhibitor of secretion. Plant Physiol. 2002;130:1102–1108. doi: 10.1104/pp.011569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 20.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li DP, Zhu LH, Pachuau J, Lee HA, Pan HL. mGluR5 upregulation increases excitability of hypothalamic presympathetic neurons through NMDA receptor trafficking in spontaneously hypertensive rats. J Neurosci. 2014;34:4309–4317. doi: 10.1523/JNEUROSCI.4295-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, Yamashita T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16:543–551. doi: 10.1038/nn.3358. [DOI] [PubMed] [Google Scholar]
- 23.Ferri CC, Ferguson AV. Interleukin-1 beta depolarizes paraventricular nucleus parvocellular neurones. J Neuroendocrinol. 2003;15:126–133. doi: 10.1046/j.1365-2826.2003.00870.x. [DOI] [PubMed] [Google Scholar]
- 24.Olmos G, Llado J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014;2014:861231. doi: 10.1155/2014/861231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gruber-Schoffnegger D, Drdla-Schutting R, Honigsperger C, Wunderbaldinger G, Gassner M, Sandkuhler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-alpha and IL-1beta is mediated by glial cells. J Neurosci. 2013;33:6540–6551. doi: 10.1523/JNEUROSCI.5087-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wheeler D, Knapp E, Bandaru VV, Wang Y, Knorr D, Poirier C, Mattson MP, Geiger JD, Haughey NJ. Tumor necrosis factor-alpha-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J Neurochem. 2009;109:1237–1249. doi: 10.1111/j.1471-4159.2009.06038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.