

Abstract
Staphylococcus aureus is a dangerous bacterial pathogen whose clinical impact has been amplified by the emergence and rapid spread of antibiotic resistance. In the search for more effective therapeutic strategies, great effort has been placed on the study and development of staphylolytic enzymes, which benefit from high potency, activity towards drug-resistant strains, and a low inherent susceptibility to emergence of new resistance phenotypes. To date, the majority of therapeutic candidates have derived from either bacteriophage or environmental competitors of S. aureus. Little to no consideration has been given to cis-acting autolysins that represent key elements in the bacterium's endogenous cell wall maintenance and recycling machinery. In this study, five putative autolysins were cloned from the S. aureus genome, and their activities were evaluated. Four of these novel enzymes, or component domains thereof, demonstrated lytic activity towards live S. aureus cells, but their potencies were 10s to 1000s of times lower than that of the well-characterized therapeutic candidate lysostaphin. We hypothesized that their poor activities were due in part to suboptimal cell wall targeting associated with their native cell wall binding domains, and we sought to enhance their antibacterial potential via chimeragenesis with the peptidoglycan binding domain of lysostaphin. The most potent chimera exhibited a 140-fold increase in lytic rate, bringing it within 8-fold of lysostaphin. While this enzyme was sensitive to certain biologically relevant environmental factors and failed to exhibit a measurable minimal inhibitory concentration, it was able to kill lysostaphin-resistant S. aureus and ultimately proved active in lung surfactant. We conclude that the S. aureus proteome represents a rich and untapped reservoir of novel antibacterial enzymes, and we demonstrate enhanced bacteriolytic activity via improved cell wall targeting of autolysin catalytic domains.
Keywords: MRSA, Lysins, Lytic Enzyme, CHAP, Lysostaphin, Antibiotic
Introduction
Staphylococcus aureus poses a significant threat to human health, and its widespread antibiotic resistance has rendered it a top priority for both US domestic and global healthcare organizations (Centers for Disease Control and Prevention 2013; World Health Organization 2014). As with other bacterial pathogens, S. aureus has proven capable of rapidly subverting new antibacterial chemotherapies (Taubes 2008), and the increased morbidity, mortality, and costs associated with drug-resistant S. aureus infections (Cosgrove et al. 2005; Shurland et al. 2007) is motivating a search for next generation antibacterial agents.
Bacteriolytic enzymes are drawing increasing interest as potential alternatives to traditional small molecule antimicrobials (Szweda et al. 2012). These antibacterial biocatalysts target and degrade bacterial peptidoglycan, thereby compromising cell wall integrity and ultimately causing lysis and death. Staphylolytic enzymes in particular possess a number of beneficial features relevant to therapeutic applications. They generally exhibit high substrate and cellular specificity, they are active against drug-resistant strains, they elicit new resistance phenotypes at a low rate, and their catalytic modes of action render them highly potent antibacterial agents (Pastagia et al. 2013).
Lysostaphin (LST) is perhaps the most extensively studied staphylolytic enzyme, and several decades of research have shown the molecule to have potent anti-staphylococcal activity in vitro, in a variety of preclinical animal models, and in human subjects (Kokai-Kun 2012). LST is composed of an N-terminal glycylglycine zinc endopeptidase domain and a C-terminal SH3 cell wall binding domain (CWBD) (Sabala et al. 2014), which together target and degrade the pentaglycine crosslinks in S. aureus cell walls. Due to the enzyme's specificity for this pentaglycine crosslink, some S. aureus strains readily develop spontaneous resistance towards LST through cell wall modification, although this resistance comes at the cost of reduced fitness and hyper-susceptibility to beta-lactam antibiotics (Kusuma et al. 2007; Ling and Berger-Bachi 1998).
In addition to LST, phage endolysins have proven to be another productive source of staphylolytic drug candidates. Like LST, these virion-associated lysins exhibit a modular architecture containing both catalytic and CWBDs. Among the former are N-acetyl-β-D-glucosaminidases, N-acetylmuramidases, N-acetylmuramoyl-L-alanine amidases (MurNAc-LAA), cysteine/histidine dependent aminohydrolases/endopeptidases (CHAP), and a variety of other endopeptidases (Pastagia et al. 2013). Combined with these substrate-selective catalytic domains, lysins' CWBDs provide an additional element of cellular specificity. While there exists a variety of lysin targeting motifs, two prominent classes include the src homology 3 (SH3) and lysin motif domains (LysM), both of which can home to various molecular targets on the bacterial surface (Buist et al. 2008; Whisstock and Lesk 1999). Similar to LST, phage lysins exhibit fast lysis kinetics and are effective at clearing S. aureus infections in vivo; in contrast to LST, they tend to elicit bacterial resistance at an especially low rate (Rodríguez-Rubio et al. 2013; Schuch et al. 2014; Singh et al. 2014).
As opposed to the widely studied bacteriocins (e.g. LST) and phage endolysins, little to no consideration has been given to bacterial autolysins as a potential source of antibacterial enzymes. Autolysins are endogenous cell wall hydrolases involved in various aspects of bacterial physiology. They play important roles in processes such as cell division, autolysis (programmed death), peptidoglycan recycling, and biofilm formation (Thomas and Hancock 2009; Vollmer et al. 2008). Given their critical biological functions yet lethal potential, autolysin activities must be precisely regulated through tight transcriptional, translational, and post-translational controls. For example, overexpression of S. aureus autolysin LytN results in cell lysis and death, yet disruption of LytN expression results in structural damage to the cell wall, altered cellular morphology, and marked growth defects (Frankel et al. 2011). Similar effects have been seen upon knockout of the Atl autolysin in S. aureus (Takahashi et al. 2001). These observations suggest that, if autolysins could be harnessed as antibacterial agents, rates of resistance development might be exceeding low.
While the concept of autolysin based enzyme therapies is intriguing, to date the enzymes have failed to yield impressive results when used as exogenous lytic agents. The addition of the mature form of autolysin LytM to S. aureus bacterial suspensions results in marginal rates of bacterial lysis (Sabala et al. 2012), and similarly poor lytic activity is observed with recombinantly produced LytN (Frankel and Schneewind 2012). Not surprisingly, LytM also fails to exhibit therapeutic efficacy in a murine model of S. aureus skin infection (Sabala et al. 2012). The lack of potent lytic activity among autolysins is due in part to suboptimal (from a therapeutic perspective) targeting to the bacterial cell wall. For example, as part of their natural function, the S. aureus autolysins Sle1, LytN, and Atl all localize specifically to the septal cross-wall of dividing cells (Frankel and Schneewind 2012; Gotz et al. 2014; Schlag et al. 2010). While this specific localization is critical to normal bacterial physiology, it presumably limits cell wall exposure upon exogenous addition of these enzymes to bacterial suspensions. In a different example of suboptimal localization, the autolysin LytM lacks any CWBD (Sabala et al. 2012), resulting in a similar deficit in cell wall targeting. In contrast, the SH3 CWBD of LST binds specifically to the pentaglycine crosslinks in S. aureus peptidoglycan, and as a result it has been shown to bind ubiquitously throughout the bacterial cell wall (Grundling and Schneewind 2006). Notably, truncation of the LST's CWBD results in a substantial reduction in lytic activity (Gargis et al. 2010; Osipovitch and Griswold 2014), a fact that underscores the critical nature of lysin targeting to the bacterial cell surface.
The modular architecture of lysin enzymes lends itself to molecular engineering, and mix-and-match chimeragenesis has been used to great effect in improving the antibacterial activities of various phage endolysins (Fernandes et al. 2012; Singh et al. 2014). In particular, the LST CWBD has proven to be a powerful fusion partner for creating high performance synthetic lysins (Becker et al. 2009; Donovan et al. 2006; Mao et al. 2013; Rodriguez-Rubio et al. 2012; Schmelcher et al. 2012; Sundarrajan et al. 2014). Thus, the LST CWBD presents one potential solution to the issue of suboptimal autolysin targeting. Indeed, in a prior pilot study, the lytic activity of LytM, which again lacks a CWBD, was enhanced more than 500-fold upon fusion to the LST CWBD (Osipovitch and Griswold 2014). That early success prompted us to more broadly survey the S. aureus proteome for autolysins that might be leveraged as antibacterial agents. Here, we describe bioinformatic identification, cloning, and preliminary characterization of several putative S. aureus autolysins, and we demonstrate gain of function molecular engineering via chimeragenesis with the SH3 CWBD of LST.
Materials and Methods
Materials
Common disposable and chemical materials were purchased from Fisher Scientific (Pittsburgh, PA, USA) and VWR International (Radnor, PA). Enzymes and reagents for cloning were from New England BioLabs (Ipswich, MA, USA), and oligonucleotides (25 nmol scale, standard desalting) were from Integrated DNA Technology (San Diego, CA, USA). Unless noted, all strains were from American Type Culture Collection (Manassas, VA). All other materials are described.
Bioinformatics and Construct Design
The Simple Modular Architecture Research Tool, or SMART (Letunic et al. 2014; Schultz et al. 1998), was used to search for two known cell wall binding domains, LysM and SH3, within the Staphylococcus aureus subsp. aureus NCTC 8325 genome. Hits from SMART were cross-referenced with the NCBI Conserved Domain Search (Marchler-Bauer et al. 2011) to ensure proteins with the CWBDs also contained predicted catalytic domains.
For designing constructs, SignalP (Petersen et al. 2011) was used to find predicted signal sequences, which were removed during cloning. JPred3 (Cole et al. 2008) was used to predict secondary structure information about the proteins. Truncations were made in predicted unstructured regions of the proteins to increase the likelihood of stable variants.
Cloning and Expression
Whole cell lysates of S. aureus SA113 (ATCC® 35556™) were used as templates for cloning of the autolysins. Genes were amplified using the primer pairs shown in Table S1. The 5′ ends of the genes were appended with an NcoI restriction site and the 3′ ends with a BamHI restriction site. These restriction sites were then used to clone into a modified pET26b vector (EMD Millipore, Darmstadt, Germany), which coded for a 5′-hexahistidine tag followed by a serine-methionine-alanine linker (Osipovitch and Griswold 2014).
For the fusions, the LST cell wall binding domain was amplified with the LBDVector primer pair (Table S1). The 5′ primer appended a 5′ NcoI site with an internal BamHI site, and the 3′ primer appended a HindIII site. NcoI and HindIII were used to clone the LST cell wall binding domain (LBD) into the modified PET26b vector described above (Osipovitch and Griswold 2014). This vector was then used to clone all fusions using NcoI and BamHI, retaining the 5′ hexahistidine and serine-methionine-alanine linker and including a glycine-serine linker between the autolysin catalytic domain and the LBD. The primers used for specific fusions are listed in Table S1.
All ligated vectors were transformed by electroporation into E. coli DH5α [F-endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA-argF)U169, hsdR17(rK- mK+), λ−], plated onto Lysogeny Broth (Becton Dickinson, East Rutherford, NJ) supplemented with 30 μg/mL kanamycin, grown over night at 37°C, and then purified using a QIAprep Spin Miniprep Kit (Qiagen; Venlo, Netherlands). Vectors were then sequence verified before being finally transformed into the E. coli BL21(DE3) [F− ompT hsdSB (rB- mB-) gal dcm (DE3)] expression host.
Proteins were expressed and purified in the manner previously described (Osipovitch and Griswold 2014), with minor changes as noted in Table S2. Protein purity was assessed by mixing samples with 4× Laemmli Sample Buffer (Bio-Rad, Hercules, CA) and running them on a 12% SDS-PAGE gel (Bio-Rad). Gels were destained by boiling in water and then stained with GelCode Blue protein stain (Thermo Fisher Scientific), as per the instructions.
SyTox Kinetic Assay
Kinetic assays were performed essentially as previously described (Osipovitch and Griswold 2014). To maintain consistency between assays, uniform frozen starter stocks of S. aureus SA113 were made. An overnight culture of SA113 grown in Trypticase Soy Broth (TSB, Becton Dickinson) was mixed to a final percentage of 10-12% glycerol. Cells were aliquoted into sterile tubes for a total of two ODs per tube and were then stored at -80°C. Prior to the kinetic assay, one aliquot of cells was thawed, added to 25 mL of TSB in a 125 mL baffle flask, and grown at 37°C with shaking at 250 rpm for 3 hrs. After three hours of growth, the OD600 of the cells were 1.4±0.1 (mean ± st. dev.). Cells were harvested by centrifugation, washed once with PBS pH 7.3 (Green and Sambrook 2012), and then suspended in PBS pH 7.3 to a final OD600 of 2. The assay was performed in black 96-well plates, and the wells consisted of 250 μl PBS containing cells at a final OD600 of 1, SYTOX Green at 5 μM, and enzyme at the specified concentrations. Mean fluorescence intensity readings were taken every 20-30s on a SpectroMax Gemini plate reader (Molecular Devices, Sunnyvale, CA) using an excitation of 504 nm and emission of 523 nm. Kinetic data was analyzed in Microsoft Excel (Redmond, WA) by determining the slope of the steepest linear region in the lysis curve. Statistical analyses and data presentation was produced in Prism v.5 software (La Jolla, CA).
Variations of the SyTox Assay
In all variations of the assay, the total volume of 250 μl and final SYTOX Green concentration of 5 μM were maintained. For the Michaelis-Menten analyses, the wells were supplemented with 0.1% bovine serum albumin, and various dilutions of the cells were made. In the MIC Medium assay modification, PBS was replaced by Mueller Hinton II Broth (MHIIB) + 2% NaCl. In the salt dependency assay, salt stocks at pH 7.3 were diluted appropriately into the assay. In the human serum assay, wells were supplemented with the noted percentages of human serum (Sigma-Aldrich, St. Louis, MO). For the lung surfactant assay, Infasurf was kindly provided by Ony Inc. (Amherst, NY). The appropriate dilution of Infasurf was added to each well containing PBS and enzyme, and was mixed by pipet in order to suspend the surfactant.
Kinetic Assay with Resistant Cells
Two strains were kindly provided by Dr. Gary Sloan (University of Alabama): RN4220 containing an empty PLI50 vector and RN4200 containing the same vector encoding the End/Epr genes (Dehart et al. 1995; Gargis et al. 2010). Both strains were maintained in liquid or solid TSB supplemented with 34 μg/mL chloramphenicol (TSB-Cm34). For the assay, single colonies were used to inoculate 3 mL TSB-Cm34 and were then grown overnight at 37°C with shaking. Cells were then subcultured and grown to an OD600 of about 1. Cells were prepared in the same manner as described in the SYTOX Kinetic Assay section above, and the assay was run and analyzed in the same manner.
Melting Temperatures
Differential scanning fluorimetry was performed essentially as reported (Niesen et al. 2007) with modifications as described previously (Osipovitch et al. 2012).
Results
Bioinformatics Search for Novel Autolysins
In our pursuit of novel autolysins that might be co-opted as antibacterial candidates, we first performed a SMART search (Letunic et al. 2014; Schultz et al. 1998) for LysM and SH3 binding domains in the S. aureus subsp. aureus NTCC 8325 proteome. While S. aureus autolysins do not universally encode such CWBD (e.g. the LytM autolysin), we reasoned that the prevalence of these domains among known autolysins suggested a logical filter by which to identify novel candidates. The SMART search resulted in a list of ten proteins (Table S3) that were then cross-referenced with the NCBI conserved domain search (Marchler-Bauer et al. 2011) in order to remove any hits that did not also contain putative catalytic domains. Lastly, any proteins that had already been described (i.e. Sle1 and LytN) were removed. After culling the list, five putative proteins remained (Table 1).
Table 1.
Nomenclature and accession numbers for enzymes.
| Abbreviated Name | UniProt Protein Name | UniProt Accession | Genbank Accession |
|---|---|---|---|
| LytH | Probable cell wall amidase LytH | Q2FXU3 | ABD30811 |
| LytO | Autolysin | Q2FX77 | ABD31075 |
| SsaALP | Secretory antigen SsaA-like protein | Q2G0D4 | ABD29804 |
| LDP | LysM Domain Protein | Q2G278 | ABD29903 |
| PH | Petidoglycan hydrolase, putative | Q2FYD8 | ABD30597 |
While two of the candidate autolysins exhibited similarity to previously studied proteins, none of the five enzymes described here had been experimentally characterized. To avoid future ambiguity, we assigned the “Autolysin” candidate protein (Table 1) the new, unique identifier “LytO”, which is consistent with S. aureus autolysin nomenclature. LytO shares 92% sequence identity with the previously described protein LytA (UniProt ID P24556)(Wang et al. 1991). The majority of the two sequences are 100% identical, with the exception of an R13W mutation and a region between amino acids 126 and 166 that has little homology. When LytA is BLASTed against the S. aureus subsp. aureus NTCC 8325 genome, LytO is the top hit, and thus LytA itself is not encoded in the 8325 genome. We speculate that the two homologs serve the same role in different strains. SsaALP is named for its similarity to the Staphylococcal secretory antigen A protein (SsaA; UniProt ID Q2FV55) that has been associated with S. aureus pathogenicity (Martin et al. 2002; Resch et al. 2005). The two proteins share 54% identity between amino acids 141-265 of SsaALP and 131-255 of SsaA, regions that encompass their predicted CHAP domains, respectively. All five candidate proteins were BLASTed against the UniProt Knowledge Base, and each yielded at least 1000 hits with 98-100% similarity across a multitude of S. aureus strains and, in some cases, phage. Importantly, none of the candidate autolysins from Table 1 appear to have been validated beyond prediction and homology, supporting the claim of these enzymes' novelty.
At least three classes of catalytic domains were represented in the final list of autolysin candidates (Fig. 1). Most of the selected enzymes contained a CHAP domain, although LytH bore only a MurNAc-LAA domain. The PH protein contained both CHAP and MurNAc-LAA domains. Three of the five enzymes bore SH3 domains, which were arrayed both N-terminally (LytH) and C-terminally (LytO and PH). SsaALP contained two repeating LysM domains, a motif also seen in other autolysins (Frankel et al. 2011). The PGRP domain of LytO has an unknown function, although mammalian PGRP domains can exhibit both peptidoglycan binding as well as amidase properties (Dziarski and Gupta 2006). Overall, the CWBD search strategy yielded a small but diverse panel of novel autolysin candidates.
Fig. 1.
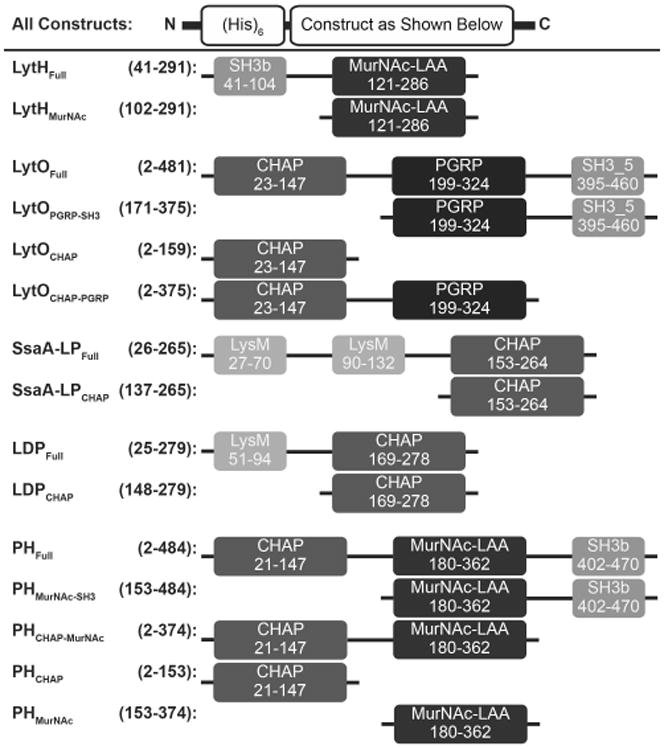
Schematic of autolysin constructs. Numbers represent amino acid positions corresponding to the full length, wild type protein from UniProt. On the left is the construct name, followed by the corresponding amino acid numbers of the truncations. On the right is the illustration of each construct. Domain names and amino acid numbers were obtained from the NCBI Conserved Domain Search and abbreviations are as follows: CHAP, cysteine/histidine-dependent amidohydrolase/peptidase; MurNAc-LAA, N-acetylmuramoyl-L-alanine amidase; PGRP, peptidoglycan recognition proteins; SH3, bacterial SRC homology 3 domain; LysM, lysin motif. Dark greys represent purported catalytic domains and light greys represent purported cell wall binding domains. The function of the PGRP domain, black, is currently unknown.
Expression, Activity, and Stability Analysis
Given the lack of experimental validation for our candidate autolysins, we next sought to assess the proteins' expressibility and lytic activity. In addition to testing the full-length constructs, we also evaluated various domain truncations (Fig. 1). The full-length and truncated coding sequences were each appended with an N-terminal His6 tag, cloned into the pET26b expression vector, and transformed into E. coli BL21(DE3). With the exception of the PH truncations, all of the expression constructs yielded soluble protein, and most of the proteins were isolated with reasonable purity in one to two chromatography steps (Table S2). Polyacrylamide gel analysis suggested that many of the proteins experienced some degree of proteolytic degradation (Fig. S1), but notably, protease inhibitors had been specifically excluded from the preparations so as to avoid inactivation of the autolysins themselves. In contrast to all other constructs, SsaALPFull failed to produce any protein of the expected molecular weight (26.9 kDa), yielding instead a single prominent band at 15 kDa. Given the metal ion affinity purification and the N-terminal location of the His6 tag, the observed SsaALPFull product would not be expected to contain the CHAP domain (see Fig. 1). In all other cases, however, the partially purified preparations were deemed suitable for preliminary testing of lytic activity.
The activities of the enzyme preparations were determined with live S. aureus SA113 cells using a fluorescence kinetic assay as previously described (Osipovitch and Griswold 2014). Six of the eleven constructs tested exhibited detectable lytic activity against S. aureus (Fig. 2a). Compared to the potent bacteriocin LST, however, the enzymes were one to three orders of magnitude less active, although LytOFull and LDPFull were significantly more active than the truncated LST catalytic domain (LSTΔCWBD, having a deleted SH3 CWBD). As predicted, truncation of the putative LytO SH3 and LDP LysM domains decreased the activities of the respective enzymes, presumably as a result of decreased cell wall localization. Additionally, the LytOPGRP-SH3 construct had no inherent lytic activity under the conditions tested, while the LytOCHAP-PRGP construct, which lacked the SH3 cell wall binding domain, exhibited higher activity than the LytOCHAP domain alone (Fig. 2a). These observations suggest that the LytO PRGP domain contributes to cell wall targeting. Finally, the degraded SsaALPFull construct displayed no lytic activity, a result consistent with the expected lack of a catalytic domain in this proteolyzed product.
Fig. 2.
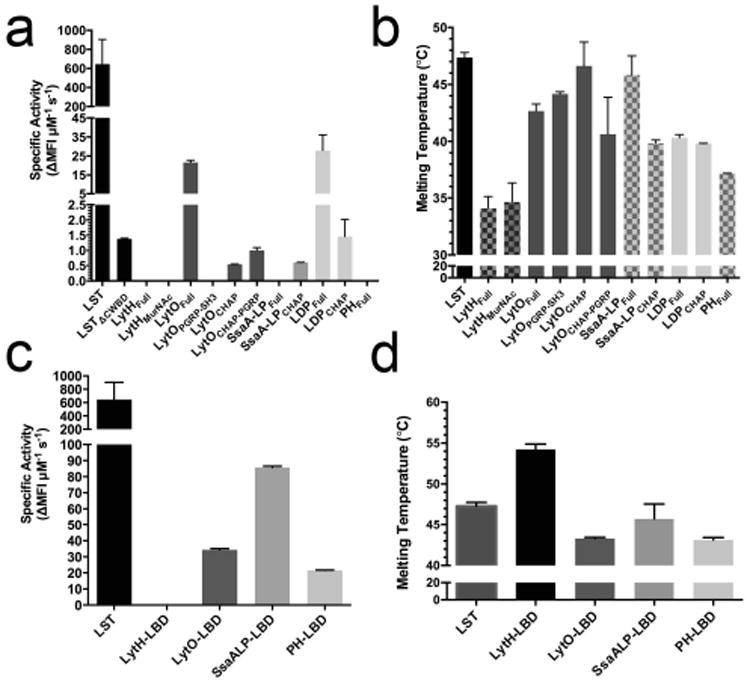
Preliminary characterization of autolysins and chimeras. A) Specific activities of the autolysins against live SA113 cells as measured by the SYTOX fluorescence kinetic assay. Lysins were tested up to 50 μg per 250 μL reaction. The LSTΔcwbd designation represents the LST's catalytic domain, for which kinetic data was previously published (Osipovitch, 2014). B) Melting temperatures of autolysins using differential scanning fluorimetry. C) Specific activity of chimeric lysins against live SA113 cells as determined by the SYTOX fluorescent assay. The LBD designation represents LST binding domain chimeras. LytH-LBD was tested up to 100 μg (14 μM), and the concentrations of the other enzymes were as follows: LytO-LBD=800 ng (100 nM), SsaALP-LBD=700 ng (100 nM), and PH-LBD=1250 ng (162 nM). D) Melting temperatures of chimeras. ΔMFI s-1 (change in mean fluorescence intensity per second) represents the slope of the steepest linear region of the lysis curve. All error bars represent standard deviations from two or more technical replicates.
The effects of domain truncation on thermostability were assessed by differential scanning fluorimetry. All constructs exhibited apparent melting temperatures (Tm) greater than 35°C (Fig. 2b), with the exception of LytH, which was found to have a realtively low Tm=34°C. In general, domain truncation did not result in substantial changes in thermostability. The SsaALPCHAP construct was the one exception, exhibiting a 6°C reduction in Tm relative to SsaALPFull, which was not in fact the originally intended full-length protein, but was instead a fragment likely containing only the LysM domains.
Chimeragenesis with the Lysostaphin SH3 Domain
Motivated by our previous success enhancing the activity of LytM via chimeragenesis with the LST CWBD (Osipovitch and Griswold 2014), we employed a similar molecular engineering strategy with candidate autolysins of the current study. Chimeric proteins were generated by genetically fusing the catalytic domains of the five autolysins (MurNAc-LAA for LytH; CHAP for the rest) to the LST CWBD (fusions referred to hereafter as LytH-LBD, LytO-LBD, SsaALP-LBD, LDP-LBD, and PH-LBD). LytH-LBD, LytO-LBD, and SsaALP-LBD expressed and purified well, while PH-LBD had low purified yields and LDP-LBD was insoluble (Fig. S2, Table S2).
Preliminary kinetic assays were run with live S. aureus to benchmark the lytic activities of the soluble chimeric lysins. Similar to the LytHFull and LytHMurNAc constructs, the LytH-LBD chimera had no detectable lytic activity (Fig. 2c). The specific activity of LytO-LBD appeared similar to that of the full-length autolysin construct LytOFull, although quantification of the latter required 5-fold higher concentrations of protein. The SsaALP-LBD chimera was 140-fold more active than the SsaALPCHAP truncation, placing it within 8-fold of the LST gold standard. While PHFull exhibited no detectable activity, the PH-LBD chimera was able to lyse S. aureus cells, providing evidence of a functional CHAP domain and suggesting that PH may in fact represent a novel autolysin. Ultimately, however, the PH-LBD construct was not carried forward into detailed characterization due to material limitations.
The thermostability of the chimeric constructs was assessed by differential scanning fluorimetry, and relative to their parent autolysins, the chimeras exhibited equivalent or higher melting temperatures (Fig. 2d). The LytH-LBD in particular showed a striking 20°C increase in Tm relative to LytHFull. Thus, fusion to the LST CWBD does not have a detrimental effect on the stability of the autolysin candidates, but rather in some cases it manifests a stabilizing effect.
Detailed Kinetics of LytO-LBD and SsaALP-LBD
Preliminary studies had identified LytO-LBD and SsaALP-LBD as the two most promising constructs, and these chimeras were therefore subjected to more rigorous kinetic analysis and comparison to the LST benchmark. Using previously described pseudo-Michaelis-Menten methods (Scanlon et al. 2010; Surovtsev et al. 2004), we leveraged our fluorescence kinetic assay to study S. aureus lysis rates in greater detail. As part of optimizing the assay conditions, measurements were initially made at two different enzyme concentrations: 200 ng/ml and 400 ng/ml (equating to 7.4 and 14.8 nM for LST and SsaALP-LBP, and 6.5 nM and 12.9 nM for LytO-LBP). The data were readily fit to saturating enzyme kinetic curves (Fig 3a), although the highest cellular substrate concentration resulted in lower than predicted rates and was therefore dropped from the analyses. This phenomenon is typical of pseudo-Michaelis-Menten studies on lytic enzymes using whole cell substrate, and as opposed to true substrate inhibition, we hypothesize that this effect reflects a “titrating out” of the enzymes across increasing numbers of cellular targets and a subsequent reduction in lysis rates for individual bacteria. Using only data that fit the expected saturation curves, apparent Vmax and Km values were extracted for each of the three enzymes (Fig. 3b). This more detailed analysis showed that the maximum reaction velocity of LytO-LBD was only 3-fold lower than that of LST, and SsaALP-LBD was only 20-30% less active than LST at Vmax. Thus, the CHAP domains of LytO and SsaALP do in fact have high inherent hydrolytic activity. On the other hand, LytO-LBD and SsaALP-LBD have higher apparent Km values, indicating that they require 3 to 4-fold greater cell concentrations to achieve 50% maximum reaction velocity. This result is somewhat counterintuitive, given that all three enzymes bear the SH3 CWBD derived from LST. However, cellular targeting and subsequent enzymatic hydrolysis of peptidoglycan is a complex and multifaceted process, and the mechanistic significance of the apparent Michaelis-Menten parameters should not be over interpreted.
Fig. 3.
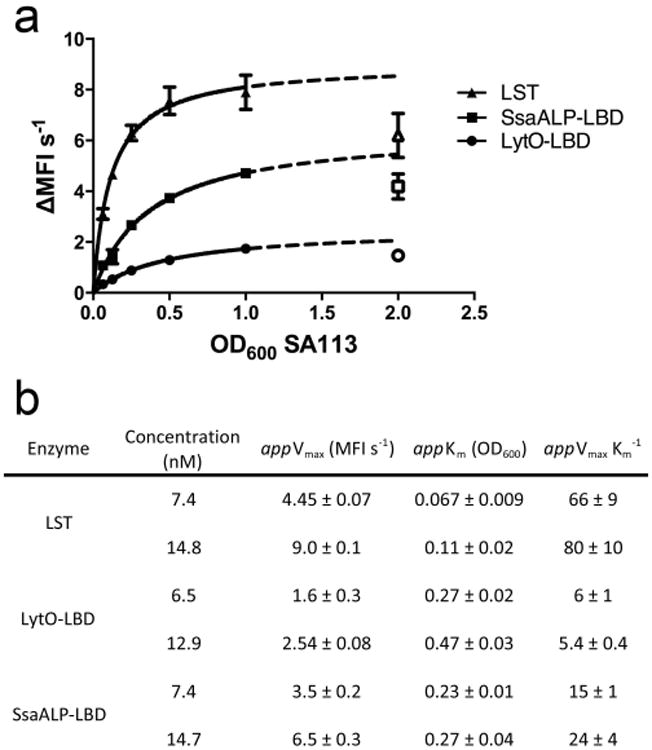
Pseudo-Michaelis-Menten kinetic analyses of LST and engineered autolysins. A) Curve fits used for determining pseudo-Michaelis-Menten constants. The dotted line represents an extrapolation of fit data, and the open shapes are values that were excluded in the curve fit. B) Table of apparent Michaelis-Menten parameters determined at two concentrations of each enzyme. Data are the means of triplicate measurements made in biological duplicate.
Interestingly, the kinetic analysis at two different enzyme concentrations yielded somewhat different results for both LytO-LBP and SsaALP-LBP. Vmax is expected to be proportional to the amount of enzyme in the assay, and doubling enzyme concentration, as done here, should result in a 2-fold higher Vmax. The expected increase was observed with LST, but Vmax increased by only 1.6-fold for LytO-LBP and 1.9-fold for SsaALP-LBP (Fig. 3b). The observed deviations were highly reproducible, which led us to probe the effect of enzyme concentration more carefully.
To more broadly assess the effect of enzyme concentration on specific rates of bacterial lysis, we performed fluorescence kinetic assays in which we fixed the concentration of bacterial cells while varying the concentration of enzyme over an order of magnitude (Fig. 4). Consistent with the initial Michaelis-Menten analysis, LST exhibited no significant difference in specific activity across the enzyme dilution series. In contrast, specific lytic rates of both LytO-LBP and SsaALP-LBP were dependent on enzyme concentration, where higher specific rates were observed with less enzyme. Again, these effects were highly reproducible. Thus, direct comparisons of lytic enzyme activities are perhaps only appropriate at identical enzyme concentrations, and more generally we conclude that richer comparisons of lytic enzyme activity might be facilitated by enzyme titration curves, as shown in Figure 4.
Fig. 4.
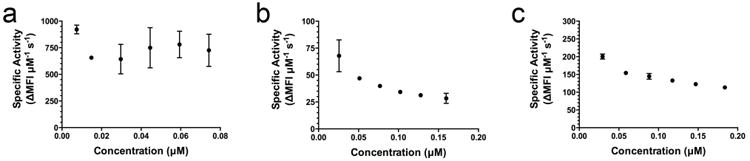
Specific activities across various concentrations of enzyme. A) LST B) LytO-LBD C) SsaALP-LBD. ΔMFI s-1 (change in mean fluorescence intensity per second) represents the slope of the steepest linear region of the lysis curve. Each data point represents the mean of duplicate measures (LST) or quadruplicate measures (LytO-LBD and SsaALP-LBD) from a representative experiment, and error bars are standard deviations. Biological replicates exhibited the same trends.
Activity in Biologic Contexts
As a more conventional analysis of LytO-LBD and SsaALP-LBD antibacterial potential, we next sought to determine minimal inhibitory concentration (MIC) in Mueller Hinton II broth supplemented with 2% NaCl (MHIIB). Interestingly, neither enzyme prevented overnight outgrowth of S. aureus strain SA113 at the highest tested concentrations (2 μM). Given the enzymes' good kinetic activities, the failure to achieve a measureable MIC was surprising, particularly for SsaALP-LBD, whose specific activity was similar to that of an analogous LytM-LBD chimera that possessed MICs as low as 15 nM (Osipovitch and Griswold 2014). This led us to test the effect of MHIIB medium on the kinetic activity of the enzymes. In MHIIB medium, LST and LytO-LBD retain 72 and 78%, respectively, of their activities as measured in PBS, whereas SsaALP-LBD was more severely inhibited, retaining only 19% of its original activity (Fig 5a). It was initially thought that the added salt in the cation adjusted medium might underlie the loss of SsaALP-LBD activity, but subsequent analysis in PBS buffer containing increasing salt concentrations showed that all three enzymes exhibited similar salt sensitivities (Fig. 5b). Thus, the nature of SsaALP-LBD sensitivity to MHIIB remains unknown.
Fig. 5.
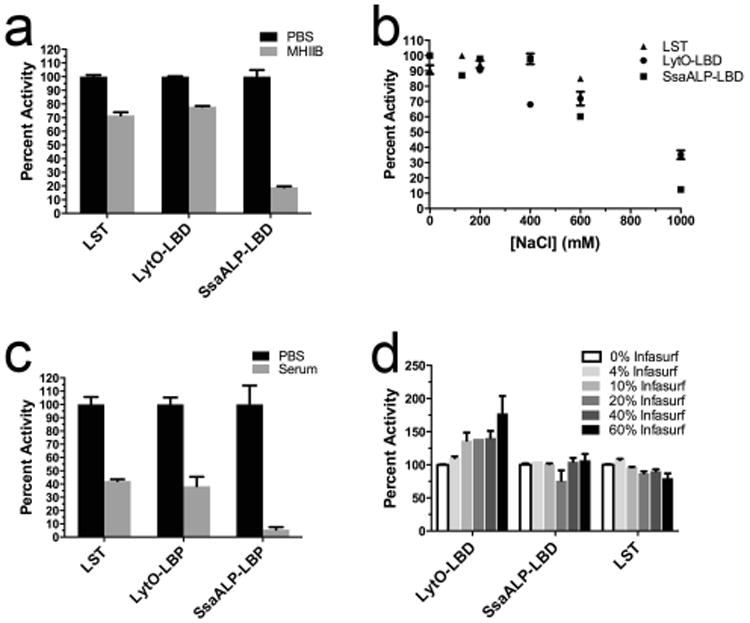
Environmental effects on the activity of LST and chimeric enzymes. Activities were measured with the SYTOX fluorescent kinetic assay and are reported as normalized percent activities. A) Activity in MHIIB medium supplemented with 2% NaCl. B) Activity in PBS supplemented with various salt concentrations. C) Activity in the presence of 60% human serum. D) Activity in the presence of the clinical-grade calf lung surfactant Infasurf. Error bars represent standard deviations, and all points include at least duplicate measurements.
As an alternative to MIC determination in MHIIB medium, we then sought to evaluate the enzymes' activities in other biologically relevant contexts. S. aureus frequently causes life-threatening bloodstream infections, and we therefore reassessed enzyme-mediated bacterial lysis in 60% human serum. Similar to the analysis in MHIIB medium, both LST and LytO-LBD retained higher activities (42% and 38%, respectively) than did SsaALP-LBD, which was only 5% active (Fig. 5c). It was subsequently found that as little as 10% human serum severely inhibited SsaALP-LBD activity (Fig. S3). S. aureus is also a major causative agent of acute pneumonias, prompting us to test the activity of the chimeric enzymes in the clinical grade lung surfactant Infasurf, which is widely used in lung surfactant replacement therapy for infants suffering from respiratory distress syndrome (Willson 2001). In PBS suspensions containing up to 60% lung surfactant, SsaALP-LBD retained full activity, while LytO-LBD exhibited a trend toward higher activity with increasing lung surfactant (Fig 5d). The LST benchmark exhibited an opposing trend, appearing to lose a small fraction of activity with increasing Infasurf concentrations.
SsaALP-LBD Kills LST-resistant S. aureus
The catalytic domains of SsaALP and LST target different bonds within the cell wall peptidoglycan, with LST's M23 peptidase domain cleaving glycylglycine bonds and SsaALP's CHAP domain cleaving either peptide bonds or peptide-sugar bonds in the peptidoglycan stem region. We hypothesized that the orthogonal selectivities of the two enzymes would be able to overcome LST resistance if they were used in combination. Using the model LST-resistant S. aureus RN4220 End:Epr cell line (Dehart et al. 1995; Gargis et al. 2010), we conducted fluorescence kinetic assays treating the bacteria with LST alone, SsaALP-LBD alone, or a combination of the two. Compared to the sensitive strain, LST exhibited a 250-fold reduction in lytic activity towards the resistant strain. Compared to LST, SsaALP-LBD was 5-fold less active against the sensitive strain, however the chimeric enzyme retained 15% of its activity when tested with the resistant strain. As a result, SsaALP-LBD had a 7-fold higher inherent activity towards the resistant RN4220 cell line. When tested against the sensitive strain, a combination of 500 ng LST with 500 ng SsaALP-LBD was nearly able to recapitulate the activity of 1000 ng of LST, retaining 80% relative activity. When testing this combination against the resistant strain, the 500 ng/500 ng combination retained 63% of the activity of a 1000 ng SsaALP-LBD single agent treatment. Thus, considering both LST-sensitive and LST-resistant bacteria, the combination treatment easily outperformed either single agent, suggesting a potential advantage with respect to mitigating emergence of new resistance phenotypes.
Discussion
One barrier to harnessing autolysins as antibiotics is the highly stringent regulatory mechanisms exerted by bacteria upon these enzymes (Brunskill and Bayles 1996; Cheung et al. 2004; Chu et al. 2013). In contrast to maintaining bacterial homeostasis, dysregulation of these stringent controls represents a prerequisite to any prospective antibacterial application. Within bacterial cells, transcription, translation, and post-translational processing are all central to regulating autolysin function, but these (largely) temporal controls are irrelevant to therapeutic use of recombinant enzymes. Instead, spatial control of subcellular localization, which is another physiological regulatory mechanism, likely constrains the antibacterial potency of exogenously applied autolysins. For example, the S. aureus enzymes Sle1, LytN, and Atl all localize specifically to the crosswall septum of dividing cells (Frankel and Schneewind 2012; Gotz et al. 2014; Schlag et al. 2010), thereby limiting total exposure of the cell wall and the lytic potential of the enzymes. To address this limitation, we contemplated using the SH3 targeting domain of LST, which has proved to be a potent targeting moiety for chimeric phage lysins (Becker et al. 2009; Donovan et al. 2006; Mao et al. 2013; Schmelcher et al. 2012; Sundarrajan et al. 2014). An earlier proof of concept study with the LytM autolysin demonstrated the feasibility of this approach (Osipovitch and Griswold 2014), and we sought here to extend this strategy by applying it to panel of novel autolysin candidates.
The literature contains detailed studies on numerous S. aureus autolysins known to be important for cell viability and virulence (Bose et al. 2012; Frankel et al. 2011; Rice and Bayles 2003; Takahashi et al. 2001), but we speculate that, generally, bacterial proteomes hold a substantial reservoir of uncharacterized cell wall hydrolases with therapeutic potential. We therefore employed bioinformatics to identify novel S. aureus autolysins based on a SMART domain search for proteins bearing the common SH3 or LysM domains. The list of candidates was filtered for those that also possessed putative catalytic domains, leaving five previously uncharacterized enzyme candidates. Expression and preliminary activity analysis showed the LytO and LDP candidates to have lytic activity towards live S. aureus, and analysis of domain truncation variants verified lytic activity in the putative CHAP domain of SsaALP, which could not be isolated in a full length format. While the full length PH enzyme had no activity here, fusion of the PH CHAP domain to the LST CWBD yielded an active lytic chimera, suggesting that PH may in fact be a functional autolysin. In all, CHAP domains from four putative proteins were functionally validated, leaving LytH, which bears a MurNAc-LAA catalytic domain, as the only unverified candidate from this study. It bears noting that only a limited range of assay conditions were evaluated here, and given the well-documented activity of other MurNAc-LAA containing proteins (Gotz et al. 2014; Mellroth et al. 2014; Tillman et al. 2013), we speculate that LytH functionality may ultimately be validated using alternative methods. More generally, our bioinformatics search successfully identified several novel autolysins, but our focus on SH3 and LysM domains was inherently constrained. Looking to the future, we anticipate that even more (and perhaps more active) S. aureus autolysins will be found using different search strategies, and these enzymes may also represent good candidates for development of novel antibacterial agents.
Although four out of five autolysins from this study yielded functional catalytic domains, their activities in native and native-like contexts were generally orders of magnitude lower than that of the well characterized therapeutic candidate LST. We had hypothesized that suboptimal cell wall targeting generally limits autolysin activity, and subsequent chimeragenesis of our newly validated CHAP catalytic domains with the LST CWBD did indeed enhanced the enzymes' lytic potential. In particular, the activity of SsaALP CHAP was increased 140-fold, and that of PH, which was non-functional in the full length native enzyme, was rendered a respectable lytic agent. Thus, we have shown here that fusion of a highly functional CWBD with autolysin catalytic domains is a general strategy for enhancing their antibacterial activity. More broadly, we expect that this approach will prove useful in targeting other microbial pathogens, as each bacterial species will encode panels of specific autolysins that might be co-opted through gain of function engineering with appropriate cell wall targeting moieties. Considering the enormous diversity of prokaryotes whose existence is dependent upon constant remodeling of peptidoglycan, we believe that autolysin engineering will open the gates to a vast reservoir of previously untapped therapeutic candidates. Leveraging appropriate molecular design and modification, we might in the future turn pathogens' endogenous cell wall maintenance machinery against them in the form of specific and potent antibacterial drugs.
Supplementary Material
Fig. 6.
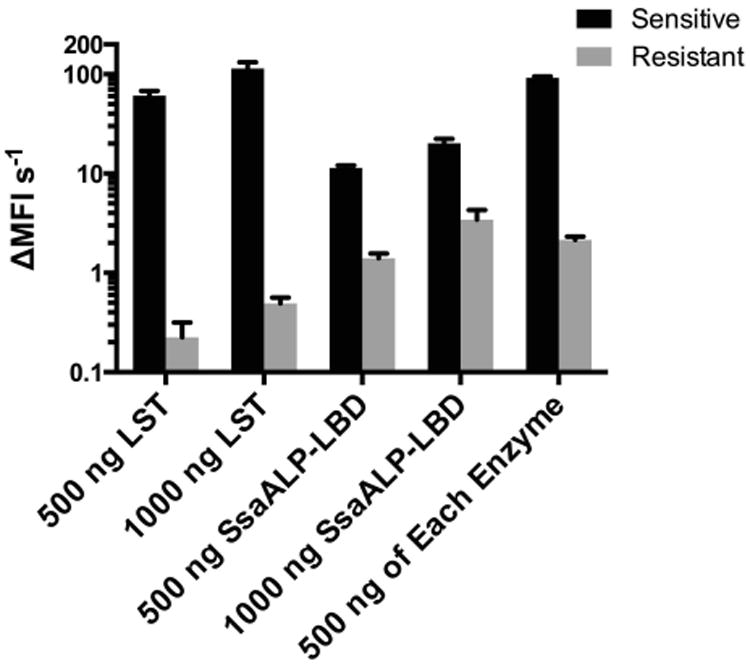
SsaALP-LBD lytic activity towards LST sensitive and resistant strains. Rate of lysis (change in mean fluorescence intensity per second), measured by a fluorescence assay, is shown for the LST-sensitive strain RN4220/PLI50 and the LST-resistant strain RN4220/PLI50∷end epr (Gargis 2010, DeHart 1995). Note the log-scale axis. For both enzymes, 500 ng and 1000 ng correspond to 0.07 and 0.15 μM, respectively. Values represent background subtracted means from triplicate measurements run in biological duplicate, with error shown as standard deviation.
Acknowledgments
We would like to thank Dr. Gary Sloan at the University of Alabama for kindly providing the RN4220 strains used in this paper. We would also like to thank Ony, Inc. for supplying the Infasurf used in this study. This work was supported in part by R21 grant 1R21AI098122 from the National Institutes of Health NIAID to KEG.
Footnotes
The authors claim no conflict of interest.
References
- Becker SC, Dong S, Baker JR, Foster-Frey J, Pritchard DG, Donovan DM. LysK CHAP endopeptidase domain is required for lysis of live staphylococcal cells. FEMS Microbiol Lett. 2009;294(1):52–60. doi: 10.1111/j.1574-6968.2009.01541.x. [DOI] [PubMed] [Google Scholar]
- Bose JL, Lehman MK, Fey PD, Bayles KW. Contribution of the Staphylococcus aureus Atl AM and GL murein hydrolase activities in cell division, autolysis, and biofilm formation. PLoS One. 2012;7(7):e42244. doi: 10.1371/journal.pone.0042244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill EW, Bayles KW. Identification of LytSR-regulated genes from Staphylococcus aureus. J Bacteriol. 1996;178(19):5810–2. doi: 10.1128/jb.178.19.5810-5812.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buist G, Steen A, Kok J, Kuipers OP. LysM, a widely distributed protein motif for binding to (peptido)glycans. Mol Microbiol. 2008;68(4):838–47. doi: 10.1111/j.1365-2958.2008.06211.x. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States. 2013 Threats Report 2013 [Google Scholar]
- Cheung AL, Bayer AS, Zhang G, Gresham H, Xiong YQ. Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus. FEMS Immunol Med Microbiol. 2004;40(1):1–9. doi: 10.1016/S0928-8244(03)00309-2. [DOI] [PubMed] [Google Scholar]
- Chu X, Xia R, He N, Fang Y. Role of Rot in bacterial autolysis regulation of Staphylococcus aureus NCTC8325. Res Microbiol. 2013;164(7):695–700. doi: 10.1016/j.resmic.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36(Web Server issue):W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove SE, Qi Y, Kaye KS, Harbarth S, Karchmer AW, Carmeli Y. The impact of methicillin resistance in Staphylococcus aureus bacteremia on patient outcomes: mortality, length of stay, and hospital charges. Infect Control Hosp Epidemiol. 2005;26(2):166–74. doi: 10.1086/502522. [DOI] [PubMed] [Google Scholar]
- Dehart HP, Heath HE, Heath LS, Leblanc PA, Sloan GL. The Lysostaphin Endopeptidase Resistance Gene (epr) Specifies Modification of Peptidoglycan Cross Bridges in Staphylococcus simulans and Staphylococcus aureus. Appl Environ Microbiol. 1995;61(7):2811. doi: 10.1128/aem.61.7.2811-2811.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan DM, Dong S, Garrett W, Rousseau GM, Moineau S, Pritchard DG. Peptidoglycan hydrolase fusions maintain their parental specificities. Appl Environ Microbiol. 2006;72(4):2988–2996. doi: 10.1128/AEM.72.4.2988-2996.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziarski R, Gupta D. The peptidoglycan recognition proteins (PGRPs) Genome Biol. 2006;7(8):232. doi: 10.1186/gb-2006-7-8-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes S, Proença D, Cantante C, Silva FA, Leandro C, Lourenço S, Milheiriço C, de Lencastre H, Cavaco-Silva P, Pimentel M, São-José C. Novel chimerical endolysins with broad antimicrobial activity against methicillin-resistant Staphylococcus aureus. Microbial drug resistance (Larchmont, NY) 2012;18(3):333–343. doi: 10.1089/mdr.2012.0025. [DOI] [PubMed] [Google Scholar]
- Frankel MB, Hendrickx AP, Missiakas DM, Schneewind O. LytN, a murein hydrolase in the cross-wall compartment of Staphylococcus aureus, is involved in proper bacterial growth and envelope assembly. J Biol Chem. 2011;286(37):32593–605. doi: 10.1074/jbc.M111.258863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel MB, Schneewind O. Determinants of murein hydrolase targeting to cross-wall of Staphylococcus aureus peptidoglycan. J Biol Chem. 2012 doi: 10.1074/jbc.M111.336404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargis SR, Heath HE, LeBlanc PA, Dekker L, Simmonds RS, Sloan GL. Inhibition of the activity of both domains of lysostaphin through peptidoglycan modification by the lysostaphin immunity protein. Appl Environ Microbiol. 2010;76(20):6944–6946. doi: 10.1128/AEM.01066-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz F, Heilmann C, Stehle T. Functional and structural analysis of the major amidase (Atl) in Staphylococcus. Int J Med Microbiol. 2014;304(2):156–63. doi: 10.1016/j.ijmm.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Green MR, Sambrook J. Molecular cloning: a laboratory manual. Molecular cloning: a laboratory manual 2012 [Google Scholar]
- Grundling A, Schneewind O. Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J Bacteriol. 2006;188(7):2463–72. doi: 10.1128/JB.188.7.2463-2472.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokai-Kun JF. Lysostaphin: a Silver Bullet for Staph. Antimicrobial Drug Discovery: Emerging Strategies 2012 [Google Scholar]
- Kusuma C, Jadanova A, Chanturiya T, Kokai-Kun JF. Lysostaphin-resistant variants of Staphylococcus aureus demonstrate reduced fitness in vitro and in vivo. Antimicrob Agents Chemother. 2007;51(2):475–82. doi: 10.1128/AAC.00786-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling B, Berger-Bachi B. Increased overall antibiotic susceptibility in Staphylococcus aureus femAB null mutants. Antimicrob Agents Chemother. 1998;42(4):936–8. doi: 10.1128/aac.42.4.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Schmelcher M, Harty WJ, Foster-Frey J, Donovan DM. Chimeric Ply187 endolysin kills Staphylococcus aureus more effectively than the parental enzyme. FEMS Microbiol Lett. 2013;342(1):30–36. doi: 10.1111/1574-6968.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39(Database issue):D225–9. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PK, Bao Y, Boyer E, Winterberg KM, McDowell L, Schmid MB, Buysse JM. Novel locus required for expression of high-level macrolide-lincosamide-streptogramin B resistance in Staphylococcus aureus. J Bacteriol. 2002;184(20):5810–3. doi: 10.1128/JB.184.20.5810-5813.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellroth P, Sandalova T, Kikhney A, Vilaplana F, Hesek D, Lee M, Mobashery S, Normark S, Svergun D, Henriques-Normark B, Achour A. Structural and functional insights into peptidoglycan access for the lytic amidase LytA of Streptococcus pneumoniae. MBio. 2014;5(1):e01120–13. doi: 10.1128/mBio.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature protocols. 2007;2(9):2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Osipovitch D, Griswold K. Fusion with a cell wall binding domain renders autolysin LytM a potent anti-Staphylococcus aureus agent. FEMS Microbiol Lett. 2014 doi: 10.1093/femsle/fnu035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipovitch DC, Parker AS, Makokha CD, Desrosiers J, Kett WC, Moise L, Bailey-Kellogg C, Griswold KE. Design and analysis of immune-evading enzymes for ADEPT therapy. Protein Eng Des Sel. 2012;25(10):613–23. doi: 10.1093/protein/gzs044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastagia M, Schuch R, Fischetti VA, Huang DB. Lysins: the arrival of pathogen-directed anti-infectives. J Med Microbiol. 2013;62(Pt 10):1506–1516. doi: 10.1099/jmm.0.061028-0. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Resch A, Rosenstein R, Nerz C, Gotz F. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl Environ Microbiol. 2005;71(5):2663–76. doi: 10.1128/AEM.71.5.2663-2676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice KC, Bayles KW. Death's toolbox: examining the molecular components of bacterial programmed cell death. Mol Microbiol. 2003;50(3):729–38. doi: 10.1046/j.1365-2958.2003.t01-1-03720.x. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Rubio L, Martínez B, Donovan DM, Rodríguez A, García P. Bacteriophage virion-associated peptidoglycan hydrolases: potential new enzybiotics. Crit Rev Microbiol. 2013;39(4):427–434. doi: 10.3109/1040841X.2012.723675. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rubio L, Martinez B, Rodriguez A, Donovan DM, Garcia P. Enhanced staphylolytic activity of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA88 HydH5 virion-associated peptidoglycan hydrolase: fusions, deletions, and synergy with LysH5. Appl Environ Microbiol. 2012;78(7):2241–8. doi: 10.1128/AEM.07621-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabala I, Jagielska E, Bardelang PT, Czapinska H, Dahms SO, Sharpe JA, James R, Than ME, Thomas NR, Bochtler M. Crystal structure of the antimicrobial peptidase lysostaphin from Staphylococcus simulans. FEBS J. 2014;281(18):4112–22. doi: 10.1111/febs.12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabala I, Jonsson IMM, Tarkowski A, Bochtler M. Anti-staphylococcal activities of lysostaphin and LytM catalytic domain. BMC Microbiol. 2012;12:97. doi: 10.1186/1471-2180-12-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon TC, Teneback CC, Gill A, Bement JL, Weiner JA, Lamppa JW, Leclair LW, Griswold KE. Enhanced antimicrobial activity of engineered human lysozyme. ACS Chem Biol. 2010;5(9):809–18. doi: 10.1021/cb1001119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlag M, Biswas R, Krismer B, Kohler T, Zoll S, Yu W, Schwarz H, Peschel A, Götz F. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol Microbiol. 2010;75(4):864–873. doi: 10.1111/j.1365-2958.2009.07007.x. [DOI] [PubMed] [Google Scholar]
- Schmelcher M, Powell AM, Becker SC, Camp MJ, Donovan DM. Chimeric phage lysins act synergistically with lysostaphin to kill mastitis-causing Staphylococcus aureus in murine mammary glands. Appl Environ Microbiol. 2012;78(7):2297–2305. doi: 10.1128/AEM.07050-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch R, Lee HM, Schneider BC, Sauve KL, Law C, Khan BK, Rotolo JA, Horiuchi Y, Couto DE, Raz A, Fischetti VA, Huang DB, Nowinski RC, Wittekind M. Combination therapy with lysin CF-301 and antibiotic is superior to antibiotic alone for treating methicillin-resistant Staphylococcus aureus-induced murine bacteremia. The Journal of infectious diseases. 2014;209(9):1469–78. doi: 10.1093/infdis/jit637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95(11):5857–64. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurland S, Zhan M, Bradham DD, Roghmann MC. Comparison of mortality risk associated with bacteremia due to methicillin-resistant and methicillin-susceptible Staphylococcus aureus. Infect Control Hosp Epidemiol. 2007;28(3):273–9. doi: 10.1086/512627. [DOI] [PubMed] [Google Scholar]
- Singh PK, Donovan DM, Kumar A. Intravitreal injection of the chimeric phage endolysin Ply187 protects mice from Staphylococcus aureus endophthalmitis. Antimicrob Agents Chemother. 2014;58(8):4621–9. doi: 10.1128/AAC.00126-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundarrajan S, Raghupatil J, Vipra A, Narasimhaswamy N, Saravanan S, Appaiah C, Poonacha N, Desai S, Nair S, Bhatt RN, Roy P, Chikkamadaiah R, Durgaiah M, Sriram B, Padmanabhan S, Sharma U. Bacteriophage-derived CHAP domain protein, P128, kills Staphylococcus cells by cleaving interpeptide cross-bridge of peptidoglycan. Microbiology. 2014;160(Pt 10):2157–69. doi: 10.1099/mic.0.079111-0. [DOI] [PubMed] [Google Scholar]
- Surovtsev VI, Fedorov TV, Borozdina MA. Michaelis-menten kinetics for determining enzymatic activity of lysostaphin. Biochemistry (Mosc) 2004;69(7):754–6. doi: 10.1023/b:biry.0000040199.64244.b4. [DOI] [PubMed] [Google Scholar]
- Szweda P, Schielmann M, Kotlowski R, Gorczyca G, Zalewska M, Milewski S. Peptidoglycan hydrolases-potential weapons against Staphylococcus aureus. Appl Microbiol Biotechnol. 2012;96(5):1157–1174. doi: 10.1007/s00253-012-4484-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi J, Komatsuzawa H, Yamada S, Nishida T, Labischinski H, Fujiwara T, Ohara M, Yamagishi Ji, Sugai M. Molecular characterization of an atl null mutant of Staphylococcus aureus. Microbiol Immunol. 2001;46(9):601–612. doi: 10.1111/j.1348-0421.2002.tb02741.x. [DOI] [PubMed] [Google Scholar]
- Taubes G. The bacteria fight back. Science. 2008;321(5887):356–61. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- Thomas VC, Hancock LE. Suicide and fratricide in bacterial biofilms. The International journal of artificial organs. 2009;32(9):537–44. doi: 10.1177/039139880903200902. [DOI] [PubMed] [Google Scholar]
- Tillman GE, Simmons M, Garrish JK, Seal BS. Expression of a Clostridium perfringens genome-encoded putative N-acetylmuramoyl-L-alanine amidase as a potential antimicrobial to control the bacterium. Arch Microbiol. 2013;195(10-11):675–81. doi: 10.1007/s00203-013-0916-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer W, Joris B, Charlier P, Foster S. Bacterial peptidoglycan (murein) hydrolases. FEMS microbiology reviews. 2008;32(2):259–86. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Wilkinson BJ, Jayaswal RK. Sequence analysis of a Staphylococcus aureus gene encoding a peptidoglycan hydrolase activity. Gene. 1991;102(1):105–9. doi: 10.1016/0378-1119(91)90547-o. [DOI] [PubMed] [Google Scholar]
- Whisstock JC, Lesk AM. SH3 domains in prokaryotes. Trends Biochem Sci. 1999;24(4):132–3. doi: 10.1016/s0968-0004(99)01366-3. [DOI] [PubMed] [Google Scholar]
- Willson D. Calfactant. Expert Opin Pharmacother. 2001;2(9):1479–93. doi: 10.1517/14656566.2.9.1479. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Antimicrobial resistance: global report on surveillance. 2014:257. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.