

Abstract
Nanoscale synthetic biology can benefit from programmable nanoliter-scale processing of DNA in microfluidic chips if they are interfaced effectively to biochemical arrays such as microwell plates. Whereas active microvalve chips require complex fabrication and operation, we show here how a passive and readily fabricated microchip can be employed for customizable nanoliter scale pipetting and reaction control involving DNA. This recently developed passive microfluidic device, supporting nanoliter scale combinatorial droplet generation and mixing, is here used to generate a DNA test library with one member per droplet exported to addressed locations on microwell plates. Standard DNA assembly techniques, such as Gibson assembly, compatible with isothermal on-chip operation, are employed and checked using off-chip PCR and assembly PCR. The control of output droplet sequences and mixing performance was verified using dyes and fluorescently labeled DNA solutions, both on-chip and in external capillary channels. Gel electrophoresis of products and DNA sequencing were employed to further verify controlled combination and functional enzymatic assembly. The scalability of the results to larger DNA libraries is also addressed by combinatorial input expansion using sequential injection plugs from a multiwell plate. Hence, the paper establishes a proof of principle of the production of functional combinatorial mixtures at the nanoliter scale for one sequence per well DNA libraries.
I. INTRODUCTION
This work addresses the application of programmable biochemical processing, using on-demand droplet generation at the nanoliter scale,1 to the specific example of generating DNA libraries on-chip by reusing DNA.2 DNA libraries play an increasing important role in Molecular and Chemical Biology, Molecular Medicine and Systems Biology, Evolutionary Biotechnology, Synthetic Biology, and a broad range of Information Technology applications ranging from tagging and security to DNA Computing and DNA nanomachines. DNA editing has been elevated from its early role in recombinant technology to the context of general computation following Adleman's work.3
Systematic automation procedures for generating ensembles of DNA date back to the 1980s, for example, with Eigen's evolution machine.4 Whereas shorter DNA can be synthesized accurately by solid phase synthesizers, for synthesizing or copying longer sequences, an error threshold occurs:5 The probability of finding a significant fraction of correct sequences drops sharply once the product of error probability per base and sequence length exceeds a constant near unity.6 In evolution, the reuse of sequences occurs through copying, recombination, and mutation processes, and Eigen's approach was to generate libraries of error mutants from existing sequences that optimally explore functionality in sequence space. In these considerations for evolutionary optimization, DNA reuse schemes such as recombination and gene doubling were also considered.7
More recently, the editing of existing DNA (either a synthesized DNA oligomer or reused DNA from natural sources or previous synthetic work) has been proposed as a word processing like basic competency required for advanced biotechnology.8 The description of a single operation, the Y operation, by which general editing can be achieved provided a simple basis for such an editor. The construction of long DNA molecules for genetic applications has reached notable attention and sophistication with the reconstruction of the genome of a whole organism by the Venter group.9 The process of Gibson assembly,10 also employed in genome assembly, allows multiple DNA assembly steps in a single one-pot isothermal reaction and has proved competitive with assembly PCR.11 We shall make use of both these standard processes for the assembly of DNA from shorter fragments in this paper. While the small library that we create is very simple, especially compared with the sophisticated genome assemblies in cells, we see it as a useful proof of principle for nanoliter scale assembly on demand in a programmable microfluidic device.
DNA library synthesis is a typical application requiring both the production of a large number of separate volumes with distinct contents and a direct interface to integrated downstream processing. The reuse of existing fragments for enzymatic synthesis of DNA has been optimized and automated.2 In particular, the number of enzymatic steps to create a library of DNA has been minimized by automated process flow optimization on liquid handling robots at μl scales. For a first proof of principle of automated DNA library processing at nl scales, we chose a rather simple combinatorial library, not requiring sophisticated process optimization.
While large combinatorial libraries of DNA can be created in a single pot by using random cassettes in solid-phase supported DNA-oligomer-synthesizers, by performing randomized ligase chain reactions, or by mutation processes in amplification reactions such as PCR, there is also a need to create physically separated libraries with many identical DNA copies in each of many samples. Although it is possible to use single molecule separation techniques (e.g., in capillaries, microwell plates (MWPs), or droplets12) and amplification to isolate subpopulations without addressing, e.g., by single molecule PCR or isothermal amplification, the mutational processes associated with enzymatic ligation create subpopulation heterogeneity which is problematic for some applications. Here, we export droplets to a microtiter plate to create a physically separated DNA library.
The on-demand droplet generation device employed here has been developed recently using readily accessible one-layer PDMS microfluidic technology1 and can be operated with macroscopic external valves. This technique, as discussed in Ref. 13, avoids the fabrication complexities of on-chip valves, which either uses multilayer micro fluidics14 or in-connection microvalves,15 by a careful optimization of downstream back-pressure control. Although major achievements have been made in nanoliter to picoliter processing,16 general purpose pipetting solutions are still predominantly integrated with liquid handling robots at the multi-microliter scale.17 Despite the emergence of more flexible integrating and programmable technologies such as “digital microfluidics,” involving droplets transported using EWOD (electrowetting on dielectrics),18 especially for diagnostic applications, these predominantly still operate at scales near to those of pipetting robots (0.3 μl).
Droplet processors in microfluidic channels have successfully bridged the gap down to nanoliter and picoliter scales, by avoiding mechanical boundaries in favor of phase boundaries, but general purpose droplet processing equivalent to pipetting robots at the nanoliter scale has proved elusive, despite reconfigurable microfluidic proposals,19,20 and significant progress in specific tasks.21 For example, work using the Fluidigm assay chip22 can perform a particular pairwise mixing between two sets of 96 samples at the scale of nanoliters, but cannot perform higher order mixtures or other processes. Schemes to create libraries of droplets with fixed flow architectures have been developed23 and other specific microfluidic systems include an oligo-synthesizer24 using microvalves and accelerated DNA recombination on a functionalized microfluidic chip.25
The passive and single layer PDMS microfluidic device1 employed in the current work is general purpose in allowing arbitrary combinations of input streams to be sampled at nanoliter scales under electronic control, allowing general input programmability of nanoliter volume mixes. We demonstrate the programmable on-chip generation of all 70 combinations of 4 out of 8 input solutions in the library construction process at the nl scale. As we shall show below, the number of input DNA solutions is by no means limited to the number of channels in the device, if segmented inputs are employed. Active on-chip, nanoliter scale, droplet on-demand (DoD) dispensing, with programmability expandable in the number of input solutions, allows for possible on-chip integrated processing prior to export from the chip. This is in contrast with direct droplet expulsion techniques. For the custom DoD system, we adopted the dynamic approach above in combination with external valves. Integrating valves increases the complexity (and cost) of devices, inhibiting rapid prototyping by necessitating two-layer- (crossing channel) PDMS-technology that is not readily compatible with thin-layer microfluidics fabrication, which is optimized for optical imaging, stiff pressure control, and low gas permeation. Programmability of the system is completed with a custom programmable sequential droplet export system.
The structure of the paper is as follows. Section II summarizes the experimental materials and methods necessary to design, operate, and evaluate the nanoliter processing system for DNA libraries. These include both the import and export (I/O) mechanisms employed for interfacing to MWPs and the fluorescence imaging, amplification, and product analysis tools employed for library characterization. Section III then presents results on in-chip nanoliter droplet production of the DNA library, assembling DNA strands using either assembly PCR post-processing or the isothermal Gibson reaction, the latter being compatible with full on-chip integration. Finally, we summarize the consequences for general programmable nanoliter scale fluid handling and future directions.
II. MATERIALS AND METHODS
A. Microfluidic system and MWP interface
1. Microfluidic design and system architecture
As developed in Ref. 1, this work employs a novel passive microfluidic chip in conjunction with commercially available off-chip pumps and valves with low dead-volume to support parallel combinatorial dispensing and mixing of nanoliter droplets in-chip. The parallel (8–10) independently controlled active in-chip dispensing nozzles do not limit the number of different solutions that can be mixed. This limitation can be overcome by using sequentially indexed plug flow to interface a microfluidic chip to array formats, such as microwell plates. Fig. 1 shows the overall concept of the input interconnection with MWPs, which can be used to generate larger combinatorial DNA libraries at the nl scale, by interfacing with translation stage control of the MWP. The control of plug movement relative to the nozzle inputs, on the timescale of seconds, is not rate limiting because many droplet combinations can be made for each set of plugs. A related extended combinatorial input concept has also been independently proposed.26
FIG. 1.

Core scheme of droplet-on-demand processor with micro-well-plate input interface. The dynamic DoD microfluidic chip is depicted on the right, with 8 flow-by on-demand inputs from sample plugs individually positioned on the chip from a sequence of such plugs shown in top left. The 96 or 384 well MWP is depicted at lower left. The core scheme is supported by an off-chip pipetting system, syringe pumps, and microvalves, and a custom export to MWP system (see Fig. 3). The basic principle of combinatorial nl-scale on-demand droplet generation is to serially program a sequence of droplets from programmed inputs. Either mixed droplets or digital droplet chains are then exported from the device.
Controlling the external pressure downstream, rather than upstream to the aqueous injection microchannels, as shown in Fig. 2, was a decisive factor in enabling a stable creation of droplets. In contrast to systems with integrated valves, the microfluidic chip employs hydrodynamic barriers (1–2 μm high shallow channels, up to 100 μm in length), which effectively decouple the flows between the carrier fluid and the water phase. This decoupling is the major reason why we are able to operate nozzle-designs with more than two nozzles (currently up to ten nozzles in operation in the same microfluidic system).
FIG. 2.

Chip design and working principle of DoD device.1 (a) An aqueous droplet (blue) is injected into the carrier stream (yellow), a hydrophobic fluid, with a short strong pressure pulse, created via a pinch-valve. A variable flow resistor adjusts a suitable working pressure. An essential innovation in Ref. 1 was to place the pinch-valve after the nozzle. (b) Schematic of actual chip layout. Inlets 2, 4, 6, 8, 14, 16, 18, 20, and 22 are used for reactants or markers and inlets 1, 10, 11, and 24 for carrier fluids, allowing for different surfactant solutions. These inlets are part of a pressure adjustment scheme. Outlet 25 transports the created droplets into an output FEP-tube (Fluorinated Ethylene Polymer, Machery-Nagel GmbH, Dueren, Germany, JR-T-6610-M10 Valco Tubing, ID: 250 μm and OD: 1/32″). Syringes are connected via adapter tubes from Roth Silicone tubes (ID/OD 0.5/2.5 mm). All other inlets are used as backpressure ports for the droplet creation. Reprinted with permission from Biomicrofluidics 9(1), 014119 (2015). Copyright 2015 AIP Publishing LLC. (c) Droplets containing DNA formed by separate nozzles from DoD device, single frame from sequence with Δt = 22 ms. (d) Droplet mixtures including Gibson mix, see Sec. II, here mixed on demand. The ends of the mixed droplet are overlaid, since blurred by the longer exposure, Δt = 140 ms. The scale bars in C and D represent 200 μm.
2. Overall system for nanoliter-scale library creation
The overall experimental system consists of syringe pumps, multiport and pinch valves, variable hydrodynamic flow resistors, two XY-tables (one for microfluidic chip and one for microwell plates), three microscopes (inverse for microfluidic chip viewing, custom tandem for larger area MWP fluorescence imaging, portable-USB for export droplet viewing), a multicolor laser illumination system, capillary connections, a motorized pipette mounted with z-axis, and solenoid push button control, all mounted on a vibration-damped laser table. The use of several powerful microscopes is convenient for analysis but two portable USB microscopes would suffice for operation. Schematics of the input and output control systems are shown in Fig. 3.
FIG. 3.

Setup of the overall DoD system. Left: simplified sketch of the overall setup. For details see text. The pipetting robot dealing with the MWP I/O is not shown. Right: Detail of MWP to microfluidic chip combinatorial interface. One channel out of eight is presented. The solenoid valve is shown in the center. From the MWP, samples are taken and injected into the VICI-valve (left upper part), pushed through the long plug-storing PTFE-tube (Machery-Nagel GmbH, Dueren Germany, JR-T-4183-M10 Valco, ID: OD 0.5 mm: 1/16"). These samples are separated by mineral oil as best separation fluid. Right image reprinted with permission from Biomicrofluidics 9(1), 014119 (2015). Copyright 2015 AIP Publishing LLC.
The optical monitoring system for microfluidic chip and droplet export can be achieved simply using a portable USB microscope (DigiMicro Profi, dnt GmbH, 27× and 100×). In order to analyze the DNA content of nanoliter droplets, two more sensitive fluorescence imaging microscopes were employed, together with laser scanning. For the on-chip measurements, a motorized inverse microscope (Olympus IX81) with three color detection (488 nm, 561 nm, and 640 nm) using polarized continuous wave excitation from DPSS and diode lasers (Coherent) and AOTF wavelength selection was employed, detected by an EMCCD camera (Andor iXion). For MWP imaging, a custom line-scan tandem-optic fluorescence microscope (described elsewhere) with two fixed magnifications was employed to image larger areas efficiently with a scientific CMOS camera (5M-pixel Andor-Zylor). Bromophenol blue (NEB, Inc., 0.33 mM, 0.02%) and white light illumination were also used in addition to fluorescence.
The core droplet-on-demand nozzle control to create programmable nanoliter combinatorial droplets has been described.1 Briefly, each aqueous-DNA input solution requires an outlet on the same side of the injection nozzle. Downstream to these outlets, electronic pinch-valves and variable flow-resistors (custom screw compression design) are placed to control the droplets. Low pitch screws are used to individually compress a silicon adapter, so that the active pressure is adjusted at each nozzle to an optimal working point prior to droplet generation. Thereafter, droplet generation by each nozzle is on demand by activating the downstream electronic pinch valves, with droplet size determined by the duration of closure.
The hardware setup for plug import and the loop-loading and loop-operation (cf. Fig. 1) is shown in Fig. 3. Loading of the plug-tube towards the T-junction is done by switching the VICI-valves appropriately and opening a three-port solenoid-valve to by-pass the microfluidic structure. After inserting the first plug, a spacer-fluid (mineral oil showed best properties) is added and then the next sample from one of the wells of the MWP is injected. Finally, an additional mineral-oil-plug completes the whole plug train. The plug train is then transported to the T-junction when the VICI- and solenoid valves are activated. After filling the loop, the VICI valve is switched back into the default position and the plugs are serially transported via the T-junction into the microfluidic structure. This particular flow scheme revealed the best plug separation properties among several variants examined. Mineral oil also displaces air-bubbles residing in T-junctions and fittings. Before actually creating the payload-plug series, we placed a long mineral-oil plug in advance into the system, because air between a nozzle and the pinch-valve makes droplet generation impossible.
Nanoliter scale combinatorial DNA processing by the chip is completed by exporting the droplets back to MWPs. After testing several more automated methods for export, using solenoid and piezoelectric spotting devices, which were not able to preserve the multiphase fluid structure, the method used in this work was to drop a larger (several μl) mixed-phase droplet containing carrier fluid and a chain of combinatorial aqueous droplets directly into the well of a MWP. This is a temporary contactless dispensing solution, to close the system, and can be complemented by more sophisticated two-phase automated MWP delivery.27 With a handheld USB microscope, we could observe the falling of the drop. An active, high-flow, ejection stream of mineral oil, inserted by an additional, low dead-volume, transparent T-junction right at the output of the system can, in addition, serve for a controlled expelling of the output-droplet. Using colored separation droplets, we could separate sequential droplet chains with different mixtures. This labeling of output droplets allows the export of mixtures without destroying droplets and a minimizing risk of contamination.28 Carefully optimizing the droplet generation procedures allowed avoiding the creation of satellite droplets. Even though a mechanical separation of the different DNA input solutions is not present in such DoD devices, the strong phase separation (oil-water-surfactant surface tension) in combination with the shallow hydrodynamic barriers makes it possible to avoid satellite droplet formation in most cases. Only with high surfactant concentrations, associated with some enzyme-mixtures to be injected into droplets, does the phase-barrier lose efficiency. Appropriate choice of droplet stabilizing surfactant and concentration is then necessary. In contrast, it was significantly more difficult to avoid contamination in the larger plug-flow system, because of satellite droplets adhering to surfaces or edges.
The hardware operating the droplet-on-demand device is distributed around the motorized inverse microscope and is controlled with custom-developed software as described in Ref. 1, which supports compiled and scripted operation.
B. Chemicals, materials, and procedures for DNA library creation and analysis
1. Separation fluids and surfactants
In most cases, mineral oil (Sigma Aldrich M5904–500ML, lot# MKBL2644V) and as a surfactant 0.02% or 0.2% Abil (EM90 from FrankenChemie GmbH & Co. KG) were used. Apart from easily forming droplets (a surfactant concentration of 0.01% up to 0.02% proved to be optimal in the carrier fluid), mineral oil is a good solvent, removing debris from aggregates and impurities in conjunction with the pulses from the pinch-valves, a requisite for long-term reuse of the microstructures. The chip supports operation with two stages of surfactant concentration: low surfactant for droplet generation and mixing and higher surfactant for stabilization and exit.
2. DNA reagents, DNA sequencing, and dyes
A DNA library was designed to test the ability of the microfluidic chip to generate library members at the nl scale. The library was designed to contain just 16 different correct members, choosing one of two variant subsequence types (a or b) at each of four locations (1, 2, 3, 4) (see Fig. 4). Either isothermal assembly (Gibson, see Fig. 4(B)) or thermocycling assembly (assembly PCR, see Fig. 4(C)) can assemble such a library of 8 different components (1a, 1b, 2a, 2b, 3a, 3b, 4a, and 4b) if one can mix in separate wells each of the 16 correct 4-component selections containing exactly one sequence at each of the four locations. Using these 8 components, 70 different 4-component subsets can be produced (binomial 8C4), and these can be used to test the ability of the microfluidic chip to generate larger combinatorial selections—only the correct 16 subsets above should yield correct full-length assembly PCR products. For this library, 150mer oligomer components were employed, as shown in the supplementary material,28 purchased as ultramers (IDT).
FIG. 4.

DNA library combinatorics, assembly and PCR amplification. The mechanism is illustrated with the example of the library member, 1b2b3a4a, from the combinatorial assembly in (A), which was assembled either isothermally or by thermocycling ((B) and (C)), amplified by PCR (D) and sequenced by cloning.28 (A) Design of combinatorial library. Four pairs ((a) and (b)) of DNA ultramers (150mers: 1, 2, 3, and 4) were designed to make the combinatorial library of 24 = 16 full length DNA sequences (525 nt) via overlap subsequences (length of 25 nt, identical for both members of pair). Sequences were otherwise chosen randomly with minimal complementarity (≤8 successive matches). The details of the sequences employed are shown in supplementary Table 1S.28 Arrows indicate the 5′ to 3′ direction of the DNA. (B) Gibson assembly mechanism. (1) The separate dsDNA inputs are shown aligned via hybridizing overlaps (grey boxes). (2) The exonuclease partially digests the strands from the 3′ ends, by sufficient bases to allow strand hybridization. (3) The newly created single stranded tails containing the 25mer overlapping sequences hybridize to form an assembly complex. (4) DNA polymerase fills the gaps between the pre-assembled strands. The 525 nt complex is formed with 6 nicks (on both strands). Exonuclease becomes inactivated before the end of the reaction and cannot compete further with the polymerase. (5) The nicks are ligated by DNA ligase to complete a full-length dsDNA assembly. (C) Assembly PCR mechanism without added primers: (1) Denaturing of the dsDNA 150 bp inputs (95 °C) or ssDNA input. The strands are shown pre-aligned according to the overlaps as in (B). (2) Assembly of pairs of the ultramers via the 25-mer overlapping sequences (50 °C). (3) Extension of the overlaps by polymerase (72 °C). (4) Following thermal cycle: The denaturing step releases the 1-2 and 3-4 strands containing the 2-3 overlap with subsequent assembly of the strands via annealing (50 °C). Formation of the assemblies 1-2-3 as well as 2-3-4 is also possible, depending on whether the initial inputs 2 and 3 are added in ds or complementary ss form. (5) Extension of the overlaps by polymerase (72 °C). (D) PCR exponential amplification of the assembled 525 bp product using primers 1F (left) and 4F (right). The primed PCR procedure included similar steps as in the assembly PCR (C): denaturing (95 °C), annealing (47 °C), as well as extension (72 °C). See Sec. II B 2 for further details.
For each pair of ultramers (e.g., 1a and 1b), we employed two primers (supplementary Table 1S28), in order to amplify (and thereby identify) particular DNA sequence components in the mixture of ultramers. The standard PCR procedure (cf. Fig. 4(D)) had five stages: (1) initial denaturing (95 °C, 5 min), (2) denaturing (95 °C, 30 s), (3) primer annealing (47 °C, 30 s), (4) primer extension (72 °C, 30 s), and (5) final extension (72 °C, 7 min) followed by holding samples at 4 °C for further research (gel analysis, purification, etc.). Gel analysis was then used to prove the composition of some combinatorial DNA mixtures via primed PCR. Additionally, all ultramers were subjected to a sequence analysis, to be sure of their identity and content.28 Sequencing was also employed to test the identity of assembled library output, checking our interpretation of direct amplification mix sequencing via clonal sequencing.
In order to show the scalability of the combinatorial construction beyond the limits of the number of input channels on the microfluidic device, using the principle in Fig. 1, we also designed and purchased two further sequences 1c and 3c (supplementary Table 1S28), expanding the potential full length combinatorial library from 16 to 36.
3. Assembly PCR, Gibson assembly, and gel analysis
Single stranded DNA ultramers 1a, 1b, 2a, 2b, 3a, 3b, 4a, and 4b of length 150 nt were amplified and purified prior to the on chip combinatorial library creation (Qiagen purification kit). The resulting DNA inputs for on-chip combination were in double stranded (ds) form, for either Gibson assembly or assembly PCR. For each full-length library DNA product, four oligonucleotide fragments (one each of a or b from 1 to 4) can produce an overlap assembly ds of 525 bp (Fig. 4). PCR amplification of assembled templates and assembly PCR of four component oligonucleotides were performed with end primers 1F and 4F (supplementary Table 1S28) and Taq polymerase (Fermentas source), using a 16-well thermocycler with heated lid. Various numbers of pre-cycles (without primer) were employed to ensure efficient annealing and elongation of components prior to amplification for assembly PCR.
The standard PCR amplification operates on a double stranded DNA template, which was created either by the Gibson reaction (see below) or at the “pre-PCR” annealing stage in assembly PCR (cf. Figs. 4(B) and 4(C)). Each set of four ultramers (for the sequence input information, see Ref. 28) had 25 nt overlaps, which allow mutual hybridization and subsequent extension of the newly formed templates (1-2, 2-3, 3-4, 1-2-3, 2-3-4, and 1-2-3-4). Assemblies 1-2 (primers 1F and 2F) and 3-4 (primers 3F and 4F) formed under the standard PCR conditions used for amplification of the single ultramers. The fully assembled product 1-2-3-4 required more cycles of annealing and extension, which was implemented by different numbers of “pre-PCR” cycles (supplementary Fig. 4S28). The annealing time for pre-PCR was extended to 2 min at the primer's annealing temperature. The primers for the following amplification of the assembled templates were added extra in the following first round of standard PCR. The reason to separate pre-PCR (2 min annealing of the ultramers) from amplification of the newly generated templates (30 s annealing of primers) was the formation of multiple products due to mis-priming events at the longer annealing times (when primers were added to the mixtures at the pre-PCR stage).
Gibson assembly29 was performed with the NEB master mix (E2611L, NEB, Inc.) under standard conditions unless otherwise specified (50 °C, 5–60 min). Products were analyzed using gel electrophoresis (for details, see supplementary Fig. 4S28).
A principle problem in proving the functionality of Gibson assembly29 is to distinguish it from assembly PCR30 during requisite amplification before gel or sequence analysis. A dilution series (as in Fig. 5) was performed to find a concentration regime for input templates in PCR (in particular, those assembled by the Gibson reaction) yielding sufficient product for gel and/or sequence analysis, but where spontaneous assembly (by assembly PCR) does not occur.
FIG. 5.

Principle of discriminating dilution series. A mixed 6 μl sample, with all four matching overlap DNA fragments (A), is portioned into two 3 μl tubes (B). From the two aliquots, half of the material (1.5 μl) is taken to be supplemented with a Gibson-mix of 1.5 μl) (C). To each half, a set of primers is added: in the first case, the primers for full assembly; in the second case, primers for the first two fragments to be ligated; in the third case, again the primers for the full assembly but without Gibson mix; and in the fourth case, primers for the final two fragments to be assembled without Gibson mix (D). The remaining lower tube at (B) is diluted with 3 μl of water and taken as the input for the next dilution round.
Gibson mix was injected on chip as well as added to the collected DNA mixtures off chip (after export the droplets were centrifuged). The reactions were incubated at 50 °C for 60 min (however, for these shorter DNA sequences, 5 min incubation time gives comparable results) and then the 525 bp outputs were amplified via standard PCR using primers 1F and 4F.
In order to simulate as closely as possible the nanoliter scale with external pipettes (minimum dosage of 100 nl), 100 nl droplet mixtures of different DNA ultramers, diluted in concentration to contain the same amount of DNA as expected from the chip, were added to mineral oil in the MWP. Then the PCR mix (3 units Taq polymerase, 1 μM primers) was added to each well. The mixtures were centrifuged (2500 rpm, 1 min) and transferred to Eppendorf-tubes and therein to the thermocycler. For initial testing of the Gibson reaction, independently of the microfluidic chip, the Gibson mix was added to the DNA mixtures in μl-scale volumes, thoroughly mixed, and pipetted into mineral oil as 100 nl droplets, so that the assembly at 50 °C happened under conditions closest to the microfluidic outputs. Further standard PCR amplification of the assembled products was then performed.
The assemblies were further purified using a Qiagen purification kit, in cases where the PCR product revealed only a pure single band, after the electrophoretic shift mobility assay (EMSA). Alternatively, the 525 bp band was cut out from the gel and DNA was eluted in 1M lithium acetate. DNA was precipitated in ethanol and washed in 80% ethanol. The concentration of DNA was measured after purification, by Nanomag as well as by EMSA, using DNA weight controls (50 bp ladder, NEB). The purified DNA samples were sent to be sequenced directly, without cloning (see supplementary Table 3S28).
The amplified PCR products were analyzed in a native 8% PAAG. The acrylamide/bis-acrylamide mixture (40%, 19:1, AppliChem) was mixed with TEMED (Roth) and ammonium peroxisulfate (Merck) in 50 mM Tris-Acetate buffer (pH 8). The buffer pH was adjusted by titration of 500 mM Tris (AppliChem) by 1M acetic acid (Roth). The gels were formed in the Biozym setup with 1 mm spacers. The gels were placed in the running 50 mM Tris-Acetate buffer and voltage 20 V/cm was applied. The xylene cyanol dye (Fermentas) in the loading buffer served as a marker of DNA mobility in the gels under running conditions. The gel running time was ca. 100 min, then removed from the electrophoresis setup and placed into 1× ethidium bromide solution, stained for 2 min, and washed. The imaging was performed using an INTAS UV light source, camera, and imaging software.
4. Droplet export to external tubing and MWPs
Experiments with earlier designs of the microfluidic structure revealed three key aspects for the successful export of the programmed (and mixed) droplets: (i) combining low surfactant for injection and mixing at the nozzles with high surfactant concentrations required for chip export without coalescence; (ii) dealing with droplet bunching in export tubes; and (iii) keeping the pressure in the droplet generation chip low enough to allow stable droplet pipetting coupled to export.
First, normal droplets without surfactant would stick to the sharp interface at the microfluidic output of the microstructure and droplets that came in contact easily coalesced. Strong droplet stabilization by surfactant interferes with droplet mixing, and had to be introduced downstream of the multi-nozzle. The microfluidic design in Fig. 2 supports this feature.
Second, in the long tube of the output-channel (Fig. 3, outlet 25, fluorinated ethylene polymer—FEP), droplet bunching prevents single droplets from dense chains from being individually exported to a MWP. In recent work,27 we have established that bunching can be resolved systematically using chains of droplets with larger lead droplets, see also Ref. 31. Here, we simply assure a high output flow current and longer pauses between newly created droplets, which of course limited throughput, but this can be resolved in future work using the chip together with the above solution.28 To adapt the droplet flow of carrier fluid with droplets to the larger inner diameter (250 μm) of the FEP output tube, two additional flow stages were incorporated in the micro-fluidic design (1 and 24 in Fig. 3). Additional reagent-mixtures and surfactants for coating can be added here to the flow. In particular, bromophenol blue solution droplets (from nozzle at inlet 22) were employed to guide visual separation of packets of droplets.
Third, to level the pressure build-up when exporting the droplets, we designed deeper output channels (ca. 115 μm). With the fluid-design shown in Fig. 3, we are able to create stable arbitrary droplet patterns if the total output flow-rate remains below 150 μl/h. With the right surfactant and the right flow conditions, droplets could be exported into a MWP as follows. The method of export we used here was to drop a larger (several μl) mixed phase drop containing carrier fluid and a chain of aqueous droplets with programmed content directly into the well of a MWP. With a portable USB microscope, we could observe the falling of the drop. Using colored separation droplets (usually between four and six droplets created with 4 to 6 s delay each), we could separate one droplet chain from another chain of droplets with a different mixture. Surplus mineral oil was removed at the tube output after each drop fell. The export of the mixtures thus proceeded without destroying the droplets and with a minimal risk of contamination. Since completing this work, a more automated droplet export has been established.27
III. RESULTS AND DISCUSSION
The principle and methods for the design and construction of the test library components has been described above. All 70 of the combinations of 4 out of 8 sequences (8C4) were generated on-chip by programming the droplet-on-demand system. Both the strategies of unmixed droplet chains of components and mixed droplet injection were used. First all combinations of DNA oligomers were produced with 1–3 nl sized droplets and exported to a MWP. Then in a second MWP, to facilitate analysis with larger quantities, 7 packets for each combination (with four source DNA droplets) were collected and exported into each well of the MWP in a single oil droplet. Each well then contained 3 × 7 × 4 = 84 nl of 2 μM DNA solution of the 150mer oligomer, with an average size of 3 nl per source droplet.
A. PCR, gel, and sequence analysis of components
To verify the efficacy of the primers employed for PCR amplification, each of the DNA ultramer building blocks for the library was amplified using its own pair of primers and sequenced. Because of the common assembly overlap sequences, the forward and reverse primers amplify both DNA building blocks (example: 1F/1R for 1a as well as 1b). PCR on DNA ultramers revealed amplification of 150-mer output only for the ultramers in the presence of their own primers. Details of gel analysis are presented in supplementary Fig. S228 and sequence analysis (supplementary Fig. S128) then confirmed the correct identity of the component oligomers.
B. Fluorescence analysis of output library mixes from microfluidic chip
To check the contents of droplets, specific DNA template input channels were additionally labeled with fluorescent dyes (Alexa-488, -647), attached to short DNA oligomers to minimize interaction with walls. The results of the fluorescence analysis of the exported mixtures were found to be 100% correct as programmed (see Fig. 6). Note that some individual droplets can still be seen in the wells, prior to fusion: as expected they often show the individual labels of premixed droplets.
FIG. 6.

Fluorescence analysis of exported nl-scale droplet-on-demand DNA library. 80 samples from the 70 possible different combinations of 8 input oligomers were assembled as discrete entities in the DoD device and exported to the MWP. The concentration of the surfactant ABIL was chosen such that droplets did not coalesce during production and export but in the MWP while centrifuged. The photographs shown are made before centrifugation. The green color is used for the 488 nm laser excitation channel and the red for the 640 nm laser excitation. Yellow is the natural color combination of red and green, indicating the presence of both labels, this happened when droplets coalesced in the MWP, a further indicator is the bigger size of the coalesced droplets. Labels were added to the components 2a and 3a only. The left image is that predicted from the DoD control script and the right image shows the assembled images of individual output wells, color recombined. Additional data is presented in supplementary Table 6S.28
DNA microarrays were designed to detect ultramers via hybridization of DNA from a mixture, directly at nanoliter volumes without amplification, using two matching sequences on the microarray for each of the 8-library component DNAs (supplementary Table 2S28). The matching sequences were 25-mers that recognize different loci of the ultramers, giving two independent hybridization events. Details are reported in supplementary Fig. 3S,28 with the general conclusion that despite careful design of controls, direct characterization of the library at the nanoliter scale without amplification was not sufficiently free of background responses to provide unambiguous characterization. Further work below concentrates on PCR amplified output.
C. Assembly PCR of DNA library
Four ultramers (1–4), each of type a or b, were assembled to a 525 base pair construction via overlapping sequences at their 3′- and 5′-ends. First, to optimize the assembly PCR off-chip, different initial amounts of input ultramers were tested, and the number of pre-PCR cycles as well as annealing time of primers during PCR was varied (see supplementary Fig. 4S).28 As a simplest control system for assembly PCR, ultramers 1 and 2 were subjected to the same tests using appropriate primers (see supplementary Fig. 4S(A)).28 Full assembly PCR of each of these four ultramers (0.4 nM) did not reveal any output under the standard PCR conditions (20 cycles) without pre-annealing, whereas the two ultramers test system formed the expected 275 bp product with comparable yield to a single 150-mer PCR. However, with appropriate pre-annealing cycles (2 min, optimally 8), prior to primer addition, full length assembly PCR for the four ultramers could be observed (supplementary Figs. 4S(B), 4S(C), and 4S(E)28). Pre-annealing allows the starting overlap assembly complexes to be stabilized by strand extension to improved templates before other competing amplification side-reactions with primers can occur.
Next, we evaluated the nanoliter scale libraries produced with the combinatorial microfluidic chip. Gel analysis (see Fig. 7) showed that the assemblies have the correct lengths of 525 bp. The 525 bp band was cut out, washed (see Sec. II), and sent to be sequenced. Direct (consensus) sequencing of these samples confirmed that DNA output contains the correct assemblies (see supplementary Tables 3S and 4S)28 for a complete sequence analysis of one manually pipetted sample for comparison. The quality of direct sequencing was limited by the amount of the DNA, the polymerase employed, as well as the quality of the amplified samples (salts, ethanol, acrylamide, etc.). Although there was some DNA contamination in negative controls, this was not unexpected in the physical development lab environment, lacking systematic decontamination control (e.g., UV lamps), and full assembly could indeed be verified in the nanoliter scale synthesis (see supplementary Table S328) by both gel analysis and sequencing. With well-tuned valves, so that satellite droplet formation is avoided, there was no evidence of contamination arising within the microfluidic device. While this is more difficult to ensure for enzyme mixes containing surfactants, these do not need to contain DNA. While amplification of DNA can potentially amplify contaminants as well and does so if no correct set of components is involved, selective primer-based amplification by PCR can actually remove contamination, so that the amount of contamination that can be tolerated in a DNA library project is dependent on post-processing and other factors.
FIG. 7.

Analysis of exported DNA library samples from the microfluidic chip, with post assembly PCR. Lanes (1) 1b2b3a4a, (2) 2b3a3b4b, (3) 1a1b4a4b, (4) 2b3a4a4b (5) 1b2a3a4b, (6) 1b3b4a4b, (7) 1a2b3b4a, (8) 2b3a4a4b, (9) 1b2a3a4a, (10) 1b2b3a4b, (11) 1a2a3b4b, (12) 1b2a2b4b, (13) 2b3a4a4b, (14) 1a1b2b3a, (15) 1a2b3b4b, and (16) 2a2b4a4b. Apart from case (9), which exhibits only a very weak band, all assemblies containing each of 1-4 components were successful, as confirmed in separate experiments by sequencing (see text).
Consensus sequencing of the assembled library products (from the microfluidic chip) following assembly PCR was performed. Only 16 of the 70 possible combinations should give full-length (525) DNA products. Sequencing results show predominantly correct library products in cases where a full-length product can be made (see Fig. 8) and amplification of a minor contaminant template (identical to the product 1b2a3a4b) otherwise. Further details of the sequence analysis are available in supplementary Tables 3S-5S.28 The direct sequencing interpretation (which is error prone towards mixtures) was confirmed by full clonal sequencing of one of the library products (supplementary Table 5S28).
FIG. 8.
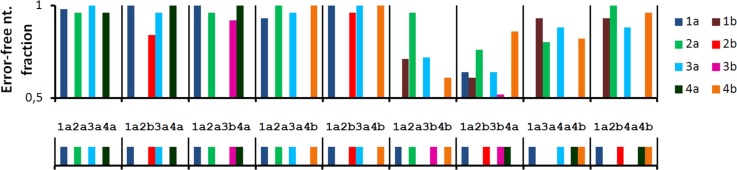
Sequence analysis of samples from nanoliter scale DNA library produced by the microfluidic chip. A selection from the nanoscale chip-based 8C4 library of 4-oligomer subsets, exported to a MWP, was incubated with assembly PCR and sequenced directly (without cloning) after desalting and purification. The figure shows the assembled product identity match (1 = 100%) to all 8 possible color-coded component building blocks, for 9 samples assembled from the 4 building blocks listed in the bottom strip. Seven samples were programmed to contain one representative of each class 1-4, while the two last (8,9) had one class missing. In 5/7 cases where all required building blocks are present, first cases, the correct sequence products are identified. In the cases where one or more of the four classes of building blocks is missing (last two cases), assembly PCR can only amplify minor species: contaminants or misprimed products. In the 2 erroneous samples (6,7), it appears that one of the correct building blocks was also missing: so that the same contaminant or a mixture with partly correct sequence was observed.
D. Discriminating Gibson assembly products from assembly PCR
PCR using end-primers can potentially lead to some overlap assembly during the amplification process, which would then prevent confirmation of nanoliter scale DNA assembly prior to amplification. Using the dilution scheme shown in Fig. 5, concentrations of DNA in the system can be found so that normal PCR can indeed be distinguished from Gibson assembly (see Fig. 9(a)). We found the optimal relation between the amount of Gibson mix and the mixed templates via a series of experiments (see supplementary Fig. 5S28). There is clear evidence that the DNA concentrations employed are in the correct range to show that the Gibson reaction is required to form full length assemblies, and assembly PCR does not interfere with this result (see Figs. 9(b) and 9(c)). Importantly, for full on chip library generation, a further investigation was undertaken to show the feasibility of injecting the Gibson mix directly into the newly created droplets on chip, as a step beyond adding Gibson mix to exported droplets on the MWP. While Gibson on-chip gave weaker bands than in post processing (as expected), the results are clearly confirmed in all 12 cases (see few examples at Fig. 9(d)), with appropriate controls. This ability to sustain on chip enzymatic processing will be an important feature in iterative process library production on-chip in future.
FIG. 9.

Dilution series to distinguish between Gibson and assembly PCR. Performance of Gibson reaction on a chip. (a) Four two-fold dilution rounds (see Fig. 5) are shown from left to right (D0 [60 nM] – D3 [7.5 nM]). For each dilution, four successive lanes show PCR primer amplification of full (525) and half-length (275) products first with and then without the Gibson mix. For details of oligomers used in each lane (see supplementary Table 7S).28 At intermediate concentrations of DNA, D1 [30 nM] (lanes 5 and 8), Gibson assembly must precede normal assembly PCR to obtain full length product, so that the two processes can be distinguished. (b) and (c) Control experiments for the dilution series with either Gibson mix added, no Gibson, or Gibson injected. The dilution series showed that an intermediate DNA concentration allows the clear distinction of Gibson assembly from assembly PCR. With the chosen parameters, this is reliably possible, compare lanes 1, 3, 5, and 7 ((a) Gibson added) with lanes 10 and 12 ((b) no Gibson) and lanes 2 and 5 ((c) no Gibson). Gibson mix injected on-chip gave weaker full-assembly bands than adding Gibson afterwards (see text). (d) Direct evidence that the Gibson reaction is working on-chip, with lanes from the same experiment distributed on two gels. Two mixtures, 1b + 2b and 3a + 4a, were mixed together with Gibson-mix in a three nozzle test structure on the microfluidic chip. Three different cases out of 12 samples in lanes (5, 8, and 11) confirm assembly of 525 bp library member 1b2b3a4a in the mixtures formed in the microfluidic device and collected for further amplification by PCR. As positive controls, in lanes 1 and 9, the results of 100 nl pipette simulated 525 bp assembly are shown, and in lane 10, the off chip assembly with extra Gibson mix added is shown. In addition, controls with primers for shorter partial assemblies also show the existence of the expected assemblies: lanes 3, 4, 6, and 7.
IV. CONCLUSION
In summary, we have employed a recently published custom microfluidic chip1 to do combinatorial generation of a DNA library at the nanoliter scale, exporting it in addressable form to a MWP. The mixtures of DNA used in the library are programmed electronically on demand and not hard wired into the device, and the sequence of exported library members is also programmed, allowing specific export of library members to external MWP locations. The verification of correct DNA library assembly has involved a complex work flow, with cross checks and comparisons (see the overview of Fig. 10). We established the 100% correct nanoliter droplet combinatorial functionality using fluorescence labeling and verified correct assembly in the majority of cases, using both assembly PCR and the on-chip compatible isothermal Gibson assembly process, via gel analysis and sequencing. Some errors arose due to minor contamination by common laboratory templates, as expected in a physical development lab. We also compared on-chip Gibson assembly in mixed nanoliter droplets with Gibson-mix post-processed combinatorial droplets from the chip, showing that although quantities are less, synthesis can indeed be completed on the chip.
FIG. 10.

Workflow employed in verifying DoD on chip library production. The top dashed enclosure contains the on-chip processing. The bottom dashed enclosure is the post-processing of the library including amplification, separation, and sequence analysis. The inputs to the chip are either ssDNA or dsDNA, depending on the assembly method (double strands can be used for both, but single strands were used in the standard assembly PCR process). The Gibson reaction mix was used as an additional input for on-chip assembly (green route) maximizing the on-chip contribution to the nl-scale library production. Combinatorial droplet sequences programmed by the DoD device were either exported as discrete droplet chains or fused and mixed on chip. The output of assembly PCR or Gibson assembly from these on-chip generated droplets was compared with 100 nl-scale pipetted droplets generated off-chip or with Gibson assembly on-chip. The results were PCR-amplified and analyzed by gel electrophoresis and different sequencing methods after cleanup. Fluorescence imaging was used both on- and off-chip to further verify correct processing.
In order to support DNA library production at the nl scale, additional microfluidic interface technology (I/O) was developed to support the transition of automated editing technology down to this scale. The import system allows the extension from eight to arbitrarily large numbers of building blocks, and the partially manual export system has already now been extended to a more automated one.27 Although special purpose (e.g., Fluidigm) commercial chips allow specific tasks to be solved and EWOD allows programmable DNA processing at the sub-μl-scale,32 a generally programmable DNA library synthesis at the nanoliter scale interfaced to MWPs is novel. While the device does not make use of the electronic on-chip processing of chemical microprocessors for iterative rounds of synthesis and product cleanup,20 it could be combined with such on chip microfluidic reaction processing to allow iterative algorithms to be employed for more complex multistage DNA library construction.
Apart from downsizing the DNA processing system, microfluidic droplet on demand technology promises to create integrated systems for DNA libraries, which can eventually be used in smart adaptive point-of-care systems. Such integrated chips can also be standardized and distributed to different labs in multiple copy numbers, to provide better-defined (statistically characterized and reproducible) biochemical environments than with classical pipetting-robots. The proposed system is relatively easy to produce, cheap, and has ample potential for further optimization and integration. The pulses applied by the pinch-valves, in combination with mineral oil, provide an inherent structure cleaning capability considerable prolonging the reusability of the microstructures.
As a further step towards fully programmable electronic control of DNA library creation, the current work pilots one approach to this using a test library, analyzing both the potential and the residual difficulties that still need to be overcome for industrial development of nanoscale automated on demand programmable library synthesis.
ACKNOWLEDGMENTS
The research leading to these results has received funding from the European Commission Seventh Framework Program (FP7/2007-2013) and Horizon 2020 (2014) under Grant Agreement No. 265505 (CADMAD). The authors from the Weizmann Institute contributed the first design of the core DNA test library and initial testing of library MWPs by PCR. The authors at Ruhr Universität Bochum wish to thank especially Jana Bagheri for technical assistance in the preparation (filling, cleaning) of microfluidic devices, solutions, and gels and Thomas Maeke for assistance with device integration and control software.
NOMENCLATURE
- MWP
Microwell plate
- DoD
Droplet on demand
- FEP
Fluorinated ethylene polymer
References
- 1. Tangen U., Sharma A., Wagler P., and McCaskill J. S., Biomicrofluidics 9(1), 014119 (2015). 10.1063/1.4907895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.See http://www.cadmad.eu for E. Shapiro, European Project CADMAD Computer aided design and manufacturing of DNA libraries.
- 3. Adleman L. M., Science 266(5187), 1021 (1994). 10.1126/science.7973651 [DOI] [PubMed] [Google Scholar]
- 4. Eigen M. and Gardiner W., Pure Appl. Chem. 56(8), 967 (1984). 10.1351/pac198456080967 [DOI] [Google Scholar]
- 5. Eigen M., Die Naturwiss. 58(10), 465 (1971). 10.1007/BF00623322 [DOI] [PubMed] [Google Scholar]
- 6. McCaskill J. S., J. Chem. Phys. 80(10), 5194 (1984). 10.1063/1.446590 [DOI] [Google Scholar]
- 7. Eigen M., McCaskill J., and Schuster P., “The molecular quasi-species,” in Advances in Chemical Physics, edited by Prigogine I. and Rice S. A. ( John Wiley & Sons, Inc., Hoboken, NJ, USA, 1989), Vol. 75. 10.1002/9780470141243.ch4 [DOI] [Google Scholar]
- 8. Shabi U., Kaplan S., Linshiz G., Benyehezkel T., Buaron H., Mazor Y., and Shapiro E., Syst. Synth. Biol. 4(3), 227 (2010); 10.1007/s11693-010-9059-y [DOI] [PMC free article] [PubMed] [Google Scholar]; Linshiz G., Yehezkel T. B., and Shapiro E., Methods Mol. Biol. 852, 151 (2012). 10.1007/978-1-61779-564-0_12 [DOI] [PubMed] [Google Scholar]
- 9. Gibson D. G., Glass J. I., Lartigue C., Noskov V. N., Chuang R. Y., Algire M. A., Benders G. A., Montague M. G., Ma L., Moodie M. M., Merryman C., Vashee S., Krishnakumar R., Assad-Garcia N., Andrews-Pfannkoch C., Denisova E. A., Young L., Qi Z. Q., Segall-Shapiro T. H., Calvey C. H., Parmar P. P., Hutchison C. A. III, Smith H. O., and Venter J. C., Science 329(5987), 52 (2010). 10.1126/science.1190719 [DOI] [PubMed] [Google Scholar]
- 10. Gibson D. G., Methods Enzymol. 498, 349 (2011). 10.1016/B978-0-12-385120-8.00015-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stemmer W. P. C., Crameri A., Ha K. D., Brennan T. M., and Heyneker H. L., Gene 164(1), 49 (1995). 10.1016/0378-1119(95)00511-4 [DOI] [PubMed] [Google Scholar]
- 12. Zhu Z., Jenkins G., Zhang W., Zhang M., Guan Z., and Yang C. J., Anal. Bioanal. Chem. 403(8), 2127 (2012); 10.1007/s00216-012-5914-x [DOI] [PubMed] [Google Scholar]; Zhang W. Y., Zhang W., Liu Z., Li C., Zhu Z., and Yang C. J., Anal. Chem. 84(1), 350 (2012). 10.1021/ac2026942 [DOI] [PubMed] [Google Scholar]
- 13. Churski K., Michalski J., and Garstecki P., Lab Chip 10, 512 (2010); 10.1039/B915155A [DOI] [PubMed] [Google Scholar]; Churski K., Nowacki M., Korczyk P. M., and Garsteck P., Lab Chip 13, 3689 (2013). 10.1039/c3lc50340b [DOI] [PubMed] [Google Scholar]
- 14. Huang Y., Castrataro P., Cheng-Chung L., and Quake S. R., Lab Chip 7, 24 (2007). 10.1039/b613923j [DOI] [PubMed] [Google Scholar]
- 15. Abate A. R., Hung T., Mary P., Agresti J. J., and Weitz D., Proc. Natl. Acad. Sci. U.S.A. 107, 19163 (2010). 10.1073/pnas.1006888107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schober A., Günther R., Schwienhorst A., and Döring M., Biotechniques 15, 324 (1993); [PubMed] [Google Scholar]; Simon M. G. and Lee A. P., “Microfluidic droplet manipulations and their applications,” in Microdroplet Technology, Principles and Emerging Applications in Biology and Chemistry, edited by Day P., Manz A., and Zhang Y. ( Springer-Verlag, New York, 2012), Chap. 2, pp. 23-50, ISBN: 978-1-4614-3264-7; [Google Scholar]; Joanicot M. and Ajdari A., Science 309(5736), 887 (2005); 10.1126/science.1112615 [DOI] [PubMed] [Google Scholar]; Seemann R., Brinkmann M., Pfohl T., and Herminghaus S., Rep. Prog. Phys. 75(1), 016601 (2012); 10.1088/0034-4885/75/1/016601 [DOI] [PubMed] [Google Scholar]; Guo M. T., Rotem A., Heyman J. A., and Weitz D. A., Lab Chip 12(12), 2146 (2012); 10.1039/c2lc21147e [DOI] [PubMed] [Google Scholar]; Tran T. M., Lan F., Thompson C. S., and Abate A. R., J. Phys. D: Appl. Phys. 46(11), 114004 (2013); 10.1088/0022-3727/46/11/114004 [DOI] [Google Scholar]; Teh S.-Y., Lin R., Hung L.-H., and Lee A. P., Lab Chip 8(2), 198 (2008). 10.1039/b715524g [DOI] [PubMed] [Google Scholar]
- 17. Gaisford W., J. Lab. Autom. 17(5), 327 (2012). 10.1177/2211068212457160 [DOI] [PubMed] [Google Scholar]
- 18. Blanc R., Castellan G., Chabrol C., David N., Dubard E., Constantin O., Fouillet Y., Jary D., Rival A., Caillat P., and Delattre C., in μTAS, San Diego (2008), p. 1696; [Google Scholar]; Pollack M. G., Fair R. B., and Shenderov A. D., Appl. Phys. Lett. 77, 1725 (2000); 10.1063/1.1308534 [DOI] [Google Scholar]; Cho S. K., Moon H., and Kim C.-J., J. Microelectromech. Syst. 12(1), 70 (2003). 10.1109/JMEMS.2002.807467 [DOI] [Google Scholar]
- 19. McCaskill J. S. and Wagler P., in Field-Programmable Logic and Applications: The Roadmap to Reconfigurable Computing, edited by Hartenstein R. W. and Grünbacher H. ( Springer, Berlin, Heidelberg, 2000), Vol. 1896, p. 286. [Google Scholar]
- 20. Wagler P. F., Tangen U., Maeke T., and McCaskill J. S., Biosystems 109(1), 2 (2012). 10.1016/j.biosystems.2012.01.005 [DOI] [PubMed] [Google Scholar]
- 21. Mark D., Haeberle S., Roth G., von Stetten F., and Zengerle R., Chem. Soc. Rev. 39(3), 1153 (2010). 10.1039/b820557b [DOI] [PubMed] [Google Scholar]
- 22. Jang J. S., Simon V. A., Feddersen R. M., Rakhshan F., Schultz D. A., Zschunke M. A., Lingle W. L., Kolbert C. P., and Jen J., BMC Genomics 12(1), 144 (2011). 10.1186/1471-2164-12-144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adamson D. N., Mustafi D., Zhang J. X. J., Zheng B., and Ismagilov R. F., Lab Chip 6(9), 1178 (2006). 10.1039/b604993a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee C.-C., Snyder T. M., and Quake S. R., Nucleic Acids Res. 38(8), 2514 (2010). 10.1093/nar/gkq092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang F., Zhang Y., Rafeah S., Ji H., Xie S., Ning Y., and Zhang G.-J., RSC Adv. 4(41), 21541 (2014). 10.1039/c4ra02076f [DOI] [Google Scholar]
- 26. Zec H., Rane T. D., and Wang T.-H., Lab Chip 12(17), 3055 (2012). 10.1039/c2lc40399d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sharma A., Tangen U., and McCaskill J. S., “Lead-controlled transport of combinatorial microdroplet trains exported from microfluidic chips” (unpublished).
- 28.See supplementary material at http://dx.doi.org/10.1063/1.4926616E-BIOMGB-9-005504 for seven supplemental tables and five supplemental figures providing further details of the nl-scale DNA library construction and its verification: Sequencing of DNA library components; primers designed and used for PCR of components and assembly PCR of library members; DNA chip oligomers for hybridization to DNA library components; sequencing of DNA library members; fluorescence confirmation of full library; summary of full assembly sequencing; and discrimination of Gibson assembly from assembly during PCR.
- 29. Gibson D. G., Young L., Chuang R.-Y., Venter J. C., Hutchison C. A., and Smith H. O., Nat. Methods 6(5), 343 (2009). 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 30. Kosuri S., Eroshenko N., LeProust E. M., Super M., Way J., Li J. B., and Church G. M., Nat. Biotechnol. 28(12), 1295 (2010); 10.1038/nbt.1716 [DOI] [PMC free article] [PubMed] [Google Scholar]; Xiong A.-S., Yao Q.-H., Peng R.-H., Li X., Fan H.-Q., Cheng Z.-M., and Li Yi., Nucleic Acids Res. 32(12), e98 (2004). 10.1093/nar/gnh094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaminski T. S., Jakiela S., Czekalska M. A., Postek W., and Garstecki P., Lab Chip 12, 3995 (2012). 10.1039/c2lc40540g [DOI] [PubMed] [Google Scholar]
- 32. Xu T. and Chakrabarty K., “Design-for-testability for digital microfluidic biochips,” in 27th IEEE VLSI Test Symposium (IEEE, 2009), pp. 309–314. 10.1109/VTS.2009.16 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- See supplementary material at http://dx.doi.org/10.1063/1.4926616E-BIOMGB-9-005504 for seven supplemental tables and five supplemental figures providing further details of the nl-scale DNA library construction and its verification: Sequencing of DNA library components; primers designed and used for PCR of components and assembly PCR of library members; DNA chip oligomers for hybridization to DNA library components; sequencing of DNA library members; fluorescence confirmation of full library; summary of full assembly sequencing; and discrimination of Gibson assembly from assembly during PCR.