


Abstract
Recent advances in next-generation sequencing technologies have revealed that cellular functional RNAs are not always expressed as single entities with fixed terminal sequences but as multiple isoforms bearing complex heterogeneity in both length and terminal sequences, such as isomiRs, the isoforms of microRNAs. Unraveling the biogenesis and biological significance of heterogenetic RNA expression requires distinctive analysis of each RNA variant. Here, we report the development of dumbbell PCR (Db-PCR), an efficient and convenient method to distinctively quantify a specific individual small RNA variant. In Db-PCR, 5′- and 3′-stem–loop adapters are specifically hybridized and ligated to the 5′- and 3′-ends of target RNAs, respectively, by T4 RNA ligase 2 (Rnl2). The resultant ligation products with ‘dumbbell-like’ structures are subsequently quantified by TaqMan RT-PCR. We confirmed that high specificity of Rnl2 ligation and TaqMan RT-PCR toward target RNAs assured both 5′- and 3′-terminal sequences of target RNAs with single nucleotide resolution so that Db-PCR specifically detected target RNAs but not their corresponding terminal variants. Db-PCR had broad applicability for the quantification of various small RNAs in different cell types, and the results were consistent with those from other quantification method. Therefore, Db-PCR provides a much-needed simple method for analyzing RNA terminal heterogeneity.
INTRODUCTION
Non-protein-coding regions of the genome are widely transcribed to produce non-coding RNAs (ncRNAs), which play crucial roles in normal biological processes and disease states (1). Within the diverse group of ncRNAs, the functional significance is particularly evident for small regulatory RNAs which direct highly specific regulation of gene expression by recognizing complementary RNA targets. Thus far, three major classes of small regulatory RNAs have been particularly studied in depth: microRNAs (miRNAs), short-interfering RNAs (siRNAs) and PIWI-interacting RNAs (piRNAs) (2–6). The defining features of these RNA classes are the short lengths of 20–31 nucleotides (nt), and interactions with Argonaute family proteins, which can be divided into AGO and PIWI subclades, to form effector ribonucleoprotein complexes. miRNAs, the best-studied class of small regulatory RNAs, are produced from stem-loop hairpin-structured precursor RNAs, which are processed by the ribonucleases Drosha and Dicer. The mature ∼22-nt mature miRNAs interact with AGO proteins and recognize complementary sequences in the target mRNAs, which are often located in the 3′-UTR. This recognition results in the deposition of the AGO/miRNA effector complex on the target mRNA, principally resulting in repression of target gene expression (7,8). For target recognition by miRNAs, full complementarity between the miRNA and its target is not required, although base pairing of nucleotides at 2–8 positions of the miRNA, the so-called seed region, is generally essential (2). The human genome encodes over 2000 miRNAs (9), which are estimated to regulate the expression of most protein-coding genes (10), thereby exhibiting a tremendous impact on normal developmental and physiological processes and disease states.
Recent advances in next-generation sequencing (NGS) technologies have revealed the complex heterogeneity of the length and terminal sequences among the majority of mature miRNAs; identical miRNA genes encode mature isomers termed isomiRs that vary in size by one or more nucleotides at the 5′- and/or 3′-end of the miRNA (5′-isomiRs and/or 3′-isomiRs, respectively) (11–13). These isomiRs can be generated by several mechanisms during miRNA biogenesis, including variable processing by Dicer or Drosha cleavage and post-transcriptional modifications, such as nontemplated nucleotide addition, exonuclease-derived trimming and RNA editing (14–19). It has been increasingly apparent that the isomiR expression is functionally significant. isomiRs have been shown to associate with AGO proteins (20–23), and terminal variations of isomiRs influence the AGO protein species on which each isomiR is loaded (24–27). Moreover, 5′-isomiRs have been reported to differentially recognize specific target mRNAs compared with canonical miRNAs due to shifts of the critical seed region (21). Variation of 5′-terminal sequences may also affect the selection of miRNA/miRNA* strands for AGO loading due to changes in the relative thermodynamic stability of the duplex ends (28). Moreover, variations to the 3′-ends of miRNAs to produce 3′-isomiRs are also crucial for gene expression regulation because post-transcriptional nontemplate 3′-end addition of uridines and adenosines markedly alters miRNA stability (29–31). Supporting the importance of terminal heterogeneity of miRNAs, isomiRs are differentially expressed across different cell and tissue types in different developmental stages (32–36). Notably, expression of human isomiRs in lymphoblastoid cells are subjected to population- and gender-based pressures (23).
To unravel the emerging complexities of small RNA heterogeneity and molecular mechanisms underlying them, accurate quantification of individual small RNA variants and easy analysis of their expression profiles are imperative. Microarray analysis of miRNAs is insufficient to completely distinguish miRNAs from their corresponding isomiRs. Although NGS can adequately capture the entire repertoire of miRNAs and their corresponding isomiRs, the required cost, time and subsequent bioinformatics analyses could preclude the use of NGS for only examining specific focused isomiRs. In northern blot analysis, distinguishing different isomiRs with the same or similar lengths is difficult. The TaqMan RT-PCR assay using stem-loop primers (37), which is widely used to quantify miRNAs, is not able to discriminate differences in miRNA terminal sequences of only 1 nt (38). Thus, a novel method for quick and efficient quantification for small RNA variants is required.
In this study, we report the development of dumbbell-PCR (Db-PCR), as an efficient and convenient TaqMan RT-PCR-based method with single-nucleotide resolution at both the 5′- and 3′-terminal sequences to specifically quantify individual small RNA variants. The Db-PCR procedure includes a nick-ligation step catalyzed by T4 RNA ligase 2 (Rnl2) which was originally identified in the bacteriophage T4 (39). Rnl2 catalyzes RNA ligation at a 3′-OH/5′-P nick in a double-stranded RNA (dsRNA) or an RNA–DNA hybrid (40–42), which makes Rnl2 an attractive tool in cDNA preparation (43) and single nucleotide polymorphism detection (44). In this study, stem–loop adapters, termed dumbbell adapters (Db-adapters), were specifically hybridized and ligated by Rnl2 to the 5′- and 3′-ends of a targeted small RNA to generate a ‘dumbbell-like’ structure. Subsequent TaqMan RT-PCR was able to quantify specific target RNA without cross-reactivity to its terminal variant even with a single-nucleotide difference at the terminal sequences. We also developed 5′-dumbbell-PCR (5′-Db-PCR) and 3′-dumbbell-PCR (3′-Db-PCR) to specifically quantify unique variations to the 5′- and 3′-ends of individual small RNAs, respectively. These methods provide a novel, efficient, and convenient technique for specific detection and differential expression analysis of small RNA variants, such as isomiRs.
MATERIALS AND METHODS
Cell culture, RNAi kockdown and total RNA isolation
HeLa, HEK293 and BT-474 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) containing 10% fetal bovine serum (FBS). SK-BR-3, DU145, PC-3 and LNCaP-FGC cell lines were cultured in RMPI1640 medium (Life Technologies) containing 10% FBS. The HepG2 cell line was cultured in minimum essential medium (MEM; Life Technologies) containing 10% FBS. The MDA-MB-231 cell line was cultured in L-15 medium (Life Technologies) containing 10% FBS, and the MCF-7 cell line was cultured in MEM medium containing 10% FBS, 1 mM sodium pyruvate, 1× non-essential amino acids solution (Life Technologies) and 10 μg/ml insulin (Sigma). Culture of the Bombyx mori BmN4 cell line and RNAi knockdown of BmPAPI were performed as previously described (45). Briefly, BmN4 cells were cultured at 27°C in Insect-Xpress medium (LONZA). For RNAi knockdown of BmPAPI, in vitro synthesized BmPAPI-targeting dsRNA or control Renilla luciferase-targeting dsRNA (5 μg) was transfected into 5 × 106 BmN4 cells using the 4D-Nucleofector System (LONZA), and cells were harvested 4 days after transfection. Total RNA from all cell lines was extracted using TRIsure reagent (Bioline) according to the manufacturer's protocol.
Synthetic miRNA and its variants
To examine detection specificities of Db-PCR, the synthetic human miR-16 (5′-P-UAGCAGCACGUAAAUAUUGGCG-3′), its 5′-variants (5′+1: 5′-P-UUAGCAGCACGUAAAUAUUGGCG-3′; 5′-1: 5′-P-AGCAGCACGUAAAUAUUGGCG-3′), its 3′-variants (3′+1: 5′-P-UAGCAGCACGUAAAUAUUGGCGG-3′; 3′-1: 5′-P-UAGCAGCACGUAAAUAUUGGC-3′) and its 5′- and 3′-variants (5′+1/3′-1: 5′-P-UUAGCAGCACGUAAAUAUUGGC-3′; 5′-1/3′+1: 5′-P-AGCAGCACGUAAAUAUUGGCGG-3′) were synthesized by Integrated DNA Technologies. Integrated DNA Technologies also synthesized all the adapters and primers used in this study described below. Northern blot analyses against these synthetic RNAs were performed as described previously (45) by using a 5′-end labeled antisense probe (5′-GCCAATATTTACGTGCTGCTA-3′).
3′-Db-PCR to quantify a specific 3′-variant of small RNAs
The sequences of adapters and primers used for 3′-Db-PCR are shown in Supplementary Table S1. To detect miR-16 by 3′-Db-PCR, a DNA 3′-dummbbell adapter (3′-Db-adapter) was designed by using mfold program (46) to form a stem-loop structure with six nucleotides protruding from the 3′-end that hybridizes with the 3′-terminal sequences of miR-16. The loop sequences of the 3′-Db-adapter were identical with those of a stem-loop primer for miRNA quantification (37). Synthetic miR-16, its variants (20 fmol each), or 1 μg of cellular total RNA was incubated with 20 pmol of the 3′-Db-adapter in a 9-μl reaction mixture at 90°C for 3 min. After adding 1 μl of 10× annealing buffer containing 50 mM Tris–HCl (pH 8.0), 5 mM ethylenediaminetetraacetic acid (EDTA) and 100 mM MgCl2, the total 10-μl mixture was annealed by incubation at 37°C for 20 min. To ligate the annealed adapter to miR-16, 10 μl of the 1× reaction buffer containing 1 U of Rnl2 (New England Biolabs) was added to the mixture. The entire mixture (20 μl) was incubated at 37°C for 1 h, followed by overnight incubation at 4°C. For reverse transcription, the ligated RNA (1 μl) was mixed with dNTP and RT primer, and the 7-μl mixture was incubated at 90°C for 2 min, followed by incubation on ice. The mixture was then subjected to reverse transcription reaction (10 μl volume) using SuperScript III Reverse Transcriptase (Life Technologies) at 55°C for 60 min. The resultant cDNA solution was diluted to 1:5 and 1.5 μl of this solution was added to the real-time PCR mixture containing 5 μl of 2× Premix Ex Taq reaction solution (Clontech Laboratories), 100 nM TaqMan probe, and 2 pmol each of the forward and reverse primers (10 μl in total). With the StepOne Plus Real-time PCR system (Applied Biosystems), the reaction mixture was incubated at 95°C for 20 s, followed by 30 or 40 cycles of 95°C for 1 s and 60°C for 20 s. For detection of Bombyx piRNA-1 (piR-1), piRNA-2 (piR-2) and their corresponding 3′-variants, piR-1-[3′+AGUC] and piR-2-[3′+ACCA], present in BmN4 cells (45), real-time PCR was performed with 400 nM of TaqMan probe, and the reaction mixture was incubated at 95°C for 20 s, followed by 40 cycles of 95°C for 1 s and 50°C or 60°C for 20 s for piR-1 or piR-2, respectively.
5′-Db-PCR to quantify a specific 5′-variant of small RNAs
The sequences of adapters and primers used for 5′-Db-PCR are shown in Supplementary Table S2. To detect miR-16 by 5′-Db-PCR, a DNA/RNA hybrid 5′-Dummbbell adapter was designed to form a stem-loop structure with six nucleotides protruding from the 5′-end to hybridize the 5′-terminal sequences of miR-16. The ligation of the 5′-Db-adapter to miR-16 or its variants and Real-time RT-PCR were performed in the same procedure of 3′-Db-PCR for miR-16 as described above. For detection of miR-26a and its 5′-variant, miR-26a-[5′+A], real-time PCR was performed with 400 nM of TaqMan probe by incubating the reaction mixture at 95°C for 20 s, followed by 40 cycles of 95°C for 1 s and 55°C for 20 s.
Db-PCR to quantify small RNA variants with distinct 5′- and 3′-terminal sequences
The sequences of adapters and primers used for Db-PCR are shown in Supplementary Table S3. To detect small RNAs by Db-PCR, a DNA/RNA hybrid 5′-dummbell adapter containing a spacer (5′-Dbs-adapter) was designed to form a stem-loop structure containing a base-lacking 1′,2′-dideoxyribose spacer in the loop region and a protruding 5′-end complementary to the 5′-terminal sequences of target RNAs. The 5′-Dbs-adapter (20 pmol) was hybridized to 20 fmol of synthetic small RNA, its variants, or 1 μg of cellular total RNA in a 10-μl mixture using the same procedure with 5′-Db-PCR as described above. After adding 10 μl of the 1× reaction buffer containing 1 U of Rnl2 (New England Biolabs), the entire mixture (20 μl) was incubated at 37°C for 30 min. Subsequently, 20 pmol 3′-Db-adapter (1 μl) was added to the reaction mixture, followed by a 30-min incubation at 16°C and then overnight incubation at 4°C. Real-time RT-PCR was performed using the same procedure as described for 3′-Db-PCR with 400 nM TaqMan probe concentration. These reaction mixtures were incubated at 95°C for 20 s, followed by 40 cycles of 95°C for 1 s and 62°C for 20 s. U6 snRNA expression was quantified for use as an internal control using SsoFast EvaGreen Supermix (BioRad) and the following primers: forward, 5′-TCGCTTCGGCAGCACATATAC-3′ and reverse, 5′-CGAATTTGCGTGTCATCCTTG-3′.
RESULTS AND DISCUSSION
Design scheme of dumbbell PCRs to quantify a specific variant of RNAs
The TaqMan RT-PCR using stem–loop primers is a standard method to quantify miRNAs (37). However, this method is inadequate to discriminate 5′-terminal variations of targeted miRNAs because it utilizes a forward primer derived from the interior sequences of the miRNAs (Supplementary Figure S1A). In addition, although stem–loop primer recognizes and hybridizes to the 3′-end of targeted miRNAs, our experiments targeting miR-16 similarly amplified the miR-16 and its 3′-variants that either contain an additional G (miR-16-[3′+G]) or lack a G (miR-16-[3′-G]) at the 3′-end (Supplementary Figure S1B). These results, in agreement with those of previous studies (38), indicate that this method is not capable of distinguishing 5′- and 3′-variants of small RNAs.
We designed methods of 3′-Db-PCR (Figure 1A) and 5′-Db-PCR (Figure 1B) for selective quantification of specific 3′- and 5′-variant of small RNAs, respectively. In 3′-Db-PCR, total RNA is extracted from cells and a 3′-Db-adapter containing protruding 3′-end is hybridized to the 3′-end of the target RNA in the total RNA. In 5′-Db-PCR, a 5′-Db-adapter containing protruding 5′-end is hybridized to the 5′-end of the target RNA. For subsequent Rnl2 ligation, the target RNA in 3′- or 5′-Db-PCR should contain a 3′-OH or 5′-P end, respectively. If the target RNA is expected to contain a different terminal structure, total RNA should be subjected to dephosphorylation/phosphorylation treatment in advance. If the hybridized RNA is the target RNA with exact terminal sequences, the hybridization unites the Db-adapters and the target RNA to form double-stranded nucleotides containing a nick, which is an efficient substrate for Rnl2 ligation (40–42). Following hybridization, Rnl2 ligates the adapter to the target RNA to generate a ligation product with a one-sided ‘dumbbell-like’ secondary structure. Rnl2 ligation efficiency becomes severely reduced when double-stranded nucleotides of the substrate contain gaps or overlaps (41), suggesting that Rnl2 ligation achieves high specificity toward target RNA. Finally, the ligation product is amplified and quantified by TaqMan RT-PCR. The TaqMan probe is designed to target the boundary of the adapter and target RNA for the PCR to exclusively quantify ‘dumbbell-like’ ligation products. Because the TaqMan probe has the ability to discriminate difference of a single nucleotide (47), the design results in highly specific detection of target RNA, which does not cross-react with its 3′- or 5′-terminal variants. Hence, 3′- or 5′-Db-PCR results in highly specific detection and quantification of specific 3′- or 5′-variant of RNAs with single-nucleotide discrimination ability.
Figure 1.
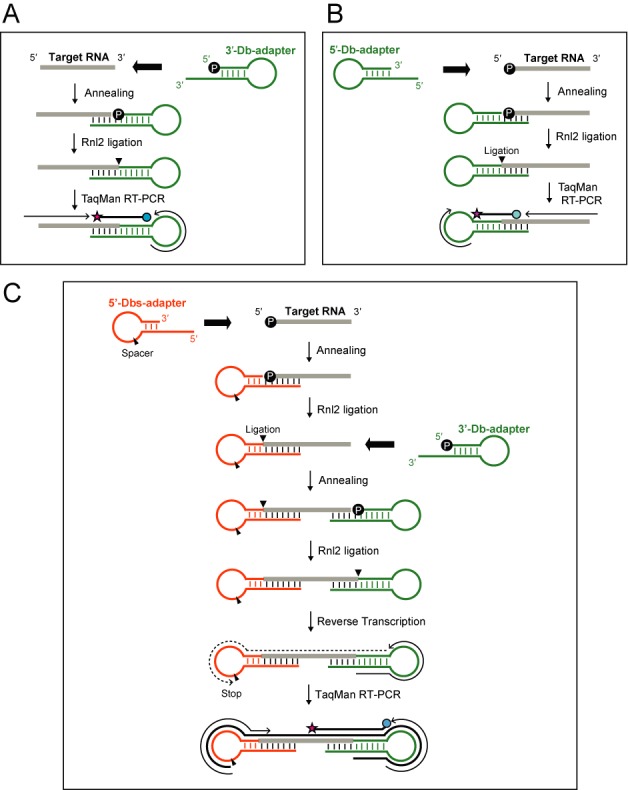
Schematic representation of 3′-Db-PCR (A), 5′-Db-PCR (B) and Db-PCR (C).
We further combined the two methods to design a Db-PCR method for selective quantification of a specific variant of small RNAs to simultaneously discriminate target RNAs from their corresponding 5′- and 3′-variants (Figure 1C). For amplification by Db-PCR, target RNA should contain the both 5′-P and 3′-OH ends. Db-PCR utilizes a 5′-Dbs-adapter (5′-dumbbell adapter with spacer) which was designed to contain a base-lacking 1′,2′-dideoxyribose spacer in the loop region. Db-PCR procedure involves hybridization and Rnl2 ligation of the 5′-Dbs-adapter to the 5′-end of target RNAs, followed by 3′-Db-adapter addition and ligation to the 3′-end. After generating the ‘dumbbell-like’ ligation product, reverse transcription is performed to synthesize cDNAs from the 3′-Db-adapter region. The reaction terminates at the nucleotide preceding a spacer in the loop region of the 5′-Dbs-adapter, which prevents the reaction from continuing to the end of the 5′-adapter and thus generating highly structured cDNAs, which may impair subsequent PCR steps. Subsequent TaqMan PCR is used to quantify the generated cDNAs. TaqMan probes are designed to target the boundary of the target RNA and 3′-Db-adapter. The forward primer is derived from the 5′-Dbs-adapter with a few 3′-terminal nucleotides corresponding to 5′-end of the target RNA, whereas the reverse primer is derived from the 3′-Db-adapter. These designs render PCR amplification completely dependent on ligation of both the 5′- and 3′-adapters to exclusively amplify ‘dumbbell-like’ ligation products. In addition, both 5′- and 3′-terminal sequences are assured by the forward primer and TaqMan probe. Hence, Db-PCR governs highly specific detection and quantification with single nucleotide discrimination ability at both the 5′- and 3′-terminal sequences of target RNAs.
Discriminative quantification of small RNAs and their 3′-variants by 3′-Db-PCR
The 3′-Db-PCR scheme was evaluated by targeting human miR-16, a widely expressed miRNA in tissues and cells (48) (Supplementary Figure S2). The 3′-Db-adapter is composed of DNA with 5′-P and 3′-OH termini, which forms a stem–loop structure containing loop sequences that are identical to those of the stem–loop primer for the standard miRNA quantification method (37) and a protruding 3′-end complementary to the 5′-end of the target RNA (Figure 2A, Supplementary Figure S2). The hybridization of the 3′-Db-adapter with the target RNA forms double-stranded DNA/RNA hybrids containing a nick of ‘RNA-OH-3′/5′-P-DNA’, which is an efficient substrate for Rnl2 ligation (40–42). The 3′-Db-PCR procedure successfully amplified synthetic miR-16 (data not shown) and endogenous miR-16 in total RNA from HeLa cells as a single amplified band (Supplementary Figure S3). The ability of the 3′-Db-PCR to discriminate target RNA from its variants that differ in sequences by a minimum of a single nucleotide was tested with the two 3′-variants containing an additional G (miR-16-[3′+G]) or lacking a G (miR-16-[3′-G]). As shown in Figure 2B, neither of the variants produced a detectable signal by 3′-Db-PCR specifically designed for authentic miR-16. Furthermore, the Ct value from miR-16 was nearly equal to those from the mixtures of miR-16 and its 3′-variants. These results indicate that 3′-Db-PCR specifically and exclusively quantifies authentic miR-16 without cross-reactivity to co-existing 3′-variants. To examine the quantification ability, 3′-Db-PCR was applied for different amounts of synthetic miR-16 (0.01–1 fmol). The quantifications showed clear linearity between the log of miR-16 input and Ct value (Supplementary Figure S4A), indicating that the 3′-Db-PCR method has a dynamic range of at least two orders of magnitude and is capable of quantifying synthetic target RNA. We further utilized this method to quantify endogenous miR-16 in HeLa total RNA (0.5–5 ng). The results showed excellent linearity between total RNA input and detected Ct value (Figure 2C), indicating the capability of 3′-Db-PCR to quantify endogenous target RNA in total RNA.
Figure 2.
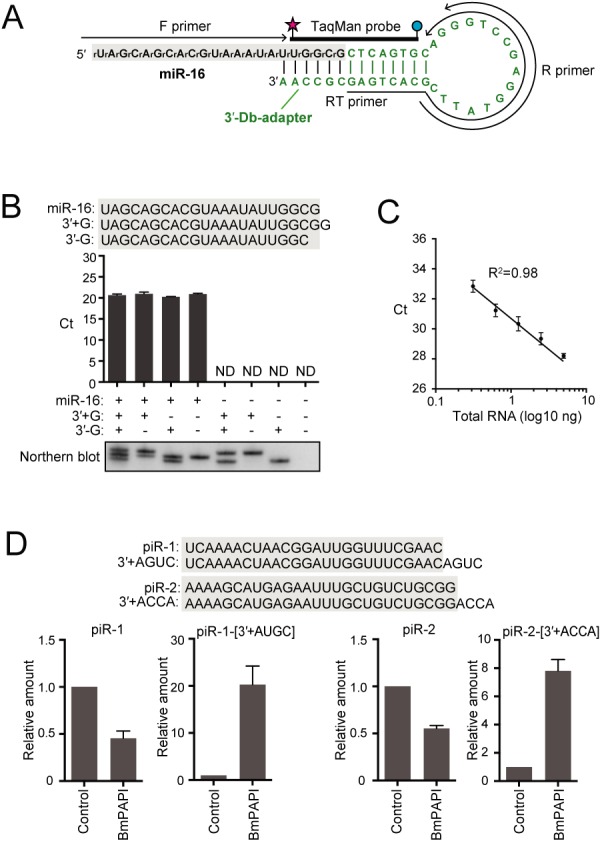
3′-Db-PCR to quantify specific 3′-variants of small RNAs. (A) Sequences and secondary structures of 3′-Db-adapter (green) and targeted human miR-16 (black). A, G, C and T designate DNA, whereas rA, rG, rC, and rU designate RNA. The regions from which the primers and TaqMan probe were derived are shown. An RT primer was used for reverse transcription, whereas forward (F) and reverse (R) primers and TaqMan probe were used for real-time PCR. (B) 3′-Db-PCR was applied for the detection of synthetic miR-16 and its 3′-variants, whose sequences are shown above. miR-16 but not its 3′-variants was specifically amplified by 3′-Db-PCR. The data represents the average Ct values from three independent experiments with bars showing the SD. Northern blot detection of the quantified RNAs is shown below. (C) Proportional correlation of HeLa total RNA input (0.313, 0.625, 1.25, 2.5 and 5 ng) to the Ct value obtained by 3′-Db-PCR. Each data set represents the average of three independent experiments with bars showing the SD. (D) 3′-Db-PCR was applied for the detection of Bombyx piR-1, piR-2, and their 3′-variants expressed in BmN4 cells treated with control or BmPAPI-targeting dsRNAs. The abundance in control cells was defined as 1 and the average relative abundance from three independent experiments are shown with SD bars.
To further examine the discriminative ability of 3′-Db-PCR, we applied the method for quantification of B. mori piRNAs and their 3′-variants expressed in BmN4 cells. BmN4 cells are B. mori ovary-derived cultured germ cells that endogenously express 24–30-nt piRNAs and their bound PIWI proteins, both of which play crucial roles in germline development, and therefore are a unique model system for piRNA research (49). We previously identified BmPAPI as a piRNA biogenesis factor shaping the 3′-end maturation step of piRNAs and found that BmPAPI-depletion causes 3′-terminal extension of mature piRNAs in BmN4 cells (45). We designed 3′-Db-adapters and primers for two BmN4-expressing mature piRNAs, piR-1 and piR-2, and their 3′-variants containing four additional nucleotides, piR-1-[3′+AGUC] and piR-2-[3′+ACCA], whose expression levels were expected to be enhanced upon BmPAPI-depletion (Supplementary Figure S5) (45). 3′-Db-PCR quantifications were then performed using total RNA extracted from BmN4 cells treated with control or BmPAPI-targeted dsRNAs. As a result, BmPAPI depletion commonly reduced piR-1 and piR-2 levels, whereas the levels of piR-1-[3′+AGUC] and piR-2-[3′+ACCA] were markedly upregulated (Figure 2D). Thus, 3′-Db-PCR allowed for discriminative quantification of small RNAs and their endogenous 3′-variants, which recapitulated our previous findings, showing the credibility and general versatility of the method to quantify specific 3′-variants of small RNAs.
Discriminative quantification of miRNAs and their 5′-variants by 5′-Db-PCR
Human miR-16 was targeted again to evaluate the 5′-Db-PCR scheme (Supplementary Figure S6). The stem–loop 5′-Db-adapter contains both 5′- and 3′-OH ends and is composed of DNA except for the last two 3′-terminal nucleotides that are designed as RNA (Figure 3A, Supplementary Figure S6). Therefore, the hybridization of the 5′-Db-adapter with the target RNA generates double-stranded DNA/RNA hybrids containing a nick of ‘RNA-OH-3′/5′-P-RNA’ between the 3′-end of the adapter and 5′-end of the target, which is an efficient substrate for Rnl2 ligation (40–42). The 5′-Db-PCR method successfully amplified synthetic miR-16 (data not shown) and HeLa endogenous miR-16 as a single band (Supplementary Figure S3). Both synthetic 5′-variants of miR-16, containing an additional U (miR-16-[5′+U]) or lacking a U (miR-16-[5′-U]) failed to show detectable signals, and the Ct value obtained from miR-16 was identical to those from the mixtures of miR-16 and its 5′-variants (Figure 3B). These results indicate that 5′-Db-PCR exclusively quantifies authentic miR-16 with the single-nucleotide resolution at 5′-terminal sequences. Clear linearity between the sample input and Ct value was observed for 5′-Db-PCR using different amounts of synthetic miR-16 (Supplementary Figure S4B) and HeLa total RNA (Figure 3C), indicating the quantification ability of 5′-Db-PCR for endogenous target RNAs as well as synthetic RNAs.
Figure 3.
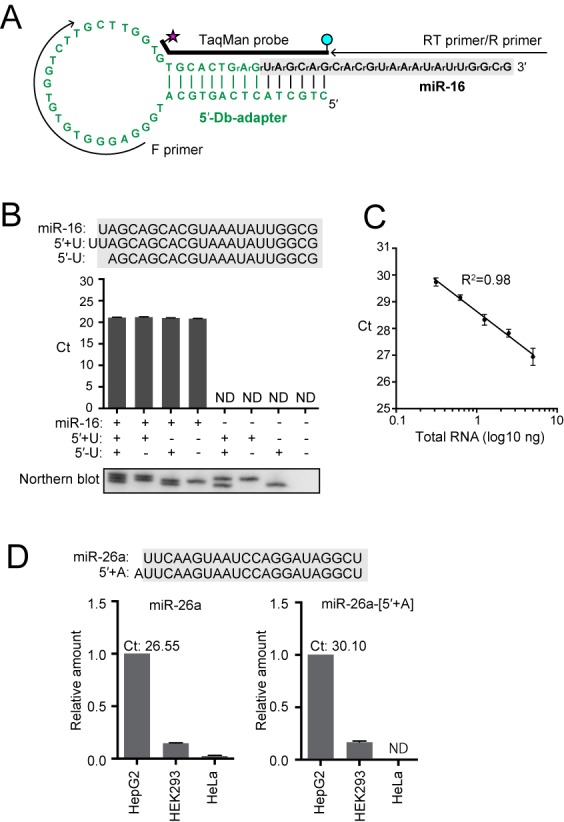
5′-Db-PCR to quantify specific 5′-variants of small RNAs. (A) Sequences and secondary structures of the 5′-Db-adapter (green) and targeted human miR-16 (black). The 5′-Db-adapter contains two 3′-terminal RNA nucleotides, whereas all other parts of the adapter consist of DNA. The regions from which primers and TaqMan probe were derived are shown. (B) 5′-Db-PCR was applied for the detection of synthetic miR-16 and its 5′-variants whose sequences are shown above. miR-16 but not its 5′-variants was specifically amplified by 5′-Db-PCR. The data represents the average Ct values from three independent experiments with bars showing the SD. Northern blot detection of the quantified RNAs is shown below. (C) Proportional correlation of HeLa total RNA input (0.313, 0.625, 1.25, 2.5 and 5 ng) to the Ct value obtained by 5′-Db-PCR. Each data set represents the average of three independent experiments with bars showing the SD. (D) 5′-Db-PCR was applied for the detection of human miR-26a and miR-26a-[5′+A], a 5′-isomiR of miR-26a containing an additional A residue at its 5′-end, in HepG2, HEK293 and HeLa cell lines. The abundance in HepG2 cells was defined as 1, and the average of the relative abundance from three independent experiments are shown with SD bars. The average Ct values of miR-26a and miR-26a-[5′+A] from HepG2 cells were 26.55 and 30.10, respectively.
To further test the credibility and applicability of 5′-Db-PCR, we targeted human miR-26a and miR-26a-[5′+A], a 5′-isomiR of miR-26a containing an additional A residue (Supplementary Figure S7). Tan et al. (32) has recently reported that HepG2 cells express both miR-26a and miR-26a-[5′+A], although expression of miR-26a-[5′+A] was much lower compared to that of miR-26a. According to northern blot analyses, HEK293 cells also expressed miR-26a and miR-26a-[5′+A]. Compared to HepG2 expression, HEK293 expression of both miRNAs seemed much lower, but there was little difference in relative expression levels of miR-26a and miR-26a-[5′+A]. HeLa cells showed an only faint band on northern blot analysis of miR-26a, and no band was detected for miR-26a-[5′+A] (32). We applied 5′-Db-PCR to quantify the two miRNAs in total RNA from HepG2, HEK293 and HeLa cell lines. As shown in Figure 2E, both miR-26a and miR-26a-[5′+A] were successfully amplified by 5′-Db-PCR from HepG2 cells with Ct values of 26.55 ± 0.27 and 30.10 ± 0.27, respectively, suggesting that 5′-Db-PCR distinctively amplified miR-26a and its much less-abundant 5′-isomiR. Relative amounts of miR-26a and miR-26a-[5′+A] in HEK293 cells versus those in HepG2 cells were revealed to be 0.145 ± 0.004 and 0.166 ± 0.011, respectively. miR-26a was quantified by 5′-Db-PCR at much lower amounts in HeLa cells than in the other two cell lines, whereas miR-26a-[5′+A] expression was undetectable in HeLa cells. These results well recapitulated those of a previous report (32).
Discriminative quantification of miR-16 and its variants by dumbbell PCR
Because both 5′- and 3′-Db-PCRs showed high specificity with single nucleotide resolution and quantification ability, we further evaluated the Db-PCR scheme, which was designed by combining the two methods, by targeting human miR-16 (Supplementary Figure S8). Db-PCR utilizes a stem-loop 5′-Dbs-adapter containing a base-lacking spacer in the loop region and the 3′-Db-adapter used in 3′-Db-PCR (Figure 4A, Supplementary Figure S8). In the process of identifying effective 5′-Dbs-adapter constructs, the length of the protruding 5′-end was found to affect detection efficiency. Three different 5′-Dbs-adapters containing 6, 12 or 16 nucleotides protruding from the 5′-end were used. As shown in Figure 4B, the longer protruding 5′-end resulted in more efficient detection, which was most likely due to the high stability of the adapter–target RNA duplex. Thus, the 5′-Dbs-adapter with a 16-nt protruding end was used in the subsequent experiments.
Figure 4.
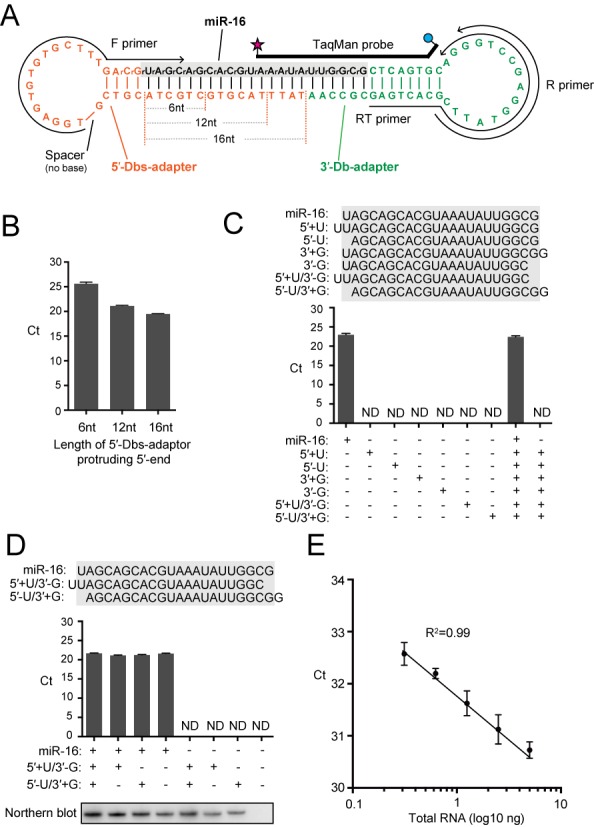
Db-PCR to specifically quantify miR-16. (A) Sequences and secondary structures of the 5′-Dbs-adapter (orange), 3′-Db-adapter (green) and targeted human miR-16 (black). The 5′-Dbs-adapter contains two 3′-terminal RNA nucleotides and a spacer in the loop region. Three different 5′-Dbs-adapters containing 6, 12 or 16 nucleotides protruding from the 5′-end were used. The regions from which primers and TaqMan probe were derived are also shown. (B) Db-PCR using a 5′-Dbs-adapter with 6, 12 or 16 nucleotides protruding from the 5′-end was applied for the detection of synthetic miR-16. The 5′-Dbs-adapter with 16-nt protruding end showed the highest detection efficiency. The data represents the average Ct value from three independent experiments with bars showing the SD. (C and D) Db-PCR was applied for the detection of synthetic miR-16 and its 5′- and 3′-variants whose sequences are shown above the graph. miR-16 was the only RNA species amplified by the Db-PCR. The data represents the average Ct value from three independent experiments with bars showing the SD. Northern blot detection of the quantified RNAs is shown below. (E) Proportional correlation of HeLa total RNA input (0.313, 0.625, 1.25, 2.5 and 5 ng) to the Ct value obtained by Db-PCR. Each data set represents the average of three independent experiments with bars showing the SD.
Db-PCR successfully amplified synthetic miR-16 (data not shown) and endogenous miR-16 in HeLa total RNA as a single amplified band (Supplementary Figure S3). As shown in Figure 4C, all tested 5′- and 3′-variants and their combinatorial variants containing an additional nucleotide or lacking a nucleotide at their termini failed to show detectable signals. In addition, the Ct value obtained from miR-16 alone was nearly equal to those from the mixture of miR-16 and six of its variants, indicating the high specificity of the method toward authentic target RNAs with single-nucleotide resolution at both the 5′- and 3′-terminal sequences. While two of the variants, miR-16-[5′+U/3′-U] and miR-16-[5′-U/3′+U], have the same length with miR-16 and were thus indistinguishable from miR-16 by northern blot, Db-PCR exclusively detected authentic miR-16 (Figure 4D), confirming the high specificity of the Db-PCR toward authentic target RNA, which is not available by northern blot. Db-PCR was further applied for different amounts of synthetic miR-16 (Supplementary Figure S4C) and endogenous miR-16 in HeLa total RNA (Figure 4E). Both cases showed linearity between the log of sample input and Ct value, indicating the capability of Db-PCR to accurately reflect the relative amount of target RNA in total RNA.
Dumbbell-PCR quantification of small RNAs in BmN4 cells and human cancer cell lines
To examine the feasibility and credibility of Db-PCR, we quantified piR-2 and piR-2-[3′+ACCA] in control and BmPAPI-depleted BmN4 cells (Supplementary Figure S9). As exactly shown by 3′-Db-PCR, upon BmPAPI depletion, Db-PCR quantification revealed a reduction in piR-2 levels, whereas the levels of piR-2-[3′+ACCA] were markedly upregulated (Figure 5A). The Db-PCR recapitulation of the results from 3′-Db-PCR suggests the credibility and general versatility of both methods. Moreover, using Db-PCR, the expression profiles of miR-16 and miR-21 were determined in identical amounts of total RNA from eight different human cancer cell lines (Figure 5B). Each cell line exhibited a distinctive signature of the expression of the two miRNAs, and each miRNA showed a distinctive expression pattern in different cell lines. The high expression of miR-21 in MCF-7 compared with those in other breast cancer cell lines was consistent with a previous study of miRNA quantification by microarray analysis (50). Among prostate cancer cell lines, in a previous study, the most abundant miR-21 expression in DU145 cells was observed using northern blot and Real-time PCR analyses (51), which is consistent with our Db-PCR results. These results indicate the credibility and potential broad applicability of Db-PCR.
Figure 5.
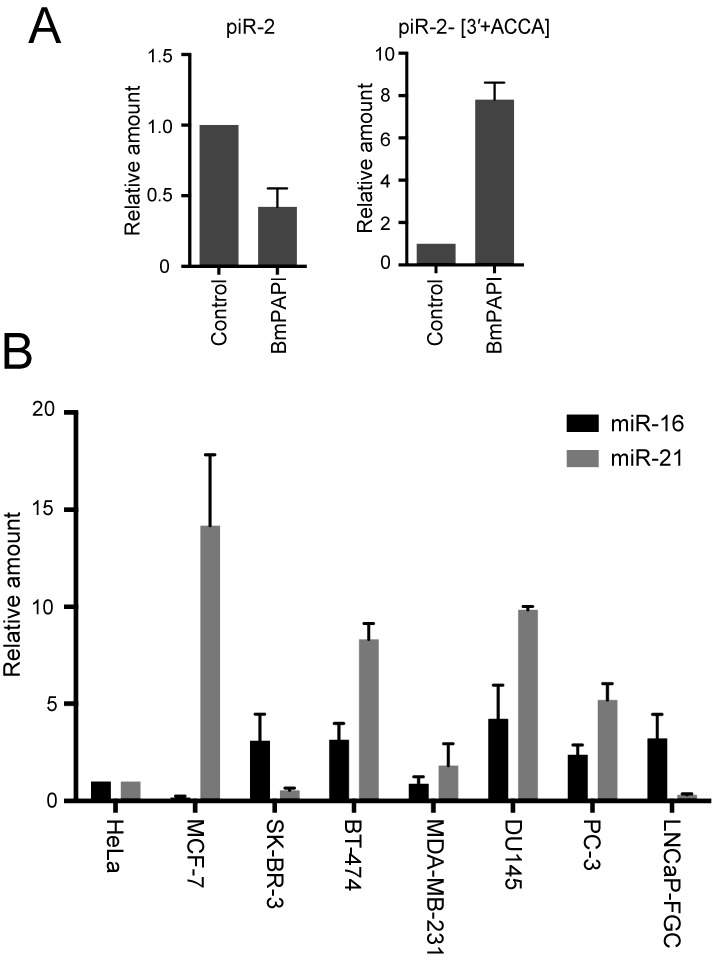
Small RNA quantifications by Db-PCR in BmN4 and human cancer cell lines. (A) piRNAs and their variants in control or BmPAPI-depleted BmN4 cells were quantified by Db-PCR. The abundance in control cells was defined as 1, and the average of relative piRNA amounts from three independent experiments are shown with SD bars. (B) Comparison of the abundance of the miR-16 and miR-21, determined by Db-PCR, in human breast cancer cell lines (MCF-7, SK-BR-3, BT-474 and MDA-MB-231), prostate cancer cell lines (DU145, PC-3 and LNCaP-FGC), and HeLa cells. The abundance in HeLa cells was defined as 1. Each data set represents the average relative abundance from three independent experiments with bars showing the SD.
CONCLUSIONS
We developed efficient and convenient methods to selectively quantify variants of small RNAs by utilizing stem–loop adapter ligations followed by TaqMan RT-PCR. The 5′- and 3′-Db-PCR methods are useful to distinctively quantify 5′- and 3′-variants of small RNAs, respectively. Db-PCR simultaneously identifies specific 5′- and 3′-terminal sequences of target RNAs and quantifies single small RNA species with specific terminal sequences. As our view of functional significance of isomiRs is being expanded, these methods will provide a much-needed simple method to analyze terminal sequence variations of small RNAs, as these factors may play important roles in various biological processes. This method can also shed light on the biological significance of small RNAs coexisting with abundant precursor RNAs in the cells, such as functional tRNA fragments (52), for which specific detection and quantification require discrimination.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to Drs. Megumi Shigematsu and Keisuke Morichika for helpful discussions.
FUNDING
National Institutes of Health (NIH) [GM106047]; institutional funds (to Y.K.). Funding for open access charge: NIH [GM106047 to Y.K.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Esteller M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 2.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siomi M.C., Sato K., Pezic D., Aravin A.A. PIWI-interacting small RNAs: the vanguard of genome defence. Nat. Rev. Mol. Cell. Biol. 2011;12:246–258. doi: 10.1038/nrm3089. [DOI] [PubMed] [Google Scholar]
- 4.Okamura K., Lai E.C. Endogenous small interfering RNAs in animals. Nat. Rev. Mol. Cell. Biol. 2008;9:673–678. doi: 10.1038/nrm2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghildiyal M., Zamore P.D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farazi T.A., Juranek S.A., Tuschl T. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development. 2008;135:1201–1214. doi: 10.1242/dev.005629. [DOI] [PubMed] [Google Scholar]
- 7.Liu X., Fortin K., Mourelatos Z. MicroRNAs: biogenesis and molecular functions. Brain Pathol. 2008;18:113–121. doi: 10.1111/j.1750-3639.2007.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pillai R.S., Bhattacharyya S.N., Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms. Trends Cell Biol. 2007;17:118–126. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 9.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neilsen C.T., Goodall G.J., Bracken C.P. IsomiRs–the overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012;28:544–549. doi: 10.1016/j.tig.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Morin R.D., O'Connor M.D., Griffith M., Kuchenbauer F., Delaney A., Prabhu A.L., Zhao Y., McDonald H., Zeng T., Hirst M., et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman M.A., Mani V., Hammond S.M. Deep sequencing of microRNA precursors reveals extensive 3′ end modification. RNA. 2011;17:1795–1803. doi: 10.1261/rna.2713611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starega-Roslan J., Krol J., Koscianska E., Kozlowski P., Szlachcic W.J., Sobczak K., Krzyzosiak W.J. Structural basis of microRNA length variety. Nucleic Acids Res. 2011;39:257–268. doi: 10.1093/nar/gkq727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu N., Abe M., Sabin L.R., Hendriks G.J., Naqvi A.S., Yu Z., Cherry S., Bonini N.M. The exoribonuclease Nibbler controls 3′ end processing of microRNAs in Drosophila. Curr. Biol. 2011;21:1888–1893. doi: 10.1016/j.cub.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han B.W., Hung J.H., Weng Z., Zamore P.D., Ameres S.L. The 3′-to-5′ exoribonuclease Nibbler shapes the 3′ ends of microRNAs bound to Drosophila Argonaute1. Curr. Biol. 2011;21:1878–1887. doi: 10.1016/j.cub.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A.O., Landthaler M., et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blow M.J., Grocock R.J., van Dongen S., Enright A.J., Dicks E., Futreal P.A., Wooster R., Stratton M.R. RNA editing of human microRNAs. Genome Biol. 2006;7:R27. doi: 10.1186/gb-2006-7-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawahara Y., Zinshteyn B., Sethupathy P., Iizasa H., Hatzigeorgiou A.G., Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burroughs A.M., Ando Y., de Hoon M.J., Tomaru Y., Suzuki H., Hayashizaki Y., Daub C.O. Deep-sequencing of human Argonaute-associated small RNAs provides insight into miRNA sorting and reveals Argonaute association with RNA fragments of diverse origin. RNA Biol. 2011;8:158–177. doi: 10.4161/rna.8.1.14300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azuma-Mukai A., Oguri H., Mituyama T., Qian Z.R., Asai K., Siomi H., Siomi M.C. Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing. Proc. Natl. Acad. Sci. U.S.A. 2008;105:7964–7969. doi: 10.1073/pnas.0800334105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cloonan N., Wani S., Xu Q., Gu J., Lea K., Heater S., Barbacioru C., Steptoe A.L., Martin H.C., Nourbakhsh E., et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011;12:R126. doi: 10.1186/gb-2011-12-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loher P., Londin E.R., Rigoutsos I. IsomiR expression profiles in human lymphoblastoid cell lines exhibit population and gender dependencies. Oncotarget. 2014;5:8790–8802. doi: 10.18632/oncotarget.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeda A., Iwasaki S., Watanabe T., Utsumi M., Watanabe Y. The mechanism selecting the guide strand from small RNA duplexes is different among argonaute proteins. Plant Cell Physiol. 2008;49:493–500. doi: 10.1093/pcp/pcn043. [DOI] [PubMed] [Google Scholar]
- 25.Montgomery T.A., Howell M.D., Cuperus J.T., Li D., Hansen J.E., Alexander A.L., Chapman E.J., Fahlgren N., Allen E., Carrington J.C. Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell. 2008;133:128–141. doi: 10.1016/j.cell.2008.02.033. [DOI] [PubMed] [Google Scholar]
- 26.Mi S., Cai T., Hu Y., Chen Y., Hodges E., Ni F., Wu L., Li S., Zhou H., Long C., et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell. 2008;133:116–127. doi: 10.1016/j.cell.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Czech B., Zhou R., Erlich Y., Brennecke J., Binari R., Villalta C., Gordon A., Perrimon N., Hannon G.J. Hierarchical rules for Argonaute loading in Drosophila. Mol. Cell. 2009;36:445–456. doi: 10.1016/j.molcel.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meijer H.A., Smith E.M., Bushell M. Regulation of miRNA strand selection: follow the leader. Biochem. Soc. Trans. 2014;42:1135–1140. doi: 10.1042/BST20140142. [DOI] [PubMed] [Google Scholar]
- 29.Lu S., Sun Y.H., Chiang V.L. Adenylation of plant miRNAs. Nucleic Acids Res. 2009;37:1878–1885. doi: 10.1093/nar/gkp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katoh T., Sakaguchi Y., Miyauchi K., Suzuki T., Kashiwabara S., Baba T., Suzuki T. Selective stabilization of mammalian microRNAs by 3′ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev. 2009;23:433–438. doi: 10.1101/gad.1761509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boele J., Persson H., Shin J.W., Ishizu Y., Newie I.S., Sokilde R., Hawkins S.M., Coarfa C., Ikeda K., Takayama K., et al. PAPD5-mediated 3′ adenylation and subsequent degradation of miR-21 is disrupted in proliferative disease. Proc. Natl. Acad. Sci. U.S.A. 2014;111:11467–11472. doi: 10.1073/pnas.1317751111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan G.C., Chan E., Molnar A., Sarkar R., Alexieva D., Isa I.M., Robinson S., Zhang S., Ellis P., Langford C.F., et al. 5′ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 2014;42:9424–9435. doi: 10.1093/nar/gku656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozubek J., Ma Z., Fleming E., Duggan T., Wu R., Shin D.G., Dadras S.S. In-depth characterization of microRNA transcriptome in melanoma. PLoS One. 2005;8:e72699. doi: 10.1371/journal.pone.0072699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Somel M., Guo S., Fu N., Yan Z., Hu H.Y., Xu Y., Yuan Y., Ning Z., Hu Y., Menzel C., et al. MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res. 2010;20:1207–1218. doi: 10.1101/gr.106849.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez-Valverde S.L., Taft R.J., Mattick J.S. Dynamic isomiR regulation in Drosophila development. RNA. 2010;16:1881–1888. doi: 10.1261/rna.2379610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bizuayehu T.T., Lanes C.F., Furmanek T., Karlsen B.O., Fernandes J.M., Johansen S.D., Babiak I. Differential expression patterns of conserved miRNAs and isomiRs during Atlantic halibut development. BMC Genomics. 2012;13:11. doi: 10.1186/1471-2164-13-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen C., Ridzon D.A., Broomer A.J., Zhou Z., Lee D.H., Nguyen J.T., Barbisin M., Xu N.L., Mahuvakar V.R., Andersen M.R., et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schamberger A., Orban T.I. 3′ IsomiR species and DNA contamination influence reliable quantification of microRNAs by stem-loop quantitative PCR. PLoS One. 2014;9:e106315. doi: 10.1371/journal.pone.0106315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho C.K., Shuman S. Bacteriophage T4 RNA ligase 2 (gp24.1) exemplifies a family of RNA ligases found in all phylogenetic domains. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12709–12714. doi: 10.1073/pnas.192184699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bullard D.R., Bowater R.P. Direct comparison of nick-joining activity of the nucleic acid ligases from bacteriophage T4. Biochem. J. 2006;398:135–144. doi: 10.1042/BJ20060313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nandakumar J., Ho C.K., Lima C.D., Shuman S. RNA substrate specificity and structure-guided mutational analysis of bacteriophage T4 RNA ligase 2. J. Biol. Chem. 2004;279:31337–31347. doi: 10.1074/jbc.M402394200. [DOI] [PubMed] [Google Scholar]
- 42.Nandakumar J., Shuman S. Dual mechanisms whereby a broken RNA end assists the catalysis of its repair by T4 RNA ligase 2. J. Biol. Chem. 2005;280:23484–23489. doi: 10.1074/jbc.M500831200. [DOI] [PubMed] [Google Scholar]
- 43.Clepet C. RNA captor: a tool for RNA characterization. PLoS One. 2011;6:e18445. doi: 10.1371/journal.pone.0018445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park K., Choi B.R., Kim Y.S., Shin S., Hah S.S., Jung W., Oh S., Kim D.E. Detection of single-base mutation in RNA using T4 RNA ligase-based nick-joining or DNAzyme-based nick-generation. Anal. Biochem. 2011;414:303–305. doi: 10.1016/j.ab.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 45.Honda S., Kirino Y., Maragkakis M., Alexiou P., Ohtaki A., Murali R., Mourelatos Z. Mitochondrial protein BmPAPI modulates the length of mature piRNAs. RNA. 2013;19:1405–1418. doi: 10.1261/rna.040428.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ranade K., Chang M.S., Ting C.T., Pei D., Hsiao C.F., Olivier M., Pesich R., Hebert J., Chen Y.D., Dzau V.J., et al. High-throughput genotyping with single nucleotide polymorphisms. Genome Res. 2001;11:1262–1268. doi: 10.1101/gr.157801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lagos-Quintana M., Rauhut R., Meyer J., Borkhardt A., Tuschl T. New microRNAs from mouse and human. RNA. 2003;9:175–179. doi: 10.1261/rna.2146903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawaoka S., Hayashi N., Suzuki Y., Abe H., Sugano S., Tomari Y., Shimada T., Katsuma S. The Bombyx ovary-derived cell line endogenously expresses PIWI/PIWI-interacting RNA complexes. RNA. 2009;15:1258–1264. doi: 10.1261/rna.1452209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riaz M., van Jaarsveld M.T., Hollestelle A., Prager-van der Smissen W.J., Heine A.A., Boersma A.W., Liu J., Helmijr J., Ozturk B., Smid M., et al. miRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation-specific miRNAs. Breast Cancer Res. 2013;15:R33. doi: 10.1186/bcr3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li T., Li D., Sha J., Sun P., Huang Y. MicroRNA-21 directly targets MARCKS and promotes apoptosis resistance and invasion in prostate cancer cells. Biochem. Biophys. Res. Commun. 2009;383:280–285. doi: 10.1016/j.bbrc.2009.03.077. [DOI] [PubMed] [Google Scholar]
- 52.Shigematsu M., Honda S., Kirino Y. Transfer RNA as a source of small functional RNA. J. Mol. Biol. Mol. Imaging. 2014;1:8. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.