


Abstract
DNA is packaged into condensed chromatin fibers by association with histones and architectural proteins such as high mobility group (HMGB) proteins. However, this DNA packaging reduces accessibility of enzymes that act on DNA, such as proteins that process DNA after double strand breaks (DSBs). Chromatin remodeling overcomes this barrier. We show here that the Saccharomyces cerevisiae HMGB protein HMO1 stabilizes chromatin as evidenced by faster chromatin remodeling in its absence. HMO1 was evicted along with core histones during repair of DSBs, and chromatin remodeling events such as histone H2A phosphorylation and H3 eviction were faster in absence of HMO1. The facilitated chromatin remodeling in turn correlated with more efficient DNA resection and recruitment of repair proteins; for example, inward translocation of the DNA-end-binding protein Ku was faster in absence of HMO1. This chromatin stabilization requires the lysine-rich C-terminal extension of HMO1 as truncation of the HMO1 C-terminal tail phenocopies hmo1 deletion. Since this is reminiscent of the need for the basic C-terminal domain of mammalian histone H1 in chromatin compaction, we speculate that HMO1 promotes chromatin stability by DNA bending and compaction imposed by its lysine-rich domain and that it must be evicted along with core histones for efficient DSB repair.
INTRODUCTION
Packaging of eukaryotic DNA into nucleosomes organizes the genome, but reduces accessibility of proteins, which are required for cellular processes such as repair of damaged DNA, replication, or transcription. To overcome this nucleosome barrier, cells have evolved mechanisms to open chromatin structures, such as the recruitment of ATP-dependent chromatin remodeling complexes. These complexes change the packaging state of chromatin by moving, destabilizing, ejecting or restructuring the nucleosome (1,2).
DNA damage and repair occurs in the context of chromatin. DNA double-strand breaks (DSBs) arise due to either exogenous factors, for example ionizing radiation, or endogenous events such as stalled replication forks. Unrepaired DSBs promote genome instability that may lead to tumorigenesis or cell death, and efficient repair is therefore essential (3). The two major DSB repair pathways are homologous recombination (HR) and nonhomologous end-joining (NHEJ). HR relies on homologous sequences to maintain the fidelity of DNA repair. In eukaryotes, homology recognition and strand exchange is mediated by the recombinase protein Rad51, which is recruited to DSBs after nucleolytic degradation to generate single-stranded 3′-ends (4). NHEJ is considered error-prone. It is initiated by Ku, a heterodimeric protein comprised of Ku70 and Ku80 subunits, which binds free DNA ends and is thought to arrive early at DSB sites. Ku facilitates binding of proteins involved in DNA end-processing and intermolecular end-joining, including Ligase IV, which is required for ligation of broken DSB ends (5).
Chromatin remodeling is an integral part of the DSB response and it is required for the sequential recruitment of DNA repair proteins at the break site. In yeast, one of the earliest events in response to DSB is phosphorylation of histone H2A on serine 129, a modification that spreads from the vicinity of the break in both directions, spanning around 50 kb (6,7). H2A is the primary yeast H2A isoform, yet the phosphorylated version is often referred-to as γ-H2AX since the equivalent phosphorylation event in mammalian cells involves the H2A isoform H2AX (which is absent in yeast) (8). This H2A phosphorylation is required for recruitment and retention of both chromatin remodeling complexes and DNA damage response proteins.
Several chromatin remodelers, including INO80, are recruited to the damage site in a γ-H2A-dependent fashion. INO80 is a conserved member of the SWI/SNF family that remodels chromatin by repositioning nucleosomes along the DNA (9). This remodeling complex contains multiple subunits, including the catalytic subunit Ino80 and three actin-related subunits Arp4, Arp5 and Arp8 (10); deletion of Arp5 and Arp8 mimics an ino80Δ phenotype and such mutants are deficient in DSB repair (10–12). INO80 participates in both HR and NHEJ pathways (11–14), and it is involved in HR-mediated recovery of stalled DNA replication forks (15). Nhp10, a high mobility group (HMGB) protein also known as HMO2, binds DNA ends and is present only in the INO80 complex and not in SWR1 or other known chromatin remodeling complexes and it is required for INO80 recruitment to γ-H2A (13,16). Major roles of INO80 include histone displacement and nucleosome disruption to enable the recruitment of repair proteins; after the completion of DNA repair, histone redeposition restores the chromatin structure (17).
HMGB proteins are non-histone DNA binding proteins with established roles in chromatin organization or dynamics (18). Saccharomyces cerevisiae contains 10 HMGB proteins, of which Nhp6 and HMO1 have been shown to affect chromatin structure. Deletion of the hmo1 gene makes the chromatin hypersensitive to nuclease (19), which indicates a general role for HMO1 in stabilizing higher order chromatin structures. In addition, hmo1Δ strains exhibit increased mutagenesis frequency (20); it was subsequently suggested that this may be explained by the ability of HMO1 to prevent lesions from entering error-prone repair pathways (21). HMO1 has two DNA binding domains, box A and box B, and a lysine-rich C-terminal extension. HMO1 bends DNA and both box A and the basic C-terminal extension is required for such changes in DNA topology (22–24). The lysine-rich C-terminal extension also confers on HMO1 the ability to compact DNA, as evidenced by enhanced DNA end-joining (23).
The nuclease-sensitive chromatin phenotype associated with hmo1 deletion is surprising by comparison to mammalian HMGB proteins, which are thought to promote flexible chromatin structures by competing with histone H1 for binding to linker DNA; by contrast, the role of yeast H1 in chromatin organization appears more limited (18,25). We show here that HMO1 stabilizes chromatin as evidenced by faster chromatin remodeling in its absence. This stabilization requires the lysine-rich C-terminus. Specifically, H2A phosphorylation, recruitment of INO80 to a DSB site, histone H3 eviction, and DNA resection is more efficient in an hmo1Δ strain, and HMO1 is evicted along with core histones during DSB repair. Furthermore, we show that these events correlate with more efficient repair by both HR and NHEJ in hmo1Δ strains, that absence of HMO1 promotes recruitment of Rad51, even in absence of induced DSBs, and that tracking of Ku protein from DNA ends correlates with efficient chromatin remodeling. We suggest that HMO1 stabilizes higher order chromatin structures, perhaps by its lysine-rich domain promoting DNA compaction, and that its eviction is important for efficient DSB repair.
MATERIALS AND METHODS
Strain construction
Strains are derived from either DDY3 or the donorless JKM179, which lacks HML and HMR loci on chromosome 3 and contains an integrated galactose-inducible HO endonuclease gene (26,27). DDY3 is isogenic to W303-1A. DDY-AB, which encodes a truncated version of HMO1 deleted for its C-terminal extension, was previously described (28). The DDY1299 derivative of DDY3 in which hmo1 is deleted was also previously described (28); strain JKM179hmo1Δ was created using the same approach, except that the selection marker URA3 was amplified from pRS426 (29). The gene encoding Ku was deleted by amplifying the URA3 marker with primers that include ∼80 nt of flanking sequence homologous to the ku gene, followed by transformation of either DDY3 or DDY1299 haploid cells to generate DDY3kuΔ and DDY3hmo1ΔkuΔ, respectively. A strain expressing HMO1-FLAG was created from DDY3 using primers amplifying the selection marker kanamycin. All strains are described in Supplementary Table S1.
ChIP and PCR analysis
Chromatin Immunoprecipitation (ChIP) was performed as described (26), with minor modifications. Yeast cells were grown at 30°C in 2% raffinose-containing YP or in synthetic defined (SD) dropout media to an optical density at 600 nm of 1.0. A 100 ml culture aliquot was removed and utilized as the uninduced sample for the ChIP assay. Galactose was added to the remaining culture to a final concentration of 2% to induce HO, and cells were collected at different time intervals for the ChIP assay. To repress HO expression and prevent further DNA damage, 2% glucose was added and cells were harvested at different time intervals for ChIP assay. Cells were fixed with formaldehyde (37%) diluted to 1.2% in the culture medium and incubated at room temperature for 20 min with gentle shaking. Cells were lysed by vortexing with glass beads for 40 min at 4°C using lysis buffer (50 mM HEPES–KOH, pH 7.5, 140 mM NaCl, 1% Triton X-100 and 0.1% sodium deoxycholate) containing protease inhibitors, pepstatin A (1 μg/ml), leupeptin (1 μg/ml) and phenyl methyl sulfonyl fluoride (PMSF 100 mM). To shear chromatin into 100–2000 bp fragments (predominant size ∼500 bp), the lysate was sonicated six times for 10 s each at 25% amplitude while keeping the samples on ice intermittently. Sheared chromatin was then aliquoted for ChIP reactions (100 μl of lysate). To reduce the non-specific binding to Sepharose beads, the lysate was precleared using protein G-Sepharose beads (GE Healthcare). For immunoprecipitation, the following antibodies were used: 5 μl of anti-FLAG (Sigma), 5 μl of antibody against phosphorylated H2A (Ser 129) (Merck Millipore), 2 μl of anti-Rad51 (Santa Cruz Biotechnology), 2 μl of anti-Arp5 (Abcam), 2 μl of anti-H3 (Abcam) and 2 μl of antibody against Ku (30).
Extracted DNA from ChIP samples or input DNA was analyzed by PCR; monitored loci included MAT (72 bp downstream of the DSB), 0.2 kb upstream, 3.1 kb downstream, 9.5 kb downstream and 29.5 kb upstream of the DSB and at POL5. PCR products were loaded on 1.4% agarose gels containing 0.01% ethidium bromide. Primer sequences are provided in Supplementary Table S2. Signal intensities from PCR data were quantified from the TIFF images by using ImageJ software (31) with some modifications. Images were first transformed to 16-bit-type images, and the threshold function was set to black and white type of image to avoid background interference. The rectangle tool was used to define the area around PCR bands. Fold enrichment was calculated as signal intensity ratio of ChIP/Input DNA. To validate the semi-quantitative PCR approach, the presence of histone H3 was also determined using quantitative real-time PCR (qRT-PCR). qPCR was conducted using an ABI Prism 7000 sequence detection system and SYBR Green for detection. Data were normalized to input control. Each experiment was repeated three times and average and standard deviations (SDs) are reported.
Survival following DSB induction
Strains of JKM179 background possess a genomic galactose inducible HO endonuclease gene. For DDY3-derived strains, the galactose-inducible HO endonuclease gene was furnished on a centromeric plasmid, with DDY3hmo1FLAG, DDY3 and DDY1299 transformed with plasmid carrying URA3 marker and DDY3-AB, DDY3kuΔ, and DDY3hmo1ΔkuΔ transformed with plasmid carrying the TRP marker. Cells of JKM179 background were grown at 30°C in raffinose-containing YP media. Transformed strains DDY3hmo1FLAG, DDY3, and DDY1299 were grown in SD drop out media minus uracil and DDY3-AB,DDY3kuΔ and DDY3hmo1ΔkuΔ were grown in SD drop out media minus tryptophan. Cells were grown at 30°C to an optical density at 600 nm of 1.0, at which point 2% galactose was added to induce HO and DSB for 4 h. To monitor survival, 0.1 ml cell culture was plated at 10−3 dilution on YPD or SD drop out agar media in replica and incubated at 30°C. Cultures to which no galactose was added were plated as a control. After 48 h colonies were counted. Each experiment was repeated three times and data reported as mean with standard deviations.
To monitor cell viability, a trypan blue exclusion assay was used. Cells were grown at 30°C to OD600 of 1.0, at which point 2% galactose was added to induce HO for 4 h. Cells were collected and mixed with 0.4% trypan blue (1:1 vol/vol), placed on a hemocytometer, and immediately examined under an inverted microscope. The fraction of dead cells is reported as the number of blue cells divided by total number of cells. The assay was repeated three times and average and standard deviations (SD) are reported.
DNA end resection
Cells were grown at 30°C to an OD600 of 1.0, and DSB was induced with 2% galactose. Cells were harvested after induction times of 20 min, 1 h, 2 h, 3 h and 4 h. Genomic DNA was extracted by vortexing cells with glass beads and phenol. Twenty microliters of genomic DNA sample (60 ng in 1× NEB exonuclease I buffer) was digested with 20 units of Escherichia coli exonuclease I at 37°C overnight. The level of DNA resection adjacent to the specific DSB was measured by qPCR using primers annealing 1.6 kb upstream of the DSB. All values were normalized to values for an independent locus on chromosome 5 (POL5). The assay was repeated three times and average and SDs are reported.
RESULTS
Efficient DSB repair in absence of HMO1
To determine how the presence of HMO1 affects DSB repair, we monitored cell survival after induction of a DSB. HO endonuclease introduces a single DSB in the mating type (MAT) locus, and repair by HR, the pathway of choice, involves one of the homologous silent mating type HM cassettes as a donor to create a MAT gene of the opposite mating type. A survival assay was performed using DDY3 (26) and DDY3hmo1Δ (28), which possess HMLα and HMRa loci and preferentially repair DSB by HR. Yeast cells were transformed with plasmid carrying galactose-inducible HO, DSB was induced by galactose, and cells were plated with glucose to allow repair. The survival assay indicated that recovery from DSB induction was ∼2-fold more efficient in the DDY3hmo1Δ strain compared to the isogenic WT (Figure 1A). No difference in plating efficiency was observed for cells not producing HO. To ensure that differential survival was not due to different efficiencies of DSB induction in the two strains, qPCR was performed using primers that flank the DSB site, revealing no significant difference in DSB induction between the two strains (Supplementary Figure S1). Since HMO1 was reported not to localize to the GAL promoter used to drive expression of HO (32), glucose repression of HO expression is unlikely to be affected by the absence of HMO1. We verified viability of cells by staining with trypan blue; while cells not induced to produce HO were viable, a significant proportion of dead cells were observed in DDY3 after continuous HO induction, whereas this fraction was significantly reduced in hmo1Δ cells (Figure 1C). This finding suggests that DDY3hmo1Δ repairs DSBs more efficiently than WT.
Figure 1.
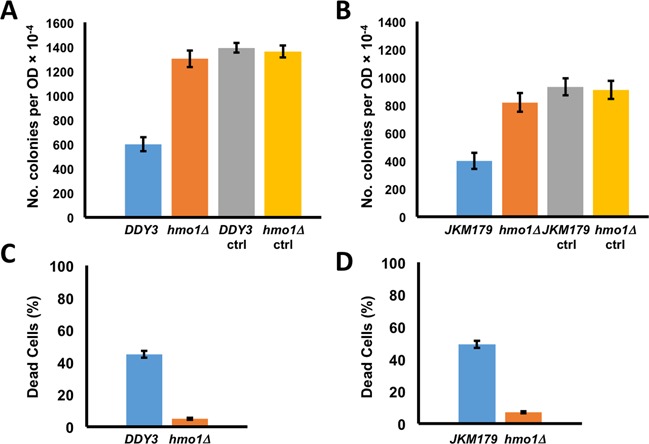
Survival of WT and hmo1Δ strains following induction of DNA double strand breaks. (A) Survival of DDY3 and corresponding hmo1Δ strain. (B) Survival of JKM179 and corresponding hmo1Δ strain. After DSB induction, cells were diluted 104-fold and plated and colonies counted. Survival is represented as colonies per OD. Cells not induced to express HO were plated as control (ctrl). (C) Viability of DDY3 and corresponding hmo1Δ strain as determined by trypan blue exclusion. (D) Viability of JKM179 and corresponding hmo1Δ strain as determined by trypan blue exclusion. Three independent experiments were performed. Error bars represent standard deviation.
To verify that the hmo1 deletion did not compromise fidelity of HR or change the preferred repair pathway, a fidelity experiment was performed with DDY3 and DDY3hmo1Δ. Result showed efficient mating type switching from MATa to MATα with no evidence of residual MATa cells (Supplementary Figure S2), indicating that repair proceeded by HR in both strains and not by NHEJ. Sequencing confirmed fidelity of the repair. These results suggest that DSB repair by DDY3hmo1Δ proceeds by HR and without compromising the fidelity.
To rigorously rule out the possibility of repair by NHEJ, the survival assay was repeated with strains in which the gene encoding Ku80, which is indispensable for NHEJ, was inactivated. Data were consistent with the previous result, showing ∼3-fold greater survival in the DDY3hmo1ΔkuΔ strain compared to DDY3kuΔ (Supplementary Figure S3A). That the hmo1 deletion resulted in even greater repair efficiency (survival) in a kuΔ background is intriguing and may reflect that competition between HR and NHEJ protein recruitment to the DSB site is attenuated in absence of Ku. The lower plating efficiency of DDY3kuΔ compared to DDY3 after HO induction is reflected in a greater proportion of inviable cells in the DDY3kuΔ strain (Supplementary Figure S3B and C).
To assess if the role of HMO1 is specific to HR, the survival assay was performed with the donorless (HMLα and HMRa deleted) JKM179 (27) and JKM179hmo1Δ, which repair DSB by NHEJ. Again, we found ∼2-fold greater survival in JKM179hmo1Δ compared to the isogenic parent JKM179 (Figure 1B) and greater viability in absence of HMO1 (Figure 1D). Measurement of DSB induction by qPCR showed no significant difference between the two strains (Supplementary Figure S1). Taken together, these data indicate that hmo1Δ strains repair DSB more efficiently than WT via both HR and NHEJ.
HMO1 localizes to the MAT locus and is evicted during DSB repair
The observation that HMO1 affects the efficiency of DSB repair by both HR and NHEJ suggests that the effect is repair pathway independent, perhaps affecting upstream events such as chromatin remodeling. To address if HMO1 localizes directly to the MAT locus, we used a Flag-tagged HMO1 strain (DDY3 background) and performed chromatin immunoprecipitation (ChIP) to monitor localization of HMO1 at MAT, 0.2 kb upstream, 9.5 kb downstream and 29.8 kb upstream from the DSB site during DSB induction and repair. HMO1 was found to localize evenly throughout the locus (Figure 2; 0 h). By comparison, genome-wide analysis of HMO1 localization revealed that HMO1 binding is variable throughout the genome, with particular enrichment of HMO1 at ribosomal protein promoters (as high as ∼11-fold above background), and 2.7-fold above background at the MAT locus (32). When DSB was induced continuously for 4 h, we observed gradual loss of HMO1 at all monitored locations beginning after 2 h of DSB induction, with complete disappearance after 4 h, even at the most distant site 29.8 kb downstream (Figure 2A and B).
Figure 2.
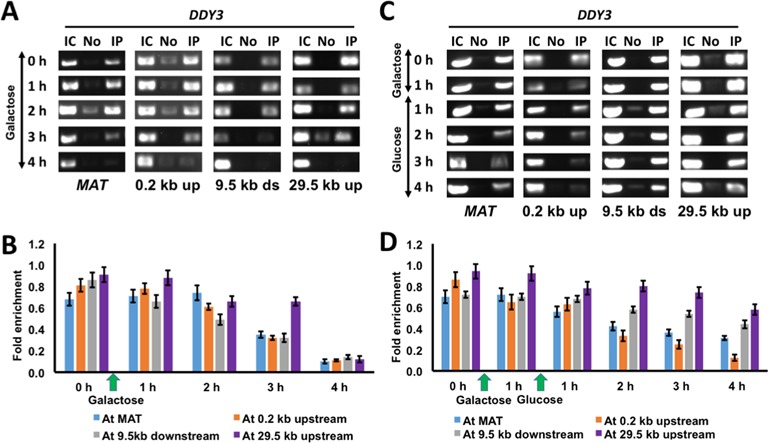
HMO1 eviction from the vicinity of DSB. (A) ChIP showing HMO1 localization at indicated loci relative to the DSB site during continuous damage induced by galactose. IC, input control; No, no antibody; IP, immunoprecipitation with antibody to FLAG-tagged HMO1. (B) Densitometric semi-quantitative analysis of ChIP data shown in (A), obtained with ImageJ software. (C) ChIP showing HMO1 localization at indicated loci relative to the DSB site after DNA damage (galactose) and during repair (glucose). (D) Densitometric analysis of ChIP data shown in (C). Fold enrichment = ChIP/Input DNA. Three independent experiments were performed. Error bars represent standard deviation.
To determine HMO1 localization during DNA repair, DSB was induced for 1 h, following which further DNA damage was prevented by the addition of glucose. We found significant loss of HMO1 after 2 h of repair, especially at sites proximal to the DSB (Figure 2C and D). To verify that differential HMO1 localization is specific to the DSB site, we monitored HMO1 localization at the POL5 gene as a control (HMO1 occupancy at the POL5 gene promoter was previously reported to be 1.5-fold above background (32); as expected, we observed no change in HMO1 binding to POL5 during DSB induction and repair at the MAT locus (Supplementary Figure S4). Evidently, HMO1 localizes to the MAT locus, and it is selectively evicted from sites proximal to the DSB during DNA repair. These results are consistent with HMO1 directly affecting DNA repair as a component of chromatin, as opposed to exerting indirect control over factors involved in repair, and they implicate HMO1 eviction as a necessary step in DNA repair.
Since HMO1 occupancy is variable across the genome, we reasoned that HMO1 localization to the MAT locus might serve a regulatory role. We therefore compared MATa transcription in DDY3 and DDY3hmo1Δ strains. Relative to a control locus (IPP1 at which HMO1 localization was reported to be below background levels (32)), transcript levels were reduced ∼50% in the hmo1Δ strain (Supplementary Figure S5).
Considering that HMO1 is localized throughout the genome, we also wondered if increased survival following DSB induction in the hmo1Δ strain is unique to DSBs induced at MAT. To explore this question, we monitored survival of wild-type and hmo1Δ strains after exposure to hydroxyurea (HU). HU stalls replication forks, an event that may lead to fork collapse and induction of DNA double strand breaks, particularly after prolonged exposure to HU (33). We found that the hmo1Δ strain exhibits an increased resistance to HU compared to the isogenic wild type strain after an 8 h exposure to HU (Supplementary Figure S6). This is consistent with the increased survival after HO-induced DSB at MAT, and it is consistent with the interpretation that the effect of HMO1 is global.
Rapid kinetics of H2A phosphorylation and dephosphorylation in absence of HMO1
DSBs elicit a DNA-damage response that includes a rapid phosphorylation of histone H2A isoforms that spreads about 50 kb on either side of the DSB (6,7). To assess if this event is affected by HMO1, we performed ChIP with DDY3 and DDY3hmo1Δ using a previously characterized antibody against γ-H2A that is specific to the phosphorylated histone variant (Ser129) (34). We monitored γ-H2A appearance at MAT and 29.8 kb upstream of the damaged site. After 20 min of DSB induction, we found a higher level of H2A phosphorylation at these loci in the hmo1Δ strain compared to DDY3, and this difference was also seen after 1 h of DNA damage (Figure 3A and B).
Figure 3.
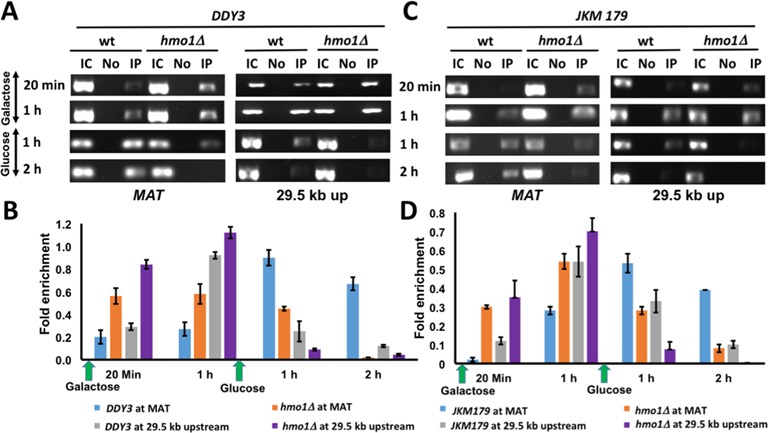
H2A phosphorylation. (A) ChIP with DDY3 WT and corresponding hmo1Δ strain using antibody to phosphorylated H2A at MAT and at 29.5 kb upstream of DSB during DNA damage (galactose) and repair (glucose). IC, input control; No, no antibody; IP, immunoprecipitation with antibody to γ-H2AX. (B) Densitometric analysis of ChIP data shown in (A), obtained with ImageJ software. (C) ChIP with JKM179 WT and corresponding hmo1Δ strain using antibody to phosphorylated H2A at MAT and at 29.5 kb upstream of DSB during DNA damage (galactose) and repair (glucose). (D) Densitometric analysis of ChIP data shown in (C). Fold enrichment = ChIP/Input DNA. Three independent experiments were performed. Error bars represent standard deviation.
Restoring chromatin after DNA repair by γ-H2A dephosphorylation is an important step that in yeast involves removal of γ-H2A followed by dephosphorylation (35). We analyzed the γ-H2A removal event by ChIP by adding glucose after 1 h of DNA damage to suppress further DSB induction. One hour after glucose addition, we observed appreciably reduced γ-H2A in DDY3hmo1Δ compared to WT and this difference was consistent after 2 h of DNA repair (Figure 3A and B).
To verify that these events are DNA repair pathway independent, we also performed the ChIP assay using JKM179 and the corresponding hmo1Δ strain, using the same time intervals and loci to monitor H2A phosphorylation. Again, we observed more efficient γ-H2A accumulation in the hmo1Δ strain, followed by its more efficient disappearance during repair (Figure 3C and D). Thus, these results reveal that the kinetics of both γ-H2A accumulation and removal are more rapid in an hmo1Δ strain compared to the isogenic WT parent strain, and that these events are independent of the repair pathway (HR or NHEJ).
H2A phosphorylation correlates with Arp5 recruitment
The chromatin remodeling complex INO80 is recruited to DSB sites in a γ-H2A-dependent process (11–14). Furthermore, association of INO80 with the MAT locus prior to DSB induction has been reported; while γ-H2A-dependent accumulation of INO80 was observed downstream of MAT after DSB induction, a pre-existing pool at the MAT locus that is involved in MAT transcription is associated with histone displacement, whereas the newly recruited pool was proposed to have a role in strand invasion (14). We performed ChIP assay with DDY3 and DDY3hmo1Δ using antibody against Arp5, a conserved subunit of INO80 in yeast and mammals. Prior to DSB induction, we observed the expected pre-existing pool of Arp5 at the MAT locus in both DDY3 and DDY3hmo1Δ strains, whereas no Arp5 was detectable 3.1 kb downstream (Figure 4A and B; 0 min). After inducing DSB for 20 min, the pre-existing pool of Arp5 at the MAT locus was reduced in DDY3hmo1Δ while no change was seen in DDY3; after 2 h of damage, the pre-existing Arp5 pool was completely lost in DDY3hmo1Δ, whereas complete loss of Arp5 in DDY3 was seen only after 4 h of damage (Figure 4A and B). Furthermore, after 2 h of DSB induction, more efficient accumulation of Arp5 was observed 3.1 kb downstream of the DSB site in DDY3hmo1Δ compared to WT (Figure 4A and B). These data show that the pre-existing pool of INO80 at the MAT locus is more rapidly displaced in the hmo1Δ strain after DSB induction, followed by its accumulation downstream of the break site. DNA repair would be expected to result in a reappearance of Arp5 at MAT and disappearance downstream. DNA repair after 4 h of DSB induction indeed resulted in the expected restoration of the INO80 localization observed prior to DNA damage (Figure 4A and B).
Figure 4.
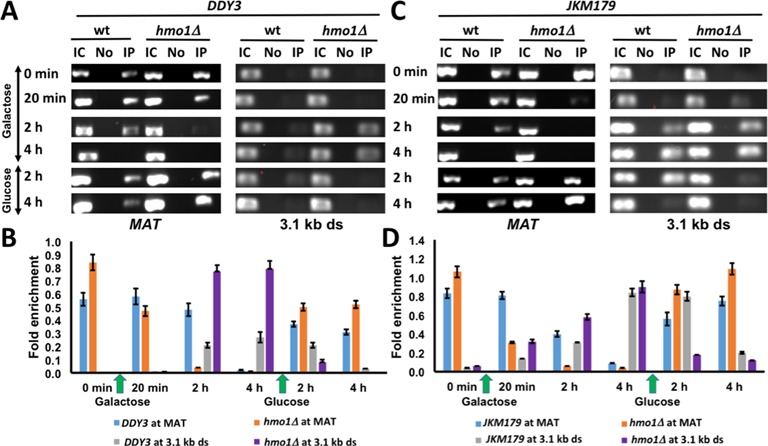
Arp5 localization. (A) ChIP with DDY3 WT and corresponding hmo1Δ strain using antibody to Arp5 at MAT and 3.1 kb downstream of DSB during DNA damage (galactose) and repair (glucose). IC, input control; No, no antibody; IP, immunoprecipitation with antibody to Arp5. (B) Densitometric analysis of ChIP data shown in (A), obtained with ImageJ software. (C) ChIP with JKM179 WT and corresponding hmo1Δ strain using antibody to Arp5 at MAT and 3.1 kb downstream of DSB of DSB during DNA damage (galactose) and repair (glucose). (D) Densitometric analysis of ChIP data shown in (C). Fold enrichment = ChIP/Input DNA. Three independent experiments were performed. Error bars represent standard deviation.
Recruitment of INO80 to DSB sites depends on the presence of γ-H2A. However, the DNA end resection that is a prerequisite for repair by HR should result in disruption of nucleosomes, including the loss of γ-H2A. We verified that γ-H2A is present 3.1 kb downstream of the DSB site in both DDY3 and the corresponding hmo1Δ strain after 2–4 h of DSB induction, consistent with the observed INO80 recruitment (Supplementary Figure S7).
We also examined these events in the JKM179 background, observing the same pattern of more rapid Arp5 displacement at MAT and enhanced accumulation downstream in the hmo1Δ strain, followed by restoration of Arp5 localization after DNA repair (Figure 4C, D). Thus, irrespective of DNA repair pathway, the pre-existing INO80 pool was displaced faster from the break site in the hmo1Δ strains, and the γ-H2A-dependent INO80 accumulation downstream was more efficient in hmo1Δ. The enhanced INO80 recruitment in hmo1Δ would be consistent with the more efficient DSB repair in hmo1Δ strains by either HR or NHEJ.
Rapid H3 eviction and redeposition in hmo1Δ strains
ChIP assay with DDY3 and DDY3hmo1Δ using antibody against histone H3 was used to monitor H3 at MAT and 0.2 kb upstream from the DSB. H3 disappearance 0.2 kb upstream of the DSB was evident in both strains after 1 h of DNA damage and became more prominent at both sites after further DNA damage, with more efficient H3 eviction in the hmo1Δ strain (Figure 5A and B). DNA repair (addition of glucose) resulted in redeposition of H3 in both strains (Figure 5A and B). In the JKM179 background, H3 eviction was also more efficient in the hmo1Δ strain after 2 h of DSB induction, and redeposition after repair occurred more efficiently (Figure 5C and D).
Figure 5.
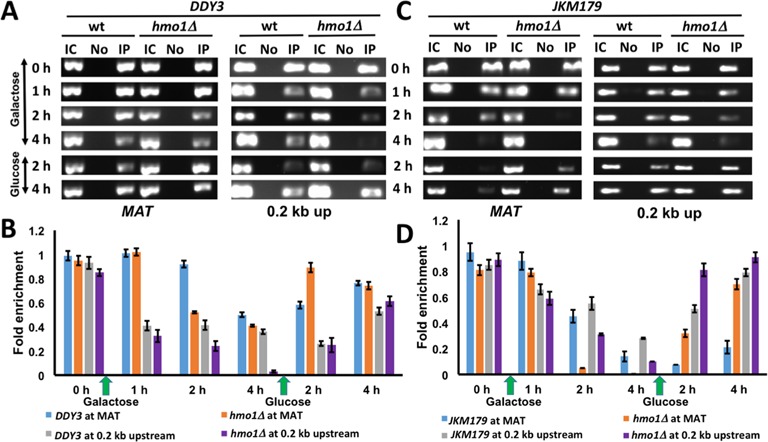
H3 localization. (A) ChIP with DDY3 WT and corresponding hmo1Δ strain using antibody to H3 at MAT and 0.2 kb upstream of DSB during DNA damage (galactose) and repair (glucose). IC, input control; No, no antibody; IP, immunoprecipitation with antibody to H3. (B) Densitometric analysis of ChIP data shown in (A), obtained with ImageJ software. (C) ChIP with JKM179 WT and corresponding hmo1Δ strain using antibody to H3 at MAT and 0.2 kb upstream of DSB during DNA damage (galactose) and repair (glucose). (D) Densitometric analysis of ChIP data shown in (C). Fold enrichment = ChIP/Input DNA. Three independent experiments were performed. Error bars represent standard deviation.
H3 eviction has been reported to parallel DNA end resection, and it has been suggested that input control DNA used for normalization may be lost as a consequence of such resection (17). We therefore verified H3 occupancy using qRT-PCR (Supplementary Figure S8). While the semi-quantitative assessment reveals some variability by comparison, the qRT-PCR analysis confirmed H3 eviction after DSB induction and redeposition following repair. Since amplification of input control DNA is constant after DSB induction (as determined by qRT-PCR), the reduced H3 occupancy observed may reflect a combination of nucleosome remodeling and displacement due to DNA resection.
DNA end resection and Rad51 recruitment
In yeast, the MRX complex (Mre11-Rad50-Xrs2) initiates DNA end resection in concert with the Sae2 endonuclease to generate short 3′-ended ssDNA overhangs. Such ssDNA ends limit Ku binding and promote more extensive resection by Exo1 and the helicase/endonuclease complex consisting of Sgs1-Top3-Rmi1 and Dna2. This extensive resection has been reported to depend on nucleosome remodeling by Fun30 (36,37). To assess if DNA end resection is affected by HMO1, we performed a DNA resection assay in which genomic DNA isolated at various times after induction of DSB was incubated with E. coli Exo I to degrade single-stranded overhangs. As shown in Figure 6, DNA resection is slower in DDY3 compared to the corresponding hmo1Δ strain, as measured by qRT-PCR using primers that anneal 1.6 kb from the DSB site.
Figure 6.
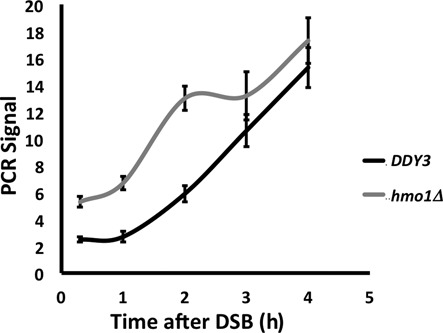
Quantification of DNA resection by qPCR. PCR products were amplified after Exonuclease I treatment of genomic DNA isolated at the indicated times following DSB induction using primers that anneal 1.6 kb upstream of the DSB. All values were normalized to that for an independent locus (POL5). DNA resection was measured in DDY3 and the corresponding hmo1Δ strain.
The homologous recombination protein Rad51 is recruited after 3′-end processing to initiate the homology search (38). We monitored Rad51 recruitment to the MAT locus in DDY3 and DDY3hmo1Δ by ChIP, observing enhanced Rad51 recruitment in the hmo1Δ strain, consistent with faster resection (Figure 7A and B). However, in absence of DSB induction, we also observed increased Rad51 localization in the hmo1Δ strain at MAT, 0.2 kb upstream, 9.5 kb downstream, and 29.8 kb upstream as well as at the unrelated POL5 locus (Supplementary Figure S9). Evidently, Rad51 association with undamaged DNA is greater in hmo1Δ and its recruitment to a DSB site is enhanced in absence of HMO1.
Figure 7.
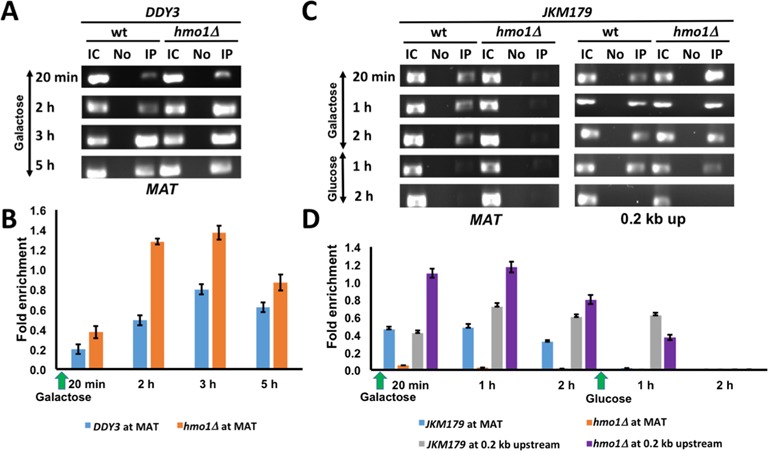
Rad51 and Ku recruitment to DSB. (A) ChIP with DDY3 WT and corresponding hmo1Δ strain using antibody to Rad51 at MAT during DNA damage induced by galactose. IC, input control; No, no antibody; IP, immunoprecipitation with antibody to Rad51. (B) Densitometric analysis of ChIP data shown in (A), obtained with ImageJ software. (C) ChIP with JKM179 WT and corresponding hmo1Δ strain using antibody to Ku at MAT and 0.2 kb upstream of DSB during DNA damage (galactose) and repair (glucose). (D) Densitometric analysis of ChIP data shown in (C). Fold enrichment = ChIP/Input DNA. Three independent experiments were performed. Error bars represent standard deviation.
Tracking of Ku from DNA ends correlates with histone eviction
The Ku heterodimer binds free DNA ends and plays an important role in NHEJ-mediated DNA repair (5). ChIP assay with JKM179 and JKM179hmo1Δ using antibodies against Ku showed the expected accumulation of Ku at the break site within the MAT locus in JKM179, followed by its rapid disappearance after DNA repair (Figure 7C, left panel). In contrast, Ku was essentially undetectable at the MAT locus in the hmo1Δ strain. Since Ku translocates from DNA ends (39), we reasoned that the absence of Ku from the break site in hmo1Δ might be a consequence of more efficient tracking. Indeed, we found that Ku was enriched 0.2 kb upstream of the break site in hmo1Δ, whereas this tracking event was less efficient in WT (Figure 7C, D). After DNA repair, accumulation of Ku at the 0.2 kb upstream site was reduced after 1 h and it was undetectable after 2 h in both strains (Figure 7C and D). These data suggest that tracking of Ku from DNA ends correlates with histone eviction and that both events are faster in hmo1Δ than in WT.
Truncation of the HMO1 C-terminal tail phenocopies HMO1 deficiency
The C-terminal tail of HMO1 is indispensable for DNA bending (23,24). To address if this architectural function of HMO1 is required for the more stable chromatin structure characteristic of WT strains, we compared DDY3 with the AB strain (28), which expresses HMO1 truncated for its C-terminal tail. Survival after induction of DSBs showed ∼2-fold increase in the AB strain, which indicates that it repairs DSBs more efficiently than WT (Supplementary Figure S10).
Consistent with more efficient repair, ChIP experiments showed more rapid H2A phosphorylation and dephosphorylation in the AB strain, both at MAT and 29.5 kb upstream (Figure 8A and B). As observed for DDY3hmo1Δ, recruitment of INO80 (Arp5) occurred more efficiently downstream of the break site in AB upon induction of DNA damage (Supplementary Figure S11A and B). Furthermore, after addition of glucose, rapid loss of Arp5 was observed downstream of MAT and a faster accumulation of Arp5 was seen at MAT in the AB strain (Supplementary Figure S11A and B). These events correlated with faster H3 eviction in AB 0.2 kb upstream of the break site and with faster redeposition following repair (Supplementary Figure S11C and D). Rad51 association with MAT was likewise increased in AB after DSB induction (Figure 8C and D). Thus, these results indicate that the AB strain phenocopies the hmo1Δ strain, featuring a higher efficiency of the chromatin remodeling events that are required for DSB repair.
Figure 8.
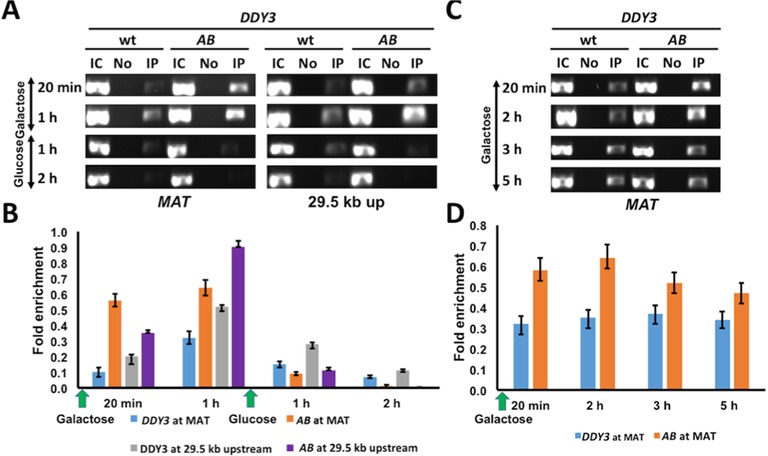
Effect of HMO1 C-terminal tail on H2A phosphorylation and Rad51 recruitment. (A) ChIP with DDY3 WT and AB strain expressing HMO1 deleted for its C-terminal tail using antibody to phosphorylated H2A at MAT and 29.5 kb upstream of DSB during DNA damage (galactose) and repair (glucose). IC, input control; No, no antibody; IP, immunoprecipitation with antibody to γ-H2AX. (B) Densitometric analysis of ChIP data shown in (A), obtained with ImageJ software. (C) ChIP with DDY3 WT and AB strain using antibody to Rad51 at MAT during DNA damage (galactose). (D) Densitometric analysis of ChIP data shown in (C). Fold enrichment = ChIP/Input DNA. Three independent experiments were performed. Error bars represent standard deviation.
DISCUSSION
The DNA damage response has to operate in the context of chromatin. After DSB, the first post-translational modification event is the H2A phosphorylation that spreads bidirectionally and creates a docking site for the chromatin remodeler INO80 and other proteins associated with DSB repair. INO80 plays a major role in chromatin dynamics around a DSB and may facilitate eviction of nucleosomes in the immediate vicinity of the DSB to allow DNA resection (11,13). Nucleosome disassembly parallels the extensive DNA end resection, which is facilitated by the chromatin remodeler Fun30 (36,37). After the completion of DNA repair, affected chromatin regions must be restored, events described by the ‘access-repair-restore' model (40). We report here that these events are modulated by the HMGB protein HMO1.
HMGB proteins bend their target DNA sites and serve architectural roles in nucleoprotein complex assembly. Vertebrate HMGB1 is thought to bind nucleosomal linker DNA to relax the chromatin structure and promote access to remodeling complexes and transcription factors (41–44). The yeast homolog HMO1 also binds DNA with little sequence specificity, bends DNA, and recognizes altered DNA conformations (19,22–24). In contrast to vertebrate homologs, deletion of HMO1 results in nuclease-sensitive chromatin (19), pointing to a role for HMO1 in stabilizing chromatin. In addition, HMO1 accumulates on ribosomal RNA genes, where it appears to prevent chromosome fragility in absence of nucleosomes; at the ribosomal DNA promoter, upstream activating factor (UAF) contains histones H3 and H4, but not H2A and H2B, suggesting that the presence of HMO1 may prevent fragility (45). At the rDNA locus, HMO1 not only stabilizes the chromosome structure, but it is associated with the open rDNA to promote transcription of ribosomal genes (46–48). HMO1 mediates DNA bridging between strands, stabilization of DNA loops, and DNA compaction by reducing the apparent DNA persistence length, which may contribute to compaction of nucleosome-free DNA (23,49).
HMO1 is localized throughout the genome, but not uniformly so. At the MAT locus, its presence appears to modulate MAT transcription and perhaps prevent chromosome fragility. A role for HMO1 in stabilizing the local chromatin structure is supported by the observation that it, like core histones, must be evicted for DSB repair. Furthermore, in the absence of HMO1, DNA damage-associated chromatin remodeling events and DNA end resection are faster. That the strain expressing HMO1 deleted for its C-terminal tail phenocopies hmo1Δ may reflect either that DNA bending is required for the stabilizing effect of HMO1 on chromatin or that the C-terminus is necessary for its recruitment. The yeast HMGB protein Nhp6A has also been shown to associate with certain chromosomal regions and to stabilize nucleosomes, and DNA bending was shown to be critical for this function (50). We also note that HMO1 contains a lysine-rich C-terminus, in contrast to vertebrate HMGB1, whose C-terminal tail is acidic. This basic extension is reminiscent of the lysine-rich domain of histone H1, which has been implicated in DNA compaction (51). It is therefore conceivable that the ability of HMO1 to stabilize chromatin may likewise rely on its lysine-rich extension, particularly in light of the more limited role of histone H1 in yeast compared to vertebrates.
Association of Rad51
The wrapping of DNA about the histone octamer as well as higher order organization protects the DNA from nucleases such as micrococcal nuclease (MNase), and the ability of MNase to digest chromatin DNA has been generally used to identify sensitive sites (52). Such nucleosome fragility has been for example associated with environmental stress response genes, where it has been suggested to render these genes more responsive to environmental signals and rapid changes in transcriptional activity (53,54). Absence of HMO1 renders chromatin nuclease sensitive, implying development of fragile chromatin structures.
In the absence of DSB induction, we observed an enrichment of Rad51 at all sites monitored in the hmo1Δ strain in addition to enhanced recruitment after DSB induction. Rad51 functions in repair of DSBs and stalled replication forks. Events that cause DSB formation or inhibit replication promote formation of Rad51 repair complexes that may be detected as foci. Accumulation of Rad51 on undamaged double stranded DNA is usually prevented by translocases such as Rad54, which prevent toxicity associated with such binding (55). Accumulation on dsDNA is rendered possible because Rad51 has little preference for ssDNA compared to dsDNA (56). We speculate that the less-stable chromatin structure associated with absence of HMO1 may promote accessibility of Rad51 to undamaged DNA sites.
The DNA damage-independent Rad51 recruitment is intriguing and may reflect formation of fragile chromatin regions on removal of HMO1 that attract surveillance complexes in preparation for eventual DNA damage. Under non-DNA damage conditions, Rad9 was shown to interact with fragile genomic regions and was suggested to facilitate genome surveillance and efficient responses in the event of DNA damage (57). It was also reported that the level of replication-independent endogenous DSBs was lower in strains lacking chromatin condensing proteins HMO1 and Sir2 (Silent Information Regulator 2), but higher in absence of DNA repair proteins such as Ku and Rad51 (58). Notably, these authors also reported lower levels of such DSBs in human cells lacking HMGB1.
Ku tracking from DNA ends correlates with chromatin remodeling
The Ku heterodimer threads onto free DNA ends by virtue of its toroidal structure and it is a key player in the NHEJ pathway of DSB repair (5). Ku in turn recruits proteins required for end-processing and ligation. However, topologically trapped Ku may interfere with cellular processes, including competition with HR-mediated repair and post-repair recovery (27,59); Ku tracks from DNA ends, and recent evidence suggests that complete removal of Ku from DNA involves ubiquitylation (39,60). Recruitment of yeast Ku to HO-induced strand breaks was previously reported to occur within 15 min of break induction and Ku started to disappear from the DSB site ∼1 h after glucose addition (59). Our results were consistent with these findings. Notably, Ku was nearly undetectable at the MAT locus during DSB induction in the hmo1Δ strain, whereas it only disappeared from the DSB site in the isogenic WT strain after ∼1 h of repair. Instead, Ku appeared faster 0.2 kb upstream from the break site in hmo1Δ compared to WT, suggesting that tracking of Ku from the free DNA end is faster in the hmo1Δ strain, an observation that is consistent with more efficient DNA repair. These data not only suggest that efficient tracking of Ku from DNA ends correlates with efficient repair, but that these events correlate with faster histone eviction.
Taken together, our results show that HMO1 stabilizes the chromatin structure and that it is evicted along with nucleosomes to facilitate recruitment of proteins involved in DSB repair. Efficient DNA resection requires a nucleosome-free region near the DSB (36,37,61), which is consistent with our observation that HR is more efficient when histone eviction near the DSB is facilitated by remodeling or end resection events. Likewise, NHEJ is faster under such conditions, as reflected by our observation that Ku tracking from the DSB site correlates with repair efficiency and chromatin remodeling. Since the presence of HMO1 protects fragile chromosomal regions, the association of HMO1 with the MAT locus is intriguing. HMO1 promotes DNA association in vitro (23), it facilitates sister chromatid junction during replication (62), and it has been shown to direct DNA lesions toward HR-mediated repair (21); it is therefore conceivable that a specific function of HMO1 at MAT is to facilitate HR-dependent mating type switching.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Jim Haber and Lorraine Symington for the JKM179 strain and HO plasmid and Sang Eun Lee and Eun Yong Shim for Ku antibody. We also appreciate many helpful discussions with David Donze, Asawari Korde, and Craig Hart.
Footnotes
Present address: LiJuan Xiao, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
FUNDING
National Science Foundation [MCB-1051610 to A.G.]. The open access publication charge for this paper has been waived by Oxford University Press - NAR.
Conflict of interest statement. None declared.
REFERENCES
- 1.Saha A., Wittmeyer J., Cairns B.R. Chromatin remodelling: the industrial revolution of DNA around histones. Nat. Rev. Mol. Cell. Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 2.Gerhold C.B., Hauer M.H., Gasser S.M. INO80-C and SWR-C: guardians of the genome. J Mol Biol. 2014;427:637–651. doi: 10.1016/j.jmb.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 3.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holthausen J.T., Wyman C., Kanaar R. Regulation of DNA strand exchange in homologous recombination. DNA Repair. 2010;9:1264–1272. doi: 10.1016/j.dnarep.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 5.Ochi T., Wu Q., Blundell T.L. The spatial organization of non-homologous end joining: from bridging to end joining. DNA Repair. 2014;17:98–109. doi: 10.1016/j.dnarep.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shroff R., Arbel-Eden A., Pilch D., Ira G., Bonner W.M., Petrini J.H., Haber J.E., Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 2004;14:1703–1711. doi: 10.1016/j.cub.2004.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Downs J.A., Allard S., Jobin-Robitaille O., Javaheri A., Auger A., Bouchard N., Kron S.J., Jackson S.P., Cote J. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol. Cell. 2004;16:979–990. doi: 10.1016/j.molcel.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 9.Ebbert R., Birkmann A., Schuller H.J. The product of the SNF2/SWI2 paralogue INO80 of Saccharomyces cerevisiae required for efficient expression of various yeast structural genes is part of a high-molecular-weight protein complex. Mol. Microbiol. 1999;32:741–751. doi: 10.1046/j.1365-2958.1999.01390.x. [DOI] [PubMed] [Google Scholar]
- 10.Shen X., Ranallo R., Choi E., Wu C. Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol. Cell. 2003;12:147–155. doi: 10.1016/s1097-2765(03)00264-8. [DOI] [PubMed] [Google Scholar]
- 11.van Attikum H., Fritsch O., Hohn B., Gasser S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–788. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 12.van Attikum H., Fritsch O., Gasser S.M. Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 2007;26:4113–4125. doi: 10.1038/sj.emboj.7601835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison A.J., Highland J., Krogan N.J., Arbel-Eden A., Greenblatt J.F., Haber J.E., Shen X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–775. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 14.Tsukuda T., Fleming A.B., Nickoloff J.A., Osley M.A. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–383. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimada K., Oma Y., Schleker T., Kugou K., Ohta K., Harata M., Gasser S.M. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr. Biol. 2008;18:566–575. doi: 10.1016/j.cub.2008.03.049. [DOI] [PubMed] [Google Scholar]
- 16.Ray S., Grove A. The yeast high mobility group protein HMO2, a subunit of the chromatin-remodeling complex INO80, binds DNA ends. Nucleic Acids Res. 2009;37:6389–6399. doi: 10.1093/nar/gkp695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen C.C., Carson J.J., Feser J., Tamburini B., Zabaronick S., Linger J., Tyler J.K. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–243. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas J.O., Stott K. H1 and HMGB1: modulators of chromatin structure. Biochem. Soc. Trans. 2012;40:341–346. doi: 10.1042/BST20120014. [DOI] [PubMed] [Google Scholar]
- 19.Lu J., Kobayashi R., Brill S.J. Characterization of a high mobility group 1/2 homolog in yeast. J. Biol. Chem. 1996;271:33678–33685. doi: 10.1074/jbc.271.52.33678. [DOI] [PubMed] [Google Scholar]
- 20.Alekseev S.Y., Kovaltsova S.V., Fedorova I.V., Gracheva L.M., Evstukhina T.A., Peshekhonov V.T., Korolev V.G. HSM2 (HMO1) gene participates in mutagenesis control in yeast Saccharomyces cerevisiae. DNA Repair. 2002;1:287–297. doi: 10.1016/s1568-7864(02)00005-8. [DOI] [PubMed] [Google Scholar]
- 21.Kim H., Livingston D.M. Suppression of a DNA polymerase delta mutation by the absence of the high mobility group protein Hmo1 in Saccharomyces cerevisiae. Curr. Genet. 2009;55:127–138. doi: 10.1007/s00294-009-0229-y. [DOI] [PubMed] [Google Scholar]
- 22.Bauerle K.T., Kamau E., Grove A. Interactions between N- and C-terminal domains of the Saccharomyces cerevisiae high-mobility group protein HMO1 are required for DNA bending. Biochemistry. 2006;45:3635–3645. doi: 10.1021/bi0522798. [DOI] [PubMed] [Google Scholar]
- 23.Xiao L., Williams A.M., Grove A. The C-terminal domain of yeast high mobility group protein HMO1 mediates lateral protein accretion and in-phase DNA bending. Biochemistry. 2010;49:4051–4059. doi: 10.1021/bi1003603. [DOI] [PubMed] [Google Scholar]
- 24.Kamau E., Bauerle K.T., Grove A. The Saccharomyces cerevisiae high mobility group box protein HMO1 contains two functional DNA binding domains. J. Biol. Chem. 2004;279:55234–55240. doi: 10.1074/jbc.M409459200. [DOI] [PubMed] [Google Scholar]
- 25.Rando O.J., Winston F. Chromatin and transcription in yeast. Genetics. 2012;190:351–387. doi: 10.1534/genetics.111.132266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simms T.A., Miller E.C., Buisson N.P., Jambunathan N., Donze D. The Saccharomyces cerevisiae TRT2 tRNAThr gene upstream of STE6 is a barrier to repression in MATalpha cells and exerts a potential tRNA position effect in MATa cells. Nucleic Acids Res. 2004;32:5206–5213. doi: 10.1093/nar/gkh858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S.E., Moore J.K., Holmes A., Umezu K., Kolodner R.D., Haber J.E. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 28.Xiao L., Kamau E., Donze D., Grove A. Expression of yeast high mobility group protein HMO1 is regulated by TOR signaling. Gene. 2011;489:55–62. doi: 10.1016/j.gene.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 29.Sikorski R.S., Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L., Trujillo K., Ramos W., Sung P., Tomkinson A.E. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol. Cell. 2001;8:1105–1115. doi: 10.1016/s1097-2765(01)00388-4. [DOI] [PubMed] [Google Scholar]
- 31.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall D.B., Wade J.T., Struhl K. An HMG protein, Hmo1, associates with promoters of many ribosomal protein genes and throughout the rRNA gene locus in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006;26:3672–3679. doi: 10.1128/MCB.26.9.3672-3679.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saintigny Y., Delacote F., Vares G., Petitot F., Lambert S., Averbeck D., Lopez B.S. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Redon C., Pilch D.R., Rogakou E.P., Orr A.H., Lowndes N.F., Bonner W.M. Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep. 2003;4:678–684. doi: 10.1038/sj.embor.embor871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keogh M.C., Kim J.A., Downey M., Fillingham J., Chowdhury D., Harrison J.C., Onishi M., Datta N., Galicia S., Emili A., et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature. 2006;439:497–501. doi: 10.1038/nature04384. [DOI] [PubMed] [Google Scholar]
- 36.Eapen V.V., Sugawara N., Tsabar M., Wu W.H., Haber J.E. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol. Cell. Biol. 2012;32:4727–4740. doi: 10.1128/MCB.00566-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X., Cui D., Papusha A., Zhang X., Chu C.D., Tang J., Chen K., Pan X., Ira G. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature. 2012;489:576–580. doi: 10.1038/nature11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renkawitz J., Lademann C.A., Kalocsay M., Jentsch S. Monitoring homology search during DNA double-strand break repair in vivo. Mol. Cell. 2013;50:261–272. doi: 10.1016/j.molcel.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 39.de Vries E., van Driel W., Bergsma W.G., Arnberg A.C., van der Vliet P.C. HeLa nuclear protein recognizing DNA termini and translocating on DNA forming a regular DNA-multimeric protein complex. J. Mol. Biol. 1989;208:65–78. doi: 10.1016/0022-2836(89)90088-0. [DOI] [PubMed] [Google Scholar]
- 40.Smerdon M.J. DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol. 1991;3:422–428. doi: 10.1016/0955-0674(91)90069-b. [DOI] [PubMed] [Google Scholar]
- 41.Cato L., Stott K., Watson M., Thomas J.O. The interaction of HMGB1 and linker histones occurs through their acidic and basic tails. J. Mol. Biol. 2008;384:1262–1272. doi: 10.1016/j.jmb.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 42.Joshi S.R., Sarpong Y.C., Peterson R.C., Scovell W.M. Nucleosome dynamics: HMGB1 relaxes canonical nucleosome structure to facilitate estrogen receptor binding. Nucleic Acids Res. 2012;40:10161–10171. doi: 10.1093/nar/gks815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watson M., Stott K., Fischl H., Cato L., Thomas J.O. Characterization of the interaction between HMGB1 and H3-a possible means of positioning HMGB1 in chromatin. Nucleic Acids Res. 2014;42:848–859. doi: 10.1093/nar/gkt950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nightingale K., Dimitrov S., Reeves R., Wolffe A.P. Evidence for a shared structural role for HMG1 and linker histones B4 and H1 in organizing chromatin. EMBO J. 1996;15:548–561. [PMC free article] [PubMed] [Google Scholar]
- 45.Keener J., Dodd J.A., Lalo D., Nomura M. Histones H3 and H4 are components of upstream activation factor required for the high-level transcription of yeast rDNA by RNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13458–13462. doi: 10.1073/pnas.94.25.13458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gadal O., Labarre S., Boschiero C., Thuriaux P. Hmo1, an HMG-box protein, belongs to the yeast ribosomal DNA transcription system. EMBO J. 2002;21:5498–5507. doi: 10.1093/emboj/cdf539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merz K., Hondele M., Goetze H., Gmelch K., Stoeckl U., Griesenbeck J. Actively transcribed rRNA genes in S. cerevisiae are organized in a specialized chromatin associated with the high-mobility group protein Hmo1 and are largely devoid of histone molecules. Genes Dev. 2008;22:1190–1204. doi: 10.1101/gad.466908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wittner M., Hamperl S., Stockl U., Seufert W., Tschochner H., Milkereit P., Griesenbeck J. Establishment and maintenance of alternative chromatin states at a multicopy gene locus. Cell. 2011;145:543–554. doi: 10.1016/j.cell.2011.03.051. [DOI] [PubMed] [Google Scholar]
- 49.Murugesapillai D., McCauley M.J., Huo R., Nelson Holte M.H., Stepanyants A., Maher L.J. 3rd, Israeloff N.E., Williams M.C. DNA bridging and looping by HMO1 provides a mechanism for stabilizing nucleosome-free chromatin. Nucleic Acids Res. 2014;42:8996–9004. doi: 10.1093/nar/gku635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dowell N.L., Sperling A.S., Mason M.J., Johnson R.C. Chromatin-dependent binding of the S. cerevisiae HMGB protein Nhp6A affects nucleosome dynamics and transcription. Genes Dev. 2010;24:2031–2042. doi: 10.1101/gad.1948910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellen T.P., van Holde K.E. Linker histone interaction shows divalent character with both supercoiled and linear DNA. Biochemistry. 2004;43:7867–7872. doi: 10.1021/bi0497704. [DOI] [PubMed] [Google Scholar]
- 52.Yuan G.C., Liu Y.J., Dion M.F., Slack M.D., Wu L.F., Altschuler S.J., Rando O.J. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- 53.Gasch A.P., Spellman P.T., Kao C.M., Carmel-Harel O., Eisen M.B., Storz G., Botstein D., Brown P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xi Y., Yao J., Chen R., Li W., He X. Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res. 2011;21:718–724. doi: 10.1101/gr.117101.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Solinger J.A., Kiianitsa K., Heyer W.D. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol. Cell. 2002;10:1175–1188. doi: 10.1016/s1097-2765(02)00743-8. [DOI] [PubMed] [Google Scholar]
- 56.Ogawa T., Yu X., Shinohara A., Egelman E.H. Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science. 1993;259:1896–1899. doi: 10.1126/science.8456314. [DOI] [PubMed] [Google Scholar]
- 57.Andreadis C., Nikolaou C., Fragiadakis G.S., Tsiliki G., Alexandraki D. Rad9 interacts with Aft1 to facilitate genome surveillance in fragile genomic sites under non-DNA damage-inducing conditions in S. cerevisiae. Nucleic Acids Res. 2014;42:12650–12667. doi: 10.1093/nar/gku915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thongsroy J., Matangkasombut O., Thongnak A., Rattanatanyong P., Jirawatnotai S., Mutirangura A. Replication-independent endogenous DNA double-strand breaks in Saccharomyces cerevisiae model. PLoS One. 2013;8:e72706. doi: 10.1371/journal.pone.0072706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu D., Topper L.M., Wilson T.E. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics. 2008;178:1237–1249. doi: 10.1534/genetics.107.083535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Postow L., Ghenoiu C., Woo E.M., Krutchinsky A.N., Chait B.T., Funabiki H. Ku80 removal from DNA through double strand break-induced ubiquitylation. J. Cell Biol. 2008;182:467–479. doi: 10.1083/jcb.200802146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adkins N.L., Niu H., Sung P., Peterson C.L. Nucleosome dynamics regulates DNA processing. Nat. Struct. Mol. Biol. 2013;20:836–842. doi: 10.1038/nsmb.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gonzalez-Huici V., Szakal B., Urulangodi M., Psakhye I., Castellucci F., Menolfi D., Rajakumara E., Fumasoni M., Bermejo R., Jentsch S., et al. DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J. 2014;33:327–340. doi: 10.1002/embj.201387425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.