


Abstract
The Escherichia coli replication terminator protein (Tus) binds to Ter sequences to block replication forks approaching from one direction. Here, we used single molecule and transient state kinetics to study responses of the heterologous phage T7 replisome to the Tus–Ter complex. The T7 replisome was arrested at the non-permissive end of Tus–Ter in a manner that is explained by a composite mousetrap and dynamic clamp model. An unpaired C(6) that forms a lock by binding into the cytosine binding pocket of Tus was most effective in arresting the replisome and mutation of C(6) removed the barrier. Isolated helicase was also blocked at the non-permissive end, but unexpectedly the isolated polymerase was not, unless C(6) was unpaired. Instead, the polymerase was blocked at the permissive end. This indicates that the Tus–Ter mechanism is sensitive to the translocation polarity of the DNA motor. The polymerase tracking along the template strand traps the C(6) to prevent lock formation; the helicase tracking along the other strand traps the complementary G(6) to aid lock formation. Our results are consistent with the model where strand separation by the helicase unpairs the GC(6) base pair and triggers lock formation immediately before the polymerase can sequester the C(6) base.
INTRODUCTION
In the circular Escherichia coli chromosome are clusters of specific replication termination (Ter) sequences, whose function is to trap the first-arriving replication fork in the terminus region to prevent its over-replication (1,2). The replication terminator protein, Tus, forms a tight complex (pM KD) with the Ter sequences (3–8). It is remarkable that the Tus–Ter complex preferentially blocks the E. coli replisome arriving from one direction (at the non-permissive face) but not the other (permissive face). High affinity binding of Tus to Ter sequences is important for efficient replication fork arrest, but high affinity by itself does not explain why the replisome is blocked in a polar manner (2). Three mechanistic models (that are not mutually exclusive) have been proposed to explain polar arrest of the replisome: (i) in the helicase interaction model, the replicative helicase DnaB interacts physically with the non-permissive face of the Tus–Ter complex to stop fork progression (9–13). (ii) In the dynamic clamp model, there is an intrinsic difference in the interactions of the Tus protein at the two ends of Ter, which leads to facile dissociation of Tus by the DnaB helicase arriving at the permissive, but not the non-permissive face (4,14). (iii) The mousetrap model proposes a highly specialized mechanism that involves a strictly conserved cytosine residue C(6) in the Ter sequence (5). In this mechanism, DnaB helicase unwinds the DNA toward the non-permissive face, exposing the conserved C(6) residue out of the double-stranded (ds)DNA to enable its binding into a specific cytosine-binding pocket in the Tus protein, forming a ‘lock’ that effectively blocks the replication complex (Figure 1A).
Figure 1.
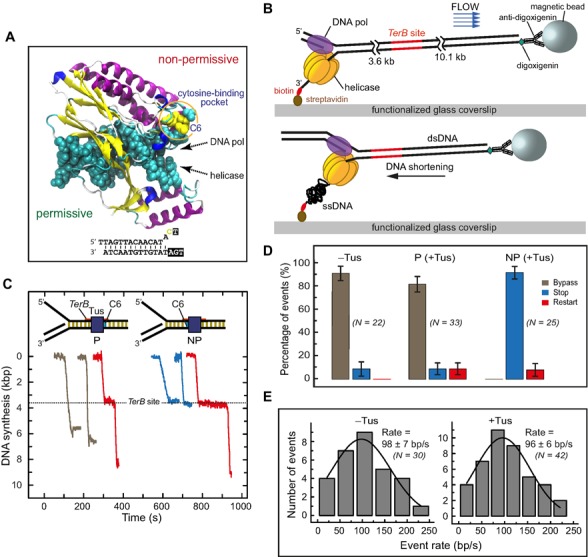
Single-molecule DNA synthesis by T7 helicase–polymerase upon encountering the permissive or non-permissive face of the Tus–TerB complex. (A) Crystal structure (PDB code: 2I06) of the locked Tus–Ter complex shows the flipped C(6) base at the non-permissive face (5). (B) A schematic representation of the single-molecule tethered-bead experimental setup for observing DNA synthesis by the T7 helicase–polymerase. DNA synthesis converts the surface-tethered dsDNA to ssDNA, which at a regime force of 2.6 pN results in shortening of the DNA and displacement of the bead in the opposite direction to the flow. (C) Representative trajectories of DNA synthesis upon encountering Tus–TerB complexes. The fork primarily bypassed Tus–TerB complex on arrival at the permissive face (P, left), while it fully stopped at the non-permissive face (NP, right). Trajectories show permanent stoppage (in blue), unimpeded bypass (gray) or restart (red) at P or NP Tus–TerB. The location of the TerB site is indicated. Traces have been offset on the time axis for clarity. (D) The percentage of populations of replication forks that bypassed, transiently stopped or fully stopped at P TerB and NP TerB are shown. A control experiment in the absence of Tus is shown for NP TerB. Error bars correspond to the standard deviation of binomial distributions. (E) Rate of leading strand synthesis using forked-λ DNA in the absence (left) or presence of Tus (right). The rate distributions were fit (lines in black) with a Gaussian distribution. The uncertainties correspond to the standard error of the distribution. Leading-strand replication reactions were carried out in the presence or in the absence of Tus protein in buffer containing 50 mM potassium glutamate.
To probe these mechanisms, we used the heterologous bacteriophage T7 replication proteins for two reasons: firstly, if DnaB helicase–Tus interactions play a dominant role in polar replication fork arrest, then a helicase or helicase–polymerase that could not have co-evolved might not be arrested in a polar manner. Secondly, unlike with the E. coli replication protein system where helicase loading is inefficient, T7 DNA replication on short DNA templates can be used to follow DNA synthesis with single base spatial resolution and on millisecond time scales, as described below. The mechanisms of coupled DNA unwinding by the T7 helicase and DNA synthesis by the T7 polymerase have been studied in detail (15–21). The T7 helicase is a ring-shaped member of the DnaB family that uses the energy of nucleotide triphosphate (NTP) hydrolysis to unwind dsDNA. Like DnaB, it translocates in the 5′ to 3′ direction on one strand and unwinds dsDNA by a strand exclusion mechanism (15,20,22–24). Thus, it separates the strands of dsTer DNA upon approach to the non-permissive face by encircling the lagging strand containing G(6) and excluding the leading strand that includes C(6). On the other hand, the T7 DNA polymerase binds on the leading strand template and translocates in the 3′ to 5′ direction to elongate a primer to copy the newly unwound DNA. The separated T7 helicase and polymerase have DNA unwinding and synthesis activities, respectively; however, strand displacement DNA synthesis is most efficiently promoted by the two enzymes working together at the replication fork (18,25). In work reported here, this fast and processive DNA synthesis has been measured at single base resolution on synthetic fork DNA substrates (26), and also at more limited resolution in real time using a single-molecule tethered-bead assay (27,28).
We show, using both of these DNA synthesis assays, that the T7 helicase–polymerase complex is effectively blocked by Tus–Ter in a polar manner at the non-permissive, but not at the permissive face, just like the E. coli replication fork in vivo. Furthermore, mutation of C(6) has clear effects on the duration (transient versus permanent) and extent of replication arrest. We found that the isolated T7 DNA polymerase, uncoupled from the helicase, is not blocked permanently at the non-permissive face of the termination complex, but is surprisingly blocked instead at the permissive end. On the other hand, isolated T7 helicase is blocked at the non-permissive but not the permissive face. These results indicate that polar arrest by Tus–Ter involves a strand-specific mechanism that is sensitive to the polarity of the DNA motor. We discuss these results in terms of the Tus–Ter interactions with the two strands of the DNA at the permissive and non-permissive faces and for the first time deduce the roles that two of the three mechanistic models play in polar replication fork arrest and the operation of the mousetrap mechanism under conditions of physiological ionic strength. The Tus–Ter block is also a great tool to probe the coupling between helicase and polymerase progressing at the replication fork.
MATERIALS AND METHODS
Proteins
T7 gp4 (gp4A’), T7 gp5 Exo- (D5A, D7A) and T7 gp5/ thioredoxin wild-type proteins were purified using published protocols (29–32). E. coli thioredoxin was purchased from Sigma Life Sciences. His6-Tus protein was purified as described (4). Protein concentration was calculated by UV absorption (in 8 M guanidine.HCl) using the extinction coefficients at 280 nm of 0.0836 μM−1cm−1 for gp4, 0.13442 μM−1cm−1 for gp5 and 0.0397 μM−1cm−1 for Tus.
Quenched flow DNA substrates
Oligodeoxynucleotides were purchased from Integrated DNA Technologies (IDT) and polyacrylamide gel electrophoresis (PAGE) purified (Figure 2 and Supplementary Table S1). The concentration of purified oligonucleotides was determined from their absorbance and extinction coefficients at 260 nm after digestion with snake venom phosphodiesterase I (33–35). For quenched flow DNA synthesis assays, a 24-nucleotide primer was labeled with fluorescein [(5/6)-FAM, SE: 5-(and-6)-carboxyfluorescein, succinimidyl ester, mixed isomers]. The 5′- and 3′-strands were mixed in equal proportions with 24-nucleotide primer in the annealing buffer (50 mM Tris–Cl, pH 7.6, 50 mM KCl, 10% glycerol) and fork annealing was carried out at 95°C for 10 min followed by slow cooling to room temperature. For the helicase unwinding assay, the 5′ or 3′ strand of the fork substrate was end labeled with fluorescein or was radiolabeled with γ-[32P]-ATP (Perkin Elmer) using T4 polynucleotide kinase (New England Biolabs).
Figure 2.
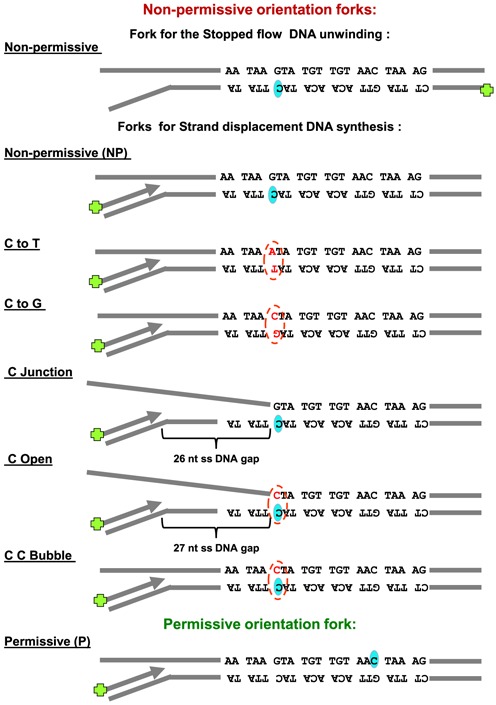
Schematic representation of the DNA fork substrates used in the bulk studies. TerB sequence orientation is shown pertaining to the DNA synthesis direction on primer. C(6) base (shaded oval shape) on Ter B sequence and any mutation of GC(6) is indicated (broken line oval shape).
Kinetics of DNA synthesis
Strand displacement DNA synthesis kinetics were measured in a rapid quenched-flow instrument (Kin Tek Instruments, Austin, TX, USA) at 18°C (18,26). T7 helicase was assembled on the fork DNA substrate (with a labeled primer) on ice for 15 min in the presence of 1 mM dTTP. T7 DNA polymerase was prepared by mixing E. coli thioredoxin and T7 gp5 Exo− (mutated in two positions D5A, D7A) (5:1) in replication buffer containing 5 mM dithiothreitol (DTT) at 22°C for 5 min. The DNA polymerase was added to the helicase and fork DNA template and further incubated for 15 min on ice followed by 10 min at the reaction temperature in the presence of the Tus protein. The protein–DNA mixture from one syringe was rapidly mixed with Mg.dVTP mixture from the second syringe. The final mixture contained the four dNTPs (1 mM each, unless specified), MgCl2 (free 4 mM), 190 nM forked DNA template, 375 nM T7 helicase hexamer, 375 nM T7 DNA polymerase, 375 nM Tus, 2 μM bovine serum albumin (BSA), 1 mM dithiothreitol and 1 mM ethylenediaminetetraacetic acid (EDTA) in the replication buffer (50 mM Tris-Cl, pH 7.6, 50–300 mM KCl, 10% glycerol). After desired time intervals the reactions were stopped with 300 mM EDTA. The quenched samples were mixed with bromophenol blue/formamide loading dye, boiled and then resolved in a 24% acrylamide/7 M urea sequencing gel with 1.5× Tris/Borate/EDTA pH 8.4 buffer (TBE buffer). The gel was imaged on a fluorescence imager and the DNA band intensities were analyzed by ImageQuant software. The percent runoff synthesis was estimated from ratio of runoff products (DNA intensity beyond the TerB sequence on template) to total DNA intensity in each lane using the following equation, which takes into account the background correction.
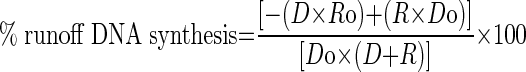 |
where R is nascent DNA intensity beyond the TerB sequence, D is nascent DNA intensity up to the TerB sequence, ‘o’ designates intensities at time zero.
Kinetics of DNA unwinding
The kinetics of DNA unwinding were measured both by real time fluorescence-based and radiometric assays as described in (26). The fluorescence-based assay was carried out in a stopped-flow instrument (Kin Tek) at 18°C. Fork DNA labeled with fluorescein on the 3′-strand was preassembled with T7 helicase and dTTP with or without Tus in the replication buffer and rapidly mixed with MgCl2 and a dT90 ssDNA trap. The final reaction mixture contained 2 mM dTTP, 5 nM DNA substrate, 100 nM T7 helicase hexamer, MgCl2 (free 4 mM), 3 mM EDTA, 6 μM dT90, 2 μM BSA, 20 nM Tus. The fluorescence at >515 nm (cut-off filter at 515 nm) was monitored after excitation with 480 nm light.
In the radiometric assay, the 5′-strand was radiolabeled and assembly with the helicase and Tus was as described above. The final reaction components were 5 nM forked substrate, 100 nM T7 helicase hexamer, 50 nM Tus, 1 mM dTTP and 4 mM MgCl2 in replication buffer. Reactions were quenched with EDTA and sodium dodecyl sulphate and resolved on a non-denaturing PAGE gel in 1.5 × TBE buffer, exposed to the phosphorscreen, and imaged on a phosphorimager. The DNA bands were analyzed using ImageQuant software. A background correction was applied to each unwound fraction value as shown in the equation used to calculate the unwound DNA strand fraction at each time point:
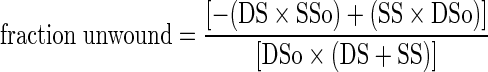 |
where DS is double-stranded DNA fraction, SS is single-stranded DNA fraction and ‘o’ designates fractions at the zero time point.
Templates for single molecule tethered bead assay
Bacteriophage λ DNA was modified by ligation of a biotinylated fork on one end and a digoxigenin (dig) moiety at the other as described previously (27). The ligated λ DNA molecules were digested with EcoRI to generate a 3.6 kb fragment from the forked end and with ApaI to generate a 10.1 kb fragment from the dig end. A 59-bp ssDNA oligonucleotide containing a single copy of wild type or variants of the 22-nt TerB site (in italics for the wild-type non-permissive example, below) and ends complementary to the overhangs in the EcoRI and ApaI digested fragments (underlined) (5′-AATTCAAGTCAC CACGACTGTGCTATAAAATAAGTATGTTGTAACTA AAGTGGTTAATATTATGGCGCGTTGGCC-3′) was first 5′-phosphorylated with T4 polynucleotide kinase. It was then annealed to the complementary oligonucleotide (5′-AACGCGCCATAATATTAACCACTTTAGTTACAA CATACTTATTTTATAGCACAGTCGTGGTGACTTG-3′), ligated to the 3.6 kb λ fork fragment, and the product was gel purified. The fragment was then 5′-phosphorylated at the ApaI site and ligated to the 10.1 kb ApaI fragment of λ. The final product was gel purified and sequenced to ensure that only a single copy of TerB site had been inserted.
The oligonucleotides used for generating the DNA template containing the permissive TerB substrate were 5′-AATTAACGCGCCATAATATTAACCACTTTAGTTAC AACATACTTATTTTATAGCACAGTCGTGGTGACTT GGGCC-3′ and 5′-CAAGTCACCACGACTGTGCTATA AAATAAGTATGTTGTAACTAAAGTGGTTAATATTAT GGCGCGTT-3′.
Single-molecule leading-strand synthesis assay
Leading strand DNA synthesis and data analysis were carried out as described previously (27) with some variation. Briefly, Tus (80 nM) was first introduced into the flow cell in 30 mM Tris–HCl pH 7.6, 50 mM NaCl, 0.5 mM EDTA, 5 mM DTT, 10 mM MgCl2 for 30 min to ensure saturation of TerB. Excess DNA-unbound protein was removed by washing with 15 flow cell volumes in replication mixture [40 mM Tris–Cl pH 7.6, 50 mM potassium glutamate, 0.1 mg/ml BSA, dNTPs (760 μM each), 2 mM DTT, 5 nM T7 helicase (hexamer) and 40 nM T7 DNA polymerase (purified 1:1 complex of gp5 and E. coli thioredoxin)]. These conditions allow the assembly of T7 proteins at the fork and synchronize the start of DNA synthesis on introduction of 10 mM MgCl2 and 80 nM Tus in replication mixture. For data analysis, after particle tracking, the traces were corrected for residual instabilities in the flow by subtracting traces corresponding to tethered DNA molecules that were not enzymatically altered. Pauses were defined as a minimum of six data points (at acquisition rate of 2 Hz) with amplitude fluctuations less than three times the standard deviation of the noise. The displacements of the beads were converted into numbers of synthesized nucleotides using a conversion factor (3.76 base/nm) derived from the difference in the length between ss and ds λ DNA (36) at the applied stretching force of 2.6 pN. Total experimental time was 30 min. To obtain statistically significant numbers of events in these multiplexed assays, experiments were repeated two or three times under each experimental condition.
RESULTS
DNA synthesis by the T7 helicase–polymerase is arrested by Tus–Ter in a polar manner
We first used single molecule imaging to investigate the encounter of the T7 helicase–polymerase complex with Tus–Ter in both the permissive or non-permissive orientations. DNA synthesis was measured by monitoring the length of individual DNA molecules in real time as described previously (27,28). Briefly, a 13.7 kb primed-forked DNA contained a single copy of the 22-base pair (bp) TerB, one of the strongest of the Ter sites, at a distance of 3.6 kb from the fork. It was attached in a microfluidic flow cell to the surface of a glass coverslip via one end and tethered to a magnetic bead at the other (Figure 1B). The DNA molecules were stretched by a laminar flow that exerted a 2.6 pico Newton (pN) drag force on the beads. Synthesis by the helicase–polymerase converts the surface-tethered dsDNA (long) to ssDNA (short) and the bead moves in the direction opposite to the buffer flow (Figure 1B) to generate real-time trajectories (Figure 1C and Supplementary Figures S1A, B and S2). The position of the TerB site at 3.6 kb could be determined with an accuracy of ±0.1 kb at 2.6 pN (Supplementary Figure S1C). We ignored events (∼50%) where DNA synthesis terminated before reaching the TerB site; under these conditions of continuous presence of all reagents in the flow, synthesis in the absence of Tus appeared to terminate at random positions (Supplementary Figure S1D) and the average processivity of the T7 replisome was 3.7 ± 0.6 kb (Supplementary Figure S1E). The remaining events (∼50%) where synthesis reached TerB were categorized into three classes: (i) those that were blocked permanently at the Ter site (e.g. Supplementary Figure S1B), where permanent stoppage (‘stop’ in Figure 1D) is defined as surviving until the end of data acquisition (10 ± 1 min); (ii) those that continued unimpeded through the Ter site (e.g. Supplementary Figure S1A; ‘bypass’ in Figure 1D); and (iii) those that paused at the Ter site and then resumed synthesis (e.g. Supplementary Figure S2; ‘restart’ in Figure 1D). Note that percentages of events in each class were calculated relative to the total numbers of events that reached or passed the Ter site, with or without pausing and standard deviations were estimated assuming binomial distributions.
In the absence of Tus, 9% (2 of 22) of the replicated molecules that reached TerB displayed full stoppage of DNA synthesis at or near TerB; we attributed this to random fork collapse in this 0.2 kb window (Figure 1D and Supplementary Figure S1D). We next introduced Tus by pre-incubating it alone with the template and also including it in the reaction with T7 DNA polymerase and T7 helicase. The Tus concentration (80 nM) was well above the reported KD values of Tus–TerB of 3.4 × 10−13 M in 150 mM potassium glutamate (3) to 3.1 × 10−9 M in 300 mM KCl (4); hence, the TerB site should always be fully saturated by the Tus protein. With Tus bound to TerB in the permissive orientation, the fork bypassed the Tus–Ter barrier just as if Tus were not present (Figure 1C and D). However, in a further 9% of the molecules, there was a brief stop before the barrier was bypassed by the helicase–polymerase to continue DNA synthesis. We attributed this bypass to the high affinity of binding of Tus to TerB acting as a temporary roadblock to DNA synthesis. In the remaining 82% of molecules, the helicase–polymerase displaced Tus without displaying even transient stops (Figure 1C, D and Supplementary Figure S1A). Furthermore, the rate of DNA synthesis on a 48.5 kb λ DNA template (27) was unaffected by the presence of Tus, suggesting that the helicase–polymerase can effectively displace any Tus that is non-specifically bound to DNA (Figure 1E).
In contrast, when the helicase–polymerase encountered Tus–TerB oriented to display the non-permissive face, full arrest of DNA synthesis was observed at TerB in 92% of the molecules and restart after pausing in the remaining 8% (Figure 1C, D and Supplementary Figure S1B). The 100% efficiency in arresting DNA synthesis at Tus–TerB confirms that Tus is bound to all TerB sites.
We next used a rapid quenched flow assay to follow progressive DNA synthesis at single base resolution, allowing us to study the encounter of the T7 helicase–polymerase with Tus–TerB at high spatial and temporal resolution. The 22-bp TerB sequence was introduced in either the permissive or non-permissive orientation at a specific position in the middle of a 60-bp dsDNA region of the synthetic replication fork substrate (Figure 2 and Supplementary Table S1). The replication fork contained a 35-nt 5′-tail for loading of the helicase and a 24-bp primer/template for loading the polymerase (Figure 3A, B). The replication fork was pre-incubated with Tus (380 nM) and the T7 DNA polymerase and helicase in the presence of dTTP, but without Mg2+. These conditions promote preassembly of the replication proteins on the DNA and allow synchronization of the DNA unwinding/synthesis reactions initiated with Mg2+ and dNTPs. The components were mixed in a chemical quenched-flow instrument and quenched after 0.004 to 600 s before analysis of primer extension at single base resolution on a DNA sequencing gel (Figure 3C).
Figure 3.
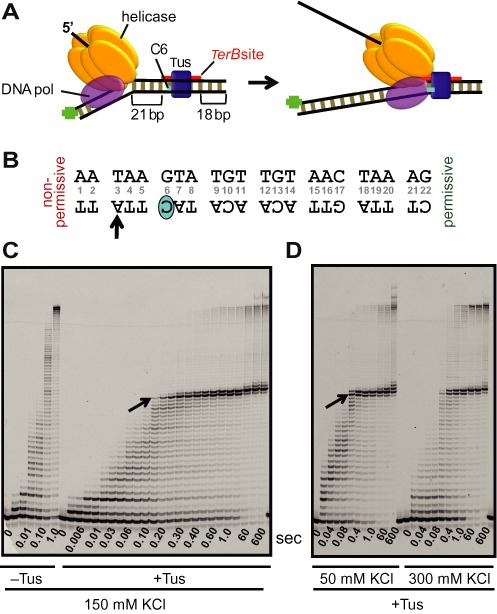
Tus–TerB arrest of DNA synthesis by T7 helicase–DNA polymerase at single-nucleotide resolution. (A) Schematic of the experimental design to study replication arrest of T7 helicase-polymerase by Tus–TerB at single-nucleotide resolution using the chemical quenched flow assay. (B) The TerB sequence and the C(6) base. The TerB sequence numbering is followed throughout. (C) High resolution DNA sequencing gel shows progressive strand displacement DNA synthesis by the T7 helicase–polymerase on a fork DNA containing TerB in the non-permissive orientation. Arrows indicate the first arrest position band corresponding to the arrow on the TerB sequence in (B). These reactions were carried out in the quenched flow apparatus (QF) in the presence or in the absence of Tus protein at 150 mM KCl at 0.1 mM dVTPs and 1 mM dTTP. (D) Sequencing gel shows the QF reactions in the presence of Tus at 50 and 300 mM KCl, with all dNTPs at 1 mM. Each time point shown here is an independent reaction. Another QF experiment is also shown in Supplementary Figure S3 and extended time scale experiments are shown in Supplementary Figures S4, S6 and S7.
Without Tus, the helicase–polymerase completed strand displacement DNA synthesis of the 60 bp DNA within 1 s at 150 mM KCl without any significant pauses (Figure 3C). However, with Tus bound in the non-permissive orientation, 24 nucleotides were incorporated up to T(3) of the TerB sequence within 0.2–0.4 s, without pausing. After incorporating T(3), DNA synthesis slowed as A(4) was added and it was effectively and permanently blocked within ∼2 s at A(4), which is two nucleotides before the GC(6). At longer times (60 s), some synthesis of A(5) was also seen, perhaps from restart as observed in the single molecule studies (Figure 1C). Overall, there was about 10% run-off synthesis of products beyond the TerB sequence on the template DNA in 60 s, resulting from displacement of Tus, which is in agreement with the efficiency of fork arrest estimated from the single molecule tethered bead experiments (Figure 1D). There were no intermediate products between A(5) and the run-off products. These results indicate that the highest barrier to the progression of the T7 helicase–polymerase occurs two to three nucleotides before GC(6).
Similar stalling behavior was observed at 40 mM NaCl (Supplementary Figure S3) and at 50 or 300 mM KCl, even at ten times higher dNTP concentration (Supplementary Figure S4, Figure 3D). The only difference was in the amount of run-off products. At 300 mM KCl, run-off synthesis at 60 s was ∼24% as compared to ∼10% at 50 mM KCl (Figure 3D). This salt dependence is most likely due to the greater ease of Tus displacement at higher ionic strength (4,5).
In contrast, similar experiments with Tus–Ter in the permissive orientation did not exhibit significant stalling or pausing of DNA synthesis by the helicase–polymerase, apart from minor pausing of some replicated molecules at the TerB site (Supplementary Figures S5–S8), which is consistent with observations of the Tus–Ter permissive orientation in the single-molecule experiments (Figure 1D). Thus, like the E. coli replisome, the heterologous T7 helicase–polymerase is arrested by Tus–Ter in a polar manner. These results argue against the necessity for a co-evolved DnaB–Tus interaction to determine polarity (i.e. the helicase interaction model).
Importance of GC(6) of Ter in replication arrest
To investigate the role of the GC(6) base pair in the operation of the mousetrap, we prepared similar fork templates containing mutant non-permissive TerB sequences (Figure 2 and Supplementary Table S1). In one set, the GC(6) base pair was replaced with AT(6) or CG(6) (C to T/G). In a second, the Ter sequence was unpaired (pre-melted) at the non-permissive end either up to AT(5) (C Junction) or GC(6) (C Open). In contrast to the GC(6) wild-type TerB, DNA synthesis by helicase–polymerase was no longer permanently arrested on the AT(6) or CG(6) Ters (C to T/G) (Figure 4A); instead, synthesis paused for 20–30 s at A(4) and A(5) and then Tus was displaced. Three times more run-off products (∼70%) were observed with these mutant forks in comparison to the GC(6) forks (∼10–20%) in 4 min (Figure 4B).
Figure 4.
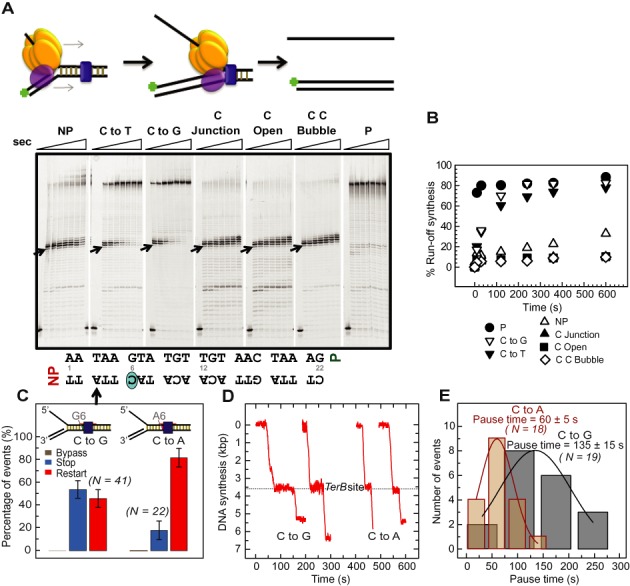
Strand displacement DNA synthesis by T7 helicase–polymerase on variant Tus–Ter fork DNAs. (A) T7 helicase and T7 DNA polymerase were preincubated with the preformed replication fork substrate and reacted with 1 mM dNTPs. DNA forks: non-permissive (NP), GC(6) to AT mutation (C to T), GC(6) to CG mutation (C to G), GC(6) at the junction (C Junction), C Open, C C Bubble and permissive (P). Reactions were quenched at 0, 10, 30, 120, 240, 360, 600, 1800 s and products were resolved in sequencing gels. Arrows indicates the first arrest position band corresponding to the arrow on the TerB sequence below. (B) Plot showing ‘% run-off synthesis’ against time, quantified from the gels in (A) as described in ‘Materials and Methods’ section. (C) Percentages of the populations of replication forks that bypassed (gray), transiently stopped (red) or fully stopped (blue) at Tus bound to TerB sites bearing the GC(6) to CG (C to G; left) or GC(6) to TA mutation (C to A; right). Error bars correspond to the standard deviation of binomial distributions. (D) Representative single molecule trajectories of the restart of DNA synthesis after transient stoppage at CG(6)-NP and TA(6)-NP TerB are shown. (E) The pause durations for events that restarted at CG(6)-NP or TA(6)-NP TerB were fit with Gaussian distributions (black and red lines, respectively). The repeat experiments carried out at 23°C are shown in Supplementary Figure S6–S9.
Similar results were obtained in tethered bead experiments. The GC(6) to CG(6) (C to G) mutant TerB resulted in five-fold increased run-off (Figure 4C and D) after a pause duration of 135 ± 15 s (Figure 4E). However, 54% of the molecules still exhibited full stoppage at TerB (Figure 4C and Supplementary Figure S2A). We attribute the lower percentage of run-off and the longer pause duration in the single molecule assay to the differences in composition of the replication buffers in the two experiments; the Tus-dsTerB complex is known to have a much lower KD in buffers containing potassium glutamate than KCl (3–5). In the case of the TA(6) substitution (C to A), we observed an eight-fold increase in run-off (Figure 4C and Supplementary Figure S2B) after a pause duration of 60 ± 5 s (Figure 4E). The GC(6) to TA(6) substitution (C to A) allows more run-off synthesis in comparison to GC(6) to CG(6) (C to G) substitution and this is consistent with a binding study that showed a modest effect on Tus binding to dsTerB when GC(6) was replaced with a CG(6) (C to G) in comparison to a TA(6) (C to A). Note that although we used TA(6) (C to A) rather that the AT(6) (C to T) TerB mutant described above, we expect the behavior of these to be similar because their affinity for Tus in both the dsTer (37) and locked (5) complexes is essentially identical, and their (reduced) efficiency of fork arrest in vivo are the same (37). Although none of these three GC(6) mutations was reported to produce more than a four-fold increase of the KD of the Tus–dsTerB interaction, they all led to a very significant reduction of fork arrest in vivo, consistent with the importance of the C(6) base in operation of the mousetrap (37). In our single-molecule experiments, the GC(6) to TA(6) (C to A) substitution resulted in less frequent permanent stoppage and more frequent restart after shorter pauses compared to the GC(6) to CG(6) (C to G) mutation (Figure 4C and D). This is not completely consistent with the in vivo data of (37), who showed that the GC(6) to CG(6) (C to G) mutation resulted in a more serious defect in fork arrest compared to the GC(6) to TA(6) or AT(6) (C to A/T) substitutions. There are clearly subtle aspects that will be resolved in future experiments. The 100% efficiency of transient pausing or stoppage of DNA synthesis in the CG(6) and AT(6) TerB sequences (C to G/T) demonstrates that the run-off DNA synthesis detected in the quenched flow assay primarily occurs after transient pausing of DNA synthesis in all of the replicated DNA molecules.
Interestingly, the histograms of the pause duration in the single molecule assays followed a Gaussian distribution (Figure 4E), indicating that the underlying mechanism of pausing and restart of DNA synthesis involves at least two steps; a single exponential decay is predicted for a single-step process. This is consistent with the observation of multiple stoppages and slow rates of DNA synthesis during the incorporation of T(3)–A(5) (Figure 3C and D). Collectively, these results indicate that the conserved GC(6) base pair is critical for permanent arrest of the helicase–polymerase fork by the mousetrap, but they also suggest contribution of a preceding dynamic clamping process that is independent of GC(6).
Unpairing the non-permissive end of TerB up to A(5) (so GC(6) is at the fork junction, C Junction) or up to C(6) (so C(6) is melted, C Open) was highly effective in completely arresting DNA synthesis. On these pre-melted Ter templates, DNA synthesis paused at A(4) for 30 s, but was completely blocked at A(5) and G(6) (Figure 4A and Supplementary Figures S6–S9); only 6–8% run-off synthesis was observed in 4 min as compared to ∼20% in the paired Ter sequence (Figure 4B). This is consistent with the expectation of the mousetrap model that a pre-melted Ter sequence with the C(6) base already bound in its pocket in Tus would present the strongest barrier to Tus displacement by replisomes (5). To narrow down the role of the C(6) base, we prepared a fork with a CC(6) mispair (C C Bubble). This single base mispair was as effective at arresting DNA synthesis as the pre-melted forks (Figure 4A and B).
The isolated DNA polymerase is not permanently arrested at the non-permissive face but is arrested at the permissive face
T7 DNA polymerase promotes limited strand displacement DNA synthesis in the absence of the helicase (21). We next probed if the Tus–Ter complex could stall the polymerase in the absence of the helicase. There are several scenarios here that depend on the relative power of the polymerase and helicase motors and whether strand separation occurs in a way that allows the C(6) base of the displaced strand to access its binding site in Tus.
We found that the isolated polymerase is not permanently arrested at the non-permissive face of Tus–TerB. DNA synthesis paused at T(3) but mostly at A(4), two nucleotides before the GC(6), but within 2–4 min the polymerase overcame the Tus barrier to make ∼55% run-off products (Figure 5 A, B and Supplementary Figure S10). The strand displacement synthesis beyond the Tus barrier is not very processive as indicated by accumulated DNA products below the run-off product and this is because of the absence of helicase. The GC(6) to AT(6) or CG(6) (C to T/G) mutant TerB sequences behaved very similarly, showing long pauses at A(4) but no permanent arrest. The pre-melted Ter sequences, on the other hand, completely arrested the isolated T7 DNA polymerase, but at A(5), which is just one nucleotide before GC(6) (Figure 5A and Supplementary Figures S9 and S10). The completely open C(6) base was most effective, permitting only ∼5% run-off in 4 min (Figure 5B). When the GC(6) is premelted, then polymerase stops one nucleotide before the C(6) base, which is similar to T7 DNA polymerase arrest one nucleotide before the interstrand crosslink in dsDNA (21). Thus, the C(6) base in the premelted forks is most likely flipped into the Tus binding pocket to form a tight barrier before polymerase approaches it.
Figure 5.
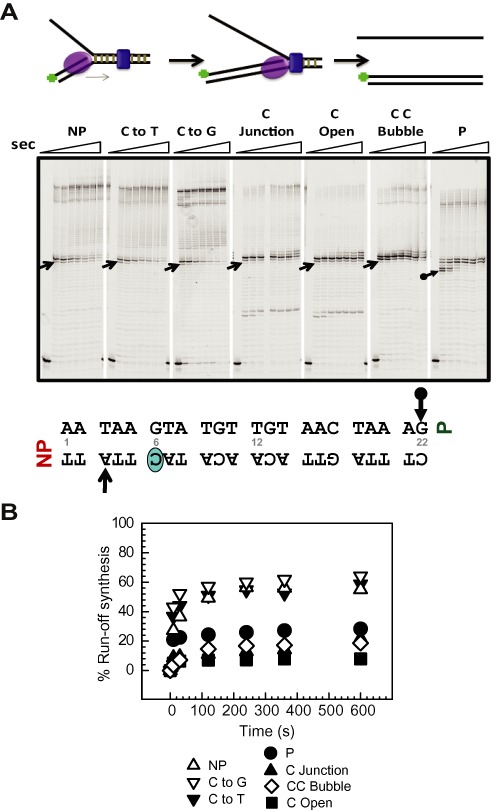
Strand displacement DNA synthesis by T7 DNA polymerase alone on various Tus–Ter fork DNAs. (A) T7 DNA polymerase was preincubated with the preformed replication fork substrate and reacted with 1 mM dNTPs. The DNA forks used here were as follows: non-permissive (NP), GC(6) to AT mutation (C to T), GC(6) to CG mutation (C to G), GC(6) at the junction (C Junction), C Open, C C Bubble and permissive (P). Reactions were quenched at 0, 10, 30, 120, 240, 360, 600, 1800 s and products resolved in sequencing gels. Arrows indicates the first arrest position band corresponding to the arrows on the TerB sequence below. (B) Plot showing ‘% run-off synthesis’ against time, quantified from the sequencing gels in (A). The duplicate sets of experiments are shown in Supplementary Figure S10 and also those conducted at 23°C in Supplementary Figure S9.
Although T7 DNA polymerase alone was not arrested at the non-permissive end, we observed pausing of DNA synthesis at the permissive end at A(23) and G(22) and a complete block at A(20) and A(19) within 10–30 s (Figure 5A). This was unexpected and indicates that arrest by Tus–Ter depends both on the polarity of the DNA motor and the strand that it occludes as it approaches the Tus–Ter complex. As corroborated below, the 5′–3′ helicase is arrested at the non-permissive but not the permissive face, whereas the 3′–5′ polymerase motor is arrested with the reverse polarity by a mechanism independent of C(6) base flipping.
The isolated T7 helicase is arrested at the non-permissive, but not the permissive face
We used a real-time fluorescence-based DNA unwinding assay on a stopped flow instrument to monitor fork separation by the T7 helicase (26,35,38). The helicase unwinds the non-permissive fork within 2.5 s in the absence of Tus, but strand separation is completely blocked when Tus is present (Figure 6A and Supplementary Figure S11). A radiometric strand separation assay (26,31) showed similar results. T7 helicase unwinds the non-permissive fork without Tus, but not with Tus present (Figure 6B and C). The permissive fork, on the other hand, is unwound by the helicase with or without Tus. The pre-melted fork bound to Tus is slightly more effective in blocking the helicase. Thus, unlike the 3′–5′ translocating DNA polymerase motor, the 5′–3′ translocating helicase is arrested at the non-permissive, but not the permissive, face of Tus–Ter (Figure 6D).
Figure 6.
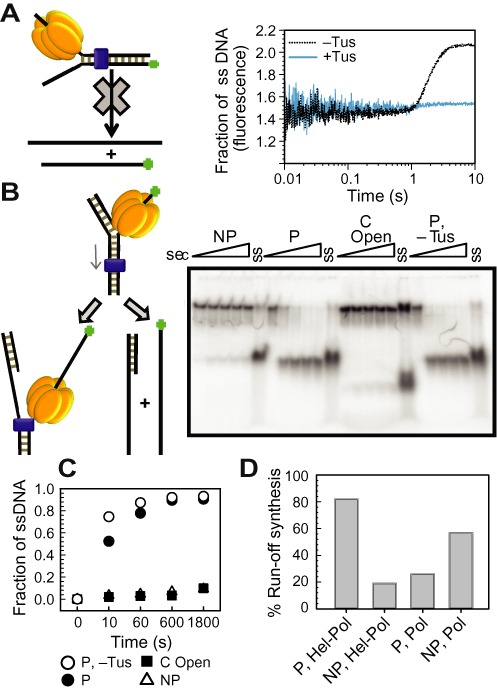
DNA unwinding by T7 helicase alone on various Tus–Ter fork DNAs. (A) Cartoon depiction of the stopped-flow fluorescence unwinding assay design (left). Fluorescence intensity traces represent the unwound ssDNA fraction as the result of DNA unwinding activity of T7 helicase on a non-permissive (NP) fork in the presence and absence of Tus (right), plotted against time. The plots are average of at least five reactions. (B) Radiometric gel unwinding assay showing helicase unwinding activity in the presence of Tus on non-permissive (NP), C Open, permissive (P) forks and on permissive (P) DNA forks in the absence of Tus. Reactions were quenched at 0, 10, 60, 600, 1800 s and then resolved in a non-denaturing PAGE gel. Lanes marked ‘ss’ represent single-stranded labeled DNA. (C) Plot showing unwound ssDNA fraction against time, quantified as described in ‘Materials and Methods’ section. (D) Comparison of run-off synthesis by helicase–DNA polymerase and DNA polymerase on fork DNAs with dsTerB in the non-permissive (NP) and permissive (P) orientations. The duplicate sets of experiments are shown in Supplementary Figure S11.
DISCUSSION
How DnaB helicase and the E. coli replisome are arrested on their approach to the non-permissive face of the Tus–Ter complex, but manage to displace Tus on approach to the permissive face, has been a subject of many studies. Despite evidence for several proposed fork arrest mechanisms, answers to this fundamental question are still controversial (39). Three models of polar arrest include the helicase interaction, mousetrap and dynamic clamp models. Our results showing that the heterologous T7 helicase is arrested in a polar manner by the E. coli Tus–Ter complex argues against obligatory Tus–DnaB interaction in determining polarity (11,13). Since T7 DNA contains no Ter sites, it seems unlikely that the heterologous T7 helicase would have evolved to show highly specific interactions with Tus and our results confirm that there is an intrinsic mechanism of replication arrest that is independent of protein–protein interactions. Nevertheless, because the T7 helicase is related to DnaB, it is plausible that there may be conserved structural features that facilitate Tus–helicase interactions in events that precede those detectable in our assays, and it is also possible that the putative Tus–DnaB interaction has an ancillary non-essential role in arrest of the E. coli replisome. Over the past decade, we have sought using sensitive surface plasmon resonance (SPR) methods in a variety of formats to detect a direct physical interaction between Tus and DnaB, but have so far been unsuccessful in doing so. Moreover, recent work showing that Tus–TerB provides a weaker, but still polar, replication fork barrier in yeast (40) is also inconsistent with a necessary role for Tus–helicase interactions.
The alternative models for polar fork arrest do not require protein–protein interactions. The mousetrap model postulates that when the helicase unwinds the GC(6) base pair of Ter, the unpaired C(6) binds in a specific binding pocket on Tus to create a tight binding interface that permanently blocks a replication complex arriving at the non-permissive face (5). The dynamic clamp model proposes that there is an intrinsic difference in the interactions of the Tus protein with the Ter site at the permissive versus the non-permissive face (4,14). Despite the elegance of the mousetrap mechanism for fork arrest at Tus-bound Ter sites, its pivotal role in replication termination has remained controversial, because of the diversity of results of in vitro strand displacement assays with various 3′–5′ and 5′–3′ helicases with different mechanisms and structures approaching Tus–Ter complexes from either direction (reviewed in (2)). In particular, reported polar arrest of DnaB sliding over dsDNA toward the non-permissive face (i.e. without strand separation) (13) is inconsistent with the mousetrap model, but is explained by the dynamic clamp. Most of these studies can now be reconciled with the operation of a dynamic clamp and/or mousetrap mechanism, depending on the inherent polarity and structure of the helicase to produce a free rather than trapped C(6), combined with an assessment of the relative power of the helicase motor under the conditions of the particular assays.
The evidence for a composite mousetrap and dynamic clamp model is the observation that the T7 replisome slows at A(4), a few nucleotides before GC(6) (dynamic clamp) even when the GC(6) is mutated to AT(6) and CG(6) (C to T/G), but permanent fork arrest only happens with the wild-type GC(6) sequence, where the C(6) base flips into its specific binding pocket (mousetrap). It should be noted that mutation of GC(6) to AT(6), TA(6) or CG(6) (C to T/A/G) severely compromises replication termination in vivo in E. coli, but has only a slight effect on the affinity of Tus for dsTerB; thus permanent fork blockage in vivo depends on the GC(6) base pair, likely involving the ultimate operation of the mousetrap.
It is notable that the KD of the locked form of Tus–TerB is only three-fold lower than the Tus–TerB duplex, but the half-life of the locked form is 40-fold longer (5). This comparison indicates that induction of C(6) base flipping may be a slow and inefficient process, but once formed the complex is kinetically stable. We showed that Tus–TerB pauses DNA synthesis by the T7 replisome for 20–30 s at A(4) and the single molecule tethered bead experiments showed that all replicated DNA molecules pause, at least transiently, even when the GC(6) base pair was mutated (Figures 1D and 4C). This indicates Tus–Ter interactions slightly upstream of GC(6) transiently stop the replisome and the run-off synthesis within the first 20–30 s is due to failure of C(6) base flipping in some of the replicated molecules. However, premelting the C(6) base converts the transient pause to a complete arrest of DNA synthesis just before the C(6) base. Indeed, even a single mismatched CC(6) bubble was sufficient to do this. We showed previously (5) using SPR that this structure in an otherwise fully dsTerB does not lead to formation of a locked complex with Tus. However, here the situation is quite different; pausing of the replisome (Figure 4A) or the polymerase alone (Figure 5A) at A(4) would leave only one base pair of dsDNA before the bubble, which would be expected to readily separate thermally to facilitate entry of C(6) into its binding pocket. These results are all consistent with the critical involvement of C(6) in the mousetrap mechanism of polar arrest, operating under physiological conditions of ionic strength. We propose that transient pausing of the fork before C(6) provides a mechanism that enables Tus–Ter to deal with the inefficient base flipping process.
DNA unwinding by T7 helicase alone was arrested by Tus–Ter in the non-permissive but not the permissive orientation. However, the Tus–Ter complex failed to permanently arrest T7 DNA polymerase at its non-permissive face when the helicase was absent. Thus, the Tus–Ter mediated blockage of DNA motors depends on whether the motor approaching the non-permissive face is traveling along the 5′-non-template strand (as is the T7 helicase) or the 3′-template strand (the polymerase). The 5′–3′ translocating helicase motor is effectively blocked at the non-permissive face, but the 3′–5′ translocating polymerase motor is not effectively arrested. We rationalize these results as follows: the polymerase moves on its template 3′-strand that contains the C(6) base and traps the C(6) nucleotide within its template binding channel (41). This prevents flipping and binding of C(6) into the cytosine binding pocket of Tus. On the other hand, the helicase moves on the opposite 5′-strand, trapping G(6) within its central DNA binding channel and effectively excluding C(6) so that it is free to bind to Tus. Thus, at the non-permissive end, helicase alone is arrested but polymerase alone is not. Only the pre-melted C(6) base that can bind in the template binding pocket of the DNA polymerase can effectively arrest the polymerase coming toward the non-permissive face. That the polymerase alone is blocked efficiently when C(6) is premelted further reinforces the argument that specific helicase–Tus interactions are unnecessary to provide an effective block to motor proteins, and indicate that the C(6) in normal dsTer is exposed by the T7 helicase before it is trapped by the polymerase.
Nevertheless, the very close proximity of the polymerase active site that can approach to within a few nucleotides of the strand separation ‘pin’ of the helicase–polymerase is evident from our single base resolution assays that show that DNA synthesis proceeds without impediment to two nucleotides before the conserved GC(6) base pair of the Ter sequence. This is consistent with our recent study which showed that the T7 helicase and polymerase move together at the fork to unwind and copy the DNA in single-nucleotide steps and the ‘pin’ of strand separation by the helicase–polymerase (21), which is the leading subunit of the helicase hexamer (19), is within two nucleotides of the polymerase active site (21) as shown in Figure 7. Polymerase and helicase proximity was apparently also observed with the E. coli replisome, which was arrested at the fourth base pair before GC(6) (42,43). While the E. coli studies were at much lower time resolution and may have been complicated by completion of synthesis occurring after dissociation of the helicase (2), this is clearly not the case here where the polymerase reaches at least to TA(3) without any sign of pausing. The helicase must normally be involved in this pre-melting step to explain the effective arrest of the helicase–polymerase complex at the non-permissive face. The helicase, a few nucleotides ahead of the polymerase can capture the G(6) nucleotide of Ter within its central channel, while pausing of the replisome upon encounter with the dynamic clamp provides time for the exposed complementary C(6) base of Ter to flip and lock into the cytosine binding pocket of Tus to spring the mousetrap. The helicase thus promotes permanent arrest of the replication fork just before the polymerase is able to capture the exposed C(6) by DNA synthesis; this would otherwise lead to displacement of Tus. Thus, a kinetic competition between trapping of C(6) by lock formation and DNA synthesis dictates the efficiency of fork arrest. On the other hand, an isolated DNA polymerase, although slowed by the dynamic clamp, can eventually trap C(6) by DNA synthesis, thus disabling the mousetrap mechanism and dissociating Tus for continued DNA synthesis.
Figure 7.
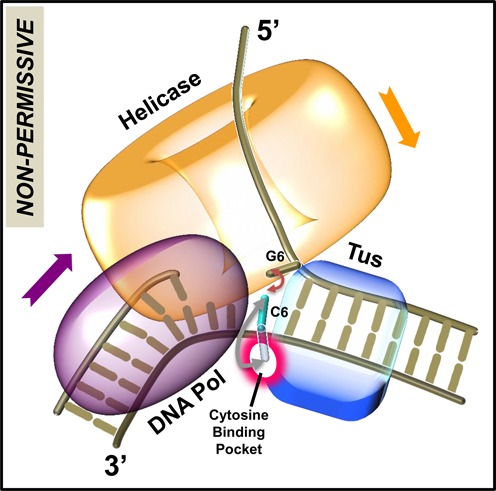
Model for replication fork arrest. The helicase ring and DNA polymerase are bound at the fork junction containing the Tus–Ter complex. After the Ter sequence is unwound up to the GC(6) base pair, the unpaired C(6) base can flip out and bind into the cytosine-specific pocket on Tus to form the C(6) Tus–Ter lock, or reanneal back with its complementary base G(6), or become base-paired through DNA synthesis. The ‘pin’ for strand separation occurs when C(6) on the template strand is already within three or fewer nucleotides of the polymerase active site. When the helicase-polymerase complex unwinds DNA, the helicase traps the G(6) base to prevent reannealing of GC(6) base pair, aiding formation of the locked complex. If GC(6) base pair is unwound by the strand displacement activity of the isolated polymerase, the C(6) is captured by the polymerase through DNA synthesis, which prevents the formation of the locked complex.
Another unanticipated observation was that the isolated T7 DNA polymerase was arrested when arriving at the other (permissive) end of the termination complex, before significant strand separation at the non-permissive end of Ter. This result is not explained by the mousetrap model, since DNA synthesis is arrested many nucleotides away from the GC(6) and in this situation the GC(6) base pair would still be intact. Examination of the Tus–Ter crystal structures (5,14) shows that even though protein–DNA interactions are distributed throughout the length of the Ter sequence, the Tus protein interacts extensively and preferentially with the 5′-end at the permissive face, i.e. the lagging strand template (Supplementary Figure S12). These interactions preferentially slow the isolated polymerase at the permissive end because the motor is traveling on the 3′-strand and hence less effective at breaking interactions that are mainly with the 5′-strand it is displacing. Essentially, the polymerase traveling along the 3′-strand cannot compete directly with the Tus interactions with the 5′-strand. On the other hand, the helicase traveling along the 5′-strand would be able to compete directly with and break Tus–Ter interactions at the permissive end, as observed. These results are consistent with a previous study that also observed partial arrest of T7 DNA polymerase at the permissive face of Tus–Ter (44). These findings imply that the Tus–Ter mechanism blocks the 3′–5′ directionality motors such as polymerases or helicases arriving at the permissive face and prevents them from colliding with the stalled replication fork, but interestingly allows the 3′–5′ directionality motors to disable the mechanism at the non-permissive end.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
Author contributions: M.P., M.M.E., S.J. and M.T. performed and analyzed the experiments; M.P., N.E.D., S.M.H. and S.S.P. conceived and designed the study, analyzed the results and wrote the manuscript; and all authors discussed and revised the manuscript.
FUNDING
National Institutes of Health (U.S.A.) [GM55310 to S.S.P.]; Australian Research Council [DP0877658 to N.E.D.]; King Abdullah University of Science and Technology Faculty Initiated Collaborative Grant [to S.M.H. and N.E.D]. Funding for open access charge: National Institutes of Health (U.S.A.) [GM55310 to S.S.P.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Hiasa H., Marians K.J. Tus prevents overreplication of oriC plasmid DNA. J. Biol. Chem. 1994;269:26959–26968. [PubMed] [Google Scholar]
- 2.Neylon C., Kralicek A.V., Hill T.M., Dixon N.E. Replication termination in Escherichia coli: structure and antihelicase activity of the Tus-Ter complex. Microbiol. Mol. Biol. Rev. 2005;69:501–526. doi: 10.1128/MMBR.69.3.501-526.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottlieb P.A., Wu S., Zhang X., Tecklenburg M., Kuempel P., Hill T.M. Equilibrium, kinetic, and footprinting studies of the Tus-Ter protein-DNA interaction. J. Biol. Chem. 1992;267:7434–7443. [PubMed] [Google Scholar]
- 4.Neylon C., Brown S.E., Kralicek A.V., Miles C.S., Love C.A., Dixon N.E. Interaction of the Escherichia coli replication terminator protein (Tus) with DNA: a model derived from DNA-binding studies of mutant proteins by surface plasmon resonance. Biochemistry. 2000;39:11989–11999. doi: 10.1021/bi001174w. [DOI] [PubMed] [Google Scholar]
- 5.Mulcair M.D., Schaeffer P.M., Oakley A.J., Cross H.F., Neylon C., Hill T.M., Dixon N.E. A molecular mousetrap determines polarity of termination of DNA replication in E. coli. Cell. 2006;125:1309–1319. doi: 10.1016/j.cell.2006.04.040. [DOI] [PubMed] [Google Scholar]
- 6.Duggin I.G., Bell S.D. Termination structures in the Escherichia coli chromosome replication fork trap. J. Mol. Biol. 2009;387:532–539. doi: 10.1016/j.jmb.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 7.Moreau M.J., Morin I., Schaeffer P.M. Quantitative determination of protein stability and ligand binding using a green fluorescent protein reporter system. Mol. Biosyst. 2010;6:1285–1292. doi: 10.1039/c002001j. [DOI] [PubMed] [Google Scholar]
- 8.Moreau M.J., Schaeffer P.M. Differential Tus-Ter binding and lock formation: implications for DNA replication termination in Escherichia coli. Mol. Biosyst. 2012;8:2783–2791. doi: 10.1039/c2mb25281c. [DOI] [PubMed] [Google Scholar]
- 9.Khatri G.S., MacAllister T., Sista P.R., Bastia D. The replication terminator protein of E. coli is a DNA sequence-specific contra-helicase. Cell. 1989;59:667–674. doi: 10.1016/0092-8674(89)90012-3. [DOI] [PubMed] [Google Scholar]
- 10.Andersen P.A., Griffiths A.A., Duggin I.G., Wake R.G. Functional specificity of the replication fork-arrest complexes of Bacillus subtilis and Escherichia coli: significant specificity for Tus-Ter functioning in E. coli. Mol. Microbiol. 2000;36:1327–1335. doi: 10.1046/j.1365-2958.2000.01945.x. [DOI] [PubMed] [Google Scholar]
- 11.Henderson T.A., Nilles A.F., Valjavec-Gratian M., Hill T.M. Site-directed mutagenesis and phylogenetic comparisons of the Escherichia coli Tus protein: DNA-protein interactions alone can not account for Tus activity. Mol. Genet. Genomics. 2001;265:941–953. doi: 10.1007/s004380100501. [DOI] [PubMed] [Google Scholar]
- 12.Mulugu S., Potnis A., Shamsuzzaman, Taylor J., Alexander K., Bastia D. Mechanism of termination of DNA replication of Escherichia coli involves helicase-contrahelicase interaction. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9569–9574. doi: 10.1073/pnas.171065898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bastia D., Zzaman S., Krings G., Saxena M., Peng X., Greenberg M.M. Replication termination mechanism as revealed by Tus-mediated polar arrest of a sliding helicase. Proc. Natl. Acad. Sci. U.S.A. 2008;105:12831–12836. doi: 10.1073/pnas.0805898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamada K., Horiuchi T., Ohsumi K., Shimamoto N., Morikawa K. Structure of a replication-terminator protein complexed with DNA. Nature. 1996;383:598–603. doi: 10.1038/383598a0. [DOI] [PubMed] [Google Scholar]
- 15.Patel S.S., Donmez I. Mechanisms of helicases. J. Biol. Chem. 2006;281:18265–18268. doi: 10.1074/jbc.R600008200. [DOI] [PubMed] [Google Scholar]
- 16.Hamdan S.M., Richardson C.C. Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 2009;78:205–243. doi: 10.1146/annurev.biochem.78.072407.103248. [DOI] [PubMed] [Google Scholar]
- 17.Hamdan S.M., van Oijen A.M. Timing, coordination, and rhythm: acrobatics at the DNA replication fork. J. Biol. Chem. 2010;285:18979–18983. doi: 10.1074/jbc.R109.022939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandey M., Syed S., Donmez I., Patel G., Ha T., Patel S.S. Coordinating DNA replication by means of priming loop and differential synthesis rate. Nature. 2009;462:940–943. doi: 10.1038/nature08611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel S.S., Pandey M., Nandakumar D. Dynamic coupling between the motors of DNA replication: hexameric helicase, DNA polymerase, and primase. Curr. Opin. Chem. Biol. 2011;15:595–605. doi: 10.1016/j.cbpa.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Syed S., Pandey M., Patel S.S., Ha T. Single-molecule fluorescence reveals the unwinding stepping mechanism of replicative helicase. Cell Rep. 2014;6:1037–1045. doi: 10.1016/j.celrep.2014.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pandey M., Patel S.S. Helicase and polymerase move together close to the fork junction and copy DNA in one-nucleotide steps. Cell Rep. 2014;6:1129–1138. doi: 10.1016/j.celrep.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hacker K.J., Johnson K.A. A hexameric helicase encircles one DNA strand and excludes the other during DNA unwinding. Biochemistry. 1997;36:14080–14087. doi: 10.1021/bi971644v. [DOI] [PubMed] [Google Scholar]
- 23.Ahnert P., Patel S.S. Asymmetric interactions of hexameric bacteriophage T7 DNA helicase with the 5′- and 3′-tails of the forked DNA substrate. J. Biol. Chem. 1997;272:32267–32273. doi: 10.1074/jbc.272.51.32267. [DOI] [PubMed] [Google Scholar]
- 24.Donmez I., Patel S.S. Mechanisms of a ring shaped helicase. Nucleic Acids Res. 2006;34:4216–4224. doi: 10.1093/nar/gkl508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stano N.M., Jeong Y.J., Donmez I., Tummalapalli P., Levin M.K., Patel S.S. DNA synthesis provides the driving force to accelerate DNA unwinding by a helicase. Nature. 2005;435:370–373. doi: 10.1038/nature03615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandey M., Levin M.K., Patel S.S. Experimental and computational analysis of DNA unwinding and polymerization kinetics. Methods Mol. Biol. 2010;587:57–83. doi: 10.1007/978-1-60327-355-8_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J.-B., Hite R.K., Hamdan S.M., Xie X.S., Richardson C.C., van Oijen A.M. DNA primase acts as a molecular brake in DNA replication. Nature. 2006;439:621–624. doi: 10.1038/nature04317. [DOI] [PubMed] [Google Scholar]
- 28.Jergic S., Horan N.P., Elshenawy M.M., Mason C.E., Urathamakul T., Ozawa K., Robinson A., Goudsmits J.M.M., Wang Y., Pan X., et al. A direct proofreader-clamp interaction stabilizes the Pol III replicase in the polymerization mode. EMBO J. 2013;32:1322–1333. doi: 10.1038/emboj.2012.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabor S., Huber H.E., Richardson C.C. Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J. Biol. Chem. 1987;262:16212–16223. [PubMed] [Google Scholar]
- 30.Patel S.S., Wong I., Johnson K.A. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 1991;30:511–525. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 31.Patel S.S., Rosenberg A.H., Studier F.W., Johnson K.A. Large scale purification and biochemical characterization of T7 primase/helicase proteins. Evidence for homodimer and heterodimer formation. J. Biol. Chem. 1992;267:15013–15021. [PubMed] [Google Scholar]
- 32.Notarnicola S.M., Mulcahy H.L., Lee J., Richardson C.C. The acidic carboxyl terminus of the bacteriophage T7 gene 4 helicase/primase interacts with T7 DNA polymerase. J. Biol. Chem. 1997;272:18425–18433. doi: 10.1074/jbc.272.29.18425. [DOI] [PubMed] [Google Scholar]
- 33.Kallansrud G., Ward B. A comparison of measured and calculated single- and double-stranded oligodeoxynucleotide extinction coefficients. Anal. Biochem. 1996;236:134–138. doi: 10.1006/abio.1996.0141. [DOI] [PubMed] [Google Scholar]
- 34.Cavaluzzi M.J., Borer P.N. Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA. Nucleic Acids Res. 2004;32:e13. doi: 10.1093/nar/gnh015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donmez I., Patel S.S. Coupling of DNA unwinding to nucleotide hydrolysis in a ring-shaped helicase. EMBO J. 2008;27:1718–1726. doi: 10.1038/emboj.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Oijen A.M., Blainey P.C., Crampton D.J., Richardson C.C., Ellenberger T., Xie X.S. Single-molecule kinetics of lambda exonuclease reveal base dependence and dynamic disorder. Science. 2003;301:1235–1238. doi: 10.1126/science.1084387. [DOI] [PubMed] [Google Scholar]
- 37.Coskun-Ari F.F., Hill T.M. Sequence-specific interactions in the Tus-Ter complex and the effect of base pair substitutions on arrest of DNA replication in Escherichia coli. J. Biol. Chem. 1997;272:26448–26456. doi: 10.1074/jbc.272.42.26448. [DOI] [PubMed] [Google Scholar]
- 38.Donmez I., Rajagopal V., Jeong Y.J., Patel S.S. Nucleic acid unwinding by hepatitis C virus and bacteriophage T7 helicases is sensitive to base pair stability. J. Biol. Chem. 2007;282:21116–21123. doi: 10.1074/jbc.M702136200. [DOI] [PubMed] [Google Scholar]
- 39.Bastia D., Zaman S. Mechanism and physiological significance of programmed replication termination. Semin. Cell Dev. Biol. 2014;30:165–173. doi: 10.1016/j.semcdb.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larsen N.B., Sass E., Suski C., Mankouri H.W., Hickson I.D. The Escherichia coli Tus-Ter replication fork barrier causes site-specific DNA replication perturbation in yeast. Nat. Commun. 2014;5:3574. doi: 10.1038/ncomms4574. [DOI] [PubMed] [Google Scholar]
- 41.Doublié S., Tabor S., Long A.M., Richardson C.C., Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 42.Hill T.M., Marians K.J. Escherichia coli Tus protein acts to arrest the progression of DNA replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2481–2485. doi: 10.1073/pnas.87.7.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohanty B.K., Sahoo T., Bastia D. Mechanistic studies on the impact of transcription on sequence-specific termination of DNA replication and vice versa. J. Biol. Chem. 1998;273:3051–3059. doi: 10.1074/jbc.273.5.3051. [DOI] [PubMed] [Google Scholar]
- 44.Lee E.H., Kornberg A. Features of replication fork blockage by the Escherichia coli terminus-binding protein. J. Biol. Chem. 1992;267:8778–8784. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.