

Abstract
Deracemization, that is, the transformation of a racemate into a single product enantiomer with theoretically 100 % conversion and 100 % ee, is an appealing but also challenging option for asymmetric synthesis. Herein a novel chemo-enzymatic deracemization concept by a cascade is described: the pathway involves two enantioselective oxidation steps and one non-stereoselective reduction step, enabling stereoinversion and a simultaneous kinetic resolution. The concept was exemplified for the transformation of rac-benzylisoquinolines to optically pure (S)-berbines. The racemic substrates were transformed to optically pure products (ee>97 %) with up to 98 % conversion and up to 88 % yield of isolated product.
Keywords: alkaloids, asymmetric synthesis, C=C coupling, deracemization, enzyme catalysis, simultaneous cascades
Several techniques have been developed to overcome the limitation of a kinetic resolution (50 % maximum yield), such as dynamic kinetic resolution (DKR),[1] dynamic kinetic asymmetric transformation (DYKAT),[2] stereoinversion,[3,4] or enantioconvergent processes (Scheme 1).[5,6] These techniques allow the process of deracemization to be achieved, that is, conversion of a racemate into an optically pure product with theoretically 100 % yield and 100 % ee.
Scheme 1.
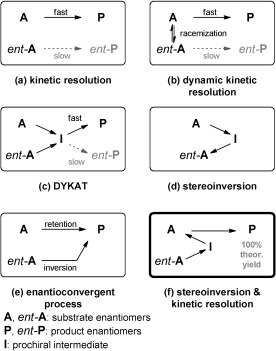
a) Kinetic resolution, b)–e) basic strategies for deracemization, and f) the designed strategy to combine stereoinversion and kinetic resolution.
The berberine bridge enzyme (BBE) catalyzes the enantioselective transformation of 1-benzyl-1,2,3,4-tetrahydroisoquinolines to yield berbines by aerobic C=H activation at the N-methyl group, forming a new intramolecular C=C bond (Scheme 2). This reaction has been shown to be suitable for the preparation of various novel optically pure berbines.[7] As the enzyme transforms only one substrate enantiomer, this reaction is a kinetic resolution.
Scheme 2.
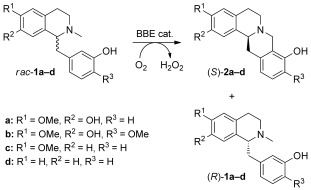
Kinetic resolution by enantioselective oxidative C=C bond formation catalyzed by the berberine bridge enzyme (BBE).
To overcome the limitation of BBE-catalyzed kinetic resolution, we first envisioned to introduce racemization of the substrate enantiomers to set up a DKR. However, this strategy proved problematic for the compounds of interest: Among the published procedures for amine racemization, which rely on homogeneous[8] or heterogeneous[9] metal catalysis, or thiyl radical-mediated reversible H-abstraction,[10] no method was suitable to act exclusively on the benzylisoquinoline substrates whilst leaving the berbine products untouched. For example, Shvo’s diruthenium complex[11] racemized benzylisoquinolines and berbines alike[7a] when employed under previously described reaction conditions.[8g] Raney nickel led to degradation of the complex alkaloids, whereas palladium on charcoal did not show any detectable racemization activity at all.
As a chemical racemization process could not be realized and because no suitable enzyme for racemization of these substrates is known,[12] an alternative was required. However, neither a DYKAT nor an enantioconvergent process seemed feasible. A stereoinversion is also unsuitable, as in this case the product structure is identical to the substrate except for its optical purity. Consequently, we designed a strategy wherein the concept of stereoinversion is combined with kinetic resolution (Scheme 1 f). Generally speaking, stereoinversion of the enantiomer ent-A will give its mirror image A, which will be transformed further in the kinetic resolution to yield exclusively product P in theoretically 100 % yield. In the present case, this strategy should allow the conversion of both substrate enantiomers of benzylisoquinolines rac-1 into optically pure berbines (S)-2.
Stereoinversion by cyclic deracemization of various amines has been described employing a monoamine oxidase (MAO) in combination with an achiral reducing agent.[3] Unfortunately, all established MAOs[3] were unsuitable for substrates 1 a–d. However, a recently developed variant of the MAO from Aspergillus niger (MAO-N variant D11) has been reported to oxidize 1-phenyl-1,2,3,4-tetrahydroisoquinoline with high (R)-selectivity.[13] The combination of this biocatalytic oxidation with non-stereoselective reduction of the resulting imine by ammonia–borane afforded the (S)-enantiomer (ee=98 %) from the racemate.
Testing the MAO-N D11 variant for the oxidation of substrates 1 a–d showed that they were indeed accepted (Figure 1). Additionally and importantly, the desired products including 2 c and 2 d were not oxidized, which is the prerequisite for their stereochemical stability under the conditions of a final simultaneous cascade process. Chiral analysis of the MAO-N catalyzed oxidations revealed that 1 a–c were converted with excellent enantioselectivity (E>200), transforming essentially only the (R)-enantiomer, while the enantioselectivity was low for 1 d (E=6.5). Consequently, 1 d was not investigated further.
Figure 1.
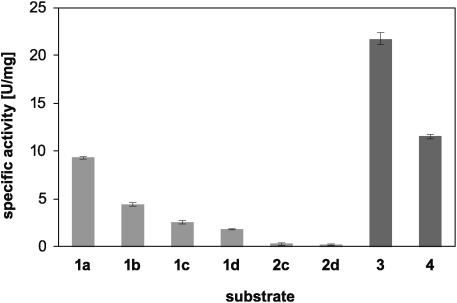
Results of substrate screening with MAO-N variant D11. Activity (criterion: mean> 5 standard deviations) was found for substrates 1 a–d (light gray), as well as the positive controls rac-phenylethylamine 3 and rac-crispine A 4 (both dark gray). Error ranges represent standard deviations of triplicate experiments.
Previous studies have found that BBE is inhibited by reducing agents,[14] as we confirmed for the ammonia–borane complex, which is most commonly employed in combination with MAO-N. We hypothesized that more bulky or less water-soluble boranes would be more compatible with BBE, as they would be prevented from entering the active site of the enzyme. Consequently, tBuNH2⋅BH3, Me3N⋅BH3, and morpholine⋅BH3 were tested as alternative reductants and proved more suitable for our system (Supporting Information, Figure S1).
Comparing the Me3N⋅BH3 and morpholine⋅BH3 complex with NH3⋅BH3 in the stereoinversion (MAO), it turned out that the former two actually performed better than the commonly employed reducing agent NH3⋅BH3, whereby morpholine⋅BH3 worked best (Supporting Information, Figure S2).
After these prerequisite investigations, a stepwise one-pot procedure was tested first (Scheme 3); thus, the stereoinversion of substrates rac-1 a–c by the MAO-N/borane-system was run to completion before BBE was added to the reaction mixture. The BBE-catalyzed ring-closure was either carried out after removal of the MAO-N biocatalyst by centrifugation, or BBE was directly added to the reaction mixture.
Scheme 3.
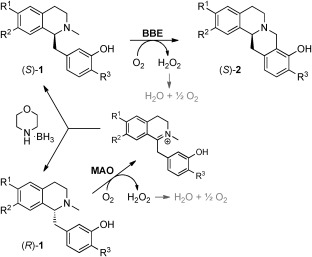
Deracemization of benzylisoquinolines rac-1 to berbines (S)-2 by a MAO/BBE/borane redox cascade (performed either in a stepwise or a concurrent fashion). MAO=monoamine oxidase MAO-N (variant D11).
The overall transformation of rac-1 to (S)-2 proceeded similarly well in both cases, resulting in high conversions (Table 1). Additionally, all compounds were obtained in enantiomerically pure form (ee>97 %, HPLC), which indicated that the enantioselectivity of BBE was not impaired by the extended reaction system.
Table 1.
Deracemization of benzylisoquinolines rac-1 to berbines (S)-2 by a stepwise transformation using MAO in the first step and BBE in the second.
| Substrate | Procedure[a] | Time[b] [h] | (S)-2[c] [%] | ee (2)[d] [%] |
|---|---|---|---|---|
| 1 a | A | 48+24 | 93 | >97 |
| 1 a | B | 48+24 | 90 | >97 |
| 1 b | A | 48+24 | 97 | >97 |
| 1 b | B | 48+24 | 95 | >97 |
| 1 c | A | 72+24 | 79[e] | >97 |
| 1 c | B | 72+24 | 79[e] | >97 |
Procedure A: direct addition of BBE to the reaction mixture after completion of the first step. Procedure B: addition of BBE after removal of MAO by centrifugation.
Reaction time for 1st+2nd step.
Determined by HPLC on an achiral stationary phase.
Determined by HPLC on a chiral stationary phase.
4 % of regioisomer formed.
Consequently, the two transformations were performed simultaneously in one pot. Thus, the two enantioselective oxidation reactions catalyzed by two different flavin-dependent enzymes (MAO and BBE) and the non-stereoselective morpholine–borane reduction were started concurrently for this cascade. It is noteworthy that in contrast to our previous studies on BBE,[7b] where catalase was required to protect the enzyme from oxidative inactivation by hydrogen peroxide, the cascade system did not profit from the addition of catalase.[15]
As the initial experiments indicated that the simultaneous reaction worked well for substrate rac-1 a and rac-1 b,[16] the synthetic applicability of the simultaneous cascade system was investigated in preparative-scale reactions (150–165 mg, 0.5 mmol). Excellent conversions were achieved for both substrates; for example, the deracemization of rac-1 b reached near-completion within 24 h (Table 2). The reaction products (S)-2 a and (S)-2 b were isolated in 88 % and 80 % yield, respectively, in optically pure form (ee>97 %).
Table 2.
Deracemization of benzylisoquinolines rac-1 to berbines (S)-2 via a cascade transformation using MAO, BBE, and morpholine-borane concurrently.[a]
| Substrate | Time [h] | Conv. (2)[b] [%] | Yield (S)-2[c] [%] | ee (2)[d] [%] |
|---|---|---|---|---|
| 1 a | 48 | 92 | 88 | >97 |
| 1 b | 24 | 98 | 80 | >97 |
Reactions were performed in buffer/DMSO=90/10, pH 7.7; 10 mm 1, 100 g L−1 E. coli/MAO-N D11, 0.05 g L−1 (1 a) or 0.02 g L−1 (1 b) BBE, 100 mm morpholine–borane, 37 °C.
Determined by HPLC on an achiral stationary phase.
Yield of islated product after column chromatography.
Determined by HPLC on a chiral stationary phase.
In summary, a new concept to transform a racemic substrate to an optically pure product was established, whereby the substrate and product differ in structure. The concept combines stereoinversion of one substrate enantiomer with a kinetic resolution. In the example presented herein, the cascade comprises an enantioselective biocatalytic amine oxidation, a chemical non-stereoselective iminium ion reduction, and an enantioselective aerobic C=C bond formation to transform benzylisoquinolines rac-1 to berbines (S)-2. The three-step system led to conversions of up to 98 % with up to 88 % yield of isolated optically pure product (S)-2 (ee>97 %, HPLC). Intriguingly, the two flavin-dependent oxidases employed act on different parts of the benzylisoquinoline substrate whereby each enzyme (MAO-N, BBE) preferentially transform the opposite enantiomer. The deracemization cascade was successfully demonstrated on preparative scale (150 mg), stimulating further developments for cascade reactions involving chemical and biocatalytic steps to avoid the disadvantages of kinetic resolution, thereby increasing the yield and making synthetic routes more economic and environmentally benign.
Experimental Section
Representative MAO-N/BBE cascade transformation on preparative scale: In an Erlenmeyer flask (250 mL), lyophilized cells of E. coli C43(DE3) expressing MAO-N D11 (5.0 g) were resuspended in phosphate buffer (45 mL; 100 mm K-phosphate, pH 7.7). Morpholine⋅BH3 (505 mg, 5 mmol), BBE solution (105 μL of a 354 μm preparation; final conc. 0.05 g L−1) and a solution of substrate 1 e (150 mg, 0.5 mmol; final conc. 10 mm) in DMSO (5 mL) were added, and the mixture was shaken at 37 °C and 150 rpm. After 24 h, additional morpholine⋅BH3 (250 mg, 2.5 mmol) and BBE solution (105 μL) were added, and shaking was continued. After 48 h, a sample (250 μL) was taken, extracted with EtOAc and analyzed for conversion. HPLC analysis showed 92 % product formation, and the reaction mixture was centrifuged (4000 rpm, 1 h, 21 °C) to remove the cell mass. The supernatant was extracted with EtOAc (3×20 mL), whereby phase separation was accelerated by centrifugation; the cell pellets were suspended in EtOAc, centrifuged again (4000 rpm, 10 min, 21 °C), and the EtOAc phase was combined with the extracts. The combined organic phases were dried over Na2SO4 and the solvent was evaporated under reduced pressure to give 0.60 g of a yellowish liquid. Column chromatography (silica gel, CH2Cl2/MeOH/NH3(aq)=96:3:1) afforded 131 mg (88 %) of (S)-2 e as a white solid (for product characterization, see the Supporting Information).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201400027.
References
- [1].For reviews, see:
- [1a].Hoyos P, Pace V, Alcántara AR. Adv. Synth. Catal. 2012;354:2585–2611. [Google Scholar]
- [1b].Pellissier H. Tetrahedron. 2011;67:3769–3802. [Google Scholar]
- [1c].Lee JH, Han K, Kim M-J, Park J. Eur. J. Org. Chem. 2010:999–1015. [Google Scholar]
- [1d].Turner NJ. Curr. Opin. Chem. Biol. 2010;14:115–121. doi: 10.1016/j.cbpa.2009.11.027. [DOI] [PubMed] [Google Scholar]
- [1e].Kamaruddin AH, Uzir MH, Aboul-Enein HY, Halim HNA. Chirality. 2009;21:449–467. doi: 10.1002/chir.20619. [DOI] [PubMed] [Google Scholar]
- [1f].Ahn Y, Ko S-B, Kim M-J, Park J. Coord. Chem. Rev. 2008;252:647–658. [Google Scholar]
- [1g].Pellissier H. Tetrahedron. 2008;64:1563–1601. [Google Scholar]
- [1h].Turner NJ. Curr. Opin. Chem. Biol. 2004;8:114–119. doi: 10.1016/j.cbpa.2004.02.001. [DOI] [PubMed] [Google Scholar]
- [1i].Pàmies O, Bäckvall J-E. Chem. Rev. 2003;103:3247–3261. doi: 10.1021/cr020029g. [DOI] [PubMed] [Google Scholar]
- [2].For a recent review, see: Steinreiber J, Faber K, Griengl H. Chem. Eur. J. 2008;14:8060–8072. doi: 10.1002/chem.200701643. [DOI] [PubMed] [Google Scholar]
- [3].Selected examples:
- [3a].Bailey KR, Ellis AJ, Reiss R, Snape T, Turner NJ. Chem. Commun. 2007:3640–3642. doi: 10.1039/b710456a. [DOI] [PubMed] [Google Scholar]
- [3b].Dunsmore CJ, Carr R, Fleming T, Turner NJ. J. Am. Chem. Soc. 2006;128:2224–2225. doi: 10.1021/ja058536d. [DOI] [PubMed] [Google Scholar]
- [3c].Carr R, Alexeeva M, Enright A, Eve TSC, Dawson MJ, Turner NJ. Angew. Chem. 115:4955–4958. doi: 10.1002/anie.200352100. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2003;42 [Google Scholar]
- [4].For reviews see:
- [4a].Schrittwieser JH, Sattler J, Resch V, Mutti FG, Kroutil W. Curr. Opin. Chem. Biol. 2011;15:249–256. doi: 10.1016/j.cbpa.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4b].Gruber CC, Lavandera I, Faber K, Kroutil W. Adv. Synth. Catal. 2006;348:1789–1805. [Google Scholar]
- [5].For a recent review, see:
- [5a].Schober M, Faber K. Trends Biotechnol. 2013;31:468–478. doi: 10.1016/j.tibtech.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].For a recent example of a chemical enantioconvergent process, see:
- [6a].Ito H, Kunii S, Sawamura M. Nat. Chem. 2010;2:972–976. doi: 10.1038/nchem.801. ; for recent examples of chemoenzymatic enantioconvergent processes, see: [DOI] [PubMed] [Google Scholar]
- [6b].Szymański W, Westerbeek A, Janssen DB, Feringa BL. Angew. Chem. 123:10900–10903. doi: 10.1002/anie.201105164. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:10900–10903. [Google Scholar]
- [6c].Schober M, Gadler P, Knaus T, Kayer H, Birner-Grünberger R, Gülly C, Macheroux P, Wagner U, Faber K. Org. Lett. 2011;13:4296–4299. doi: 10.1021/ol201635y. ; for a recent example of an enzymatic enantioconvergent process, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6d].Schober M, Toesch M, Knaus T, Strohmeier GA, van Loo B, Fuchs M, Hollfelder F, Macheroux P, Faber K. Angew. Chem. 2013;125:3359–3361. doi: 10.1002/ange.201209946. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013;52 [Google Scholar]
- [7a].Schrittwieser JH, Resch V, Sattler JH, Lienhart W-D, Durchschein K, Winkler A, Gruber K, Macheroux P, Kroutil W. Angew. Chem. 2011;123:1100–1103. doi: 10.1002/anie.201006268. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011;50 [Google Scholar]
- [7b].Resch V, Schrittwieser JH, Wallner S, Macheroux P, Kroutil W. Adv. Synth. Catal. 2011;353:2377–2383. [Google Scholar]
- [7c].Schrittwieser JH, Resch V, Wallner S, Lienhart W-D, Sattler JH, Resch J, Macheroux P, Kroutil W. J. Org. Chem. 2011;76:6703–6714. doi: 10.1021/jo201056f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7d].Resch V, Lechner H, Schrittwieser JH, Wallner S, Gruber K, Macheroux P, Kroutil W. Chem. Eur. J. 2012;18:13173–13179. doi: 10.1002/chem.201201895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7e].Schrittwieser JH, Resch V. RSC Adv. 2013;3:17602–17632. doi: 10.1039/c3ra42123f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].For examples using ruthenium complexes, see:
- [8a].Thalén LK, Hedberg MH, Bäckvall J-E. Tetrahedron Lett. 2010;51:6802–6805. [Google Scholar]
- [8b].Thalén LK, Zhao D, Sortais J-B, Paetzold J, Hoben C, Bäckvall J-E. Chem. Eur. J. 2009;15:3403–3410. doi: 10.1002/chem.200802303. [DOI] [PubMed] [Google Scholar]
- [8c].Hoben CE, Kanupp L, Bäckvall J-E. Tetrahedron Lett. 2008;49:977–979. [Google Scholar]
- [8d].Veld MAJ, Hult K, Palmans ARA, Meijer EW. Eur. J. Org. Chem. 2007:5416–5421. [Google Scholar]
- [8e].Roengpithya C, Patterson DA, Livingston AG, Taylor PC, Irwin JL, Parrett MR. Chem. Commun. 2007:3462–3463. doi: 10.1039/b709035h. [DOI] [PubMed] [Google Scholar]
- [8f].Paetzold J, Bäckvall JE. J. Am. Chem. Soc. 2005;127:17620–17621. doi: 10.1021/ja056306t. [DOI] [PubMed] [Google Scholar]
- [8g].Pàmies O, Éll AH, Samec JSM, Hermanns N, Bäckvall J-E. Tetrahedron Lett. 2002;43:4699–4702. . For examples using iridium complexes, see: [Google Scholar]
- [8h].Jerphagnon T, Gayet AJA, Berthiol F, Ritleng V, Mršić N, Meetsma A, Pfeffer M, Minnaard AJ, Feringa BL, de Vries JG. Chem. Eur. J. 2009;15:12780–12790. doi: 10.1002/chem.200902103. [DOI] [PubMed] [Google Scholar]
- [8i].Blacker AJ, Stirling MJ, Page MI. Org. Process Res. Dev. 2007;11:642–648. [Google Scholar]
- [8j].Stirling M, Blacker J, Page MI. Tetrahedron Lett. 2007;48:1247–1250. [Google Scholar]
- [9].For examples using palladium on various supports, see:
- [9a].Parvulescu AN, Jacobs PA, De Vos DE. Appl. Catal. A. 2009;368:9–16. [Google Scholar]
- [9b].Andrade LH, Silva AV, Pedrozo EC. Tetrahedron Lett. 2009;50:4331–4334. [Google Scholar]
- [9c].Parvulescu AN, Van der Eycken E, Jacobs PA, De Vos DE. J. Catal. 2008;255:206–212. [Google Scholar]
- [9d].Parvulescu AN, Jacobs PA, De Vos DE. Chem. Eur. J. 2007;13:2034–2043. doi: 10.1002/chem.200600899. [DOI] [PubMed] [Google Scholar]
- [9e].Parvulescu A, De Vos D, Jacobs P. Chem. Commun. 2005:5307–5309. doi: 10.1039/b509747a. [DOI] [PubMed] [Google Scholar]
- [9f].Reetz MT, Schimossek K. Chimia. 1996;50:668–669. ; for examples using nickel or palladium nanoparticles, see: [Google Scholar]
- [9g].Engström K, Johnston EV, Verho O, Gustafson KPJ, Shakeri M, Tai C-W, Bäckvall J-E. Angew. Chem. 125:14256–14260. doi: 10.1002/anie.201306487. Angew. Chem. Int. Ed, 2013, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9h].Geukens I, Plessers E, Seo JW, De Vos DE. Eur. J. Inorg. Chem. 2013:2623–2628. [Google Scholar]
- [9i].Shakeri M, Tai C-W, Göthelid E, Oscarsson S, Bäckvall J-E. Chem. Eur. J. 2011;17:13269–13273. doi: 10.1002/chem.201101265. [DOI] [PubMed] [Google Scholar]
- [9j].Kim Y, Park J, Kim M-J. Tetrahedron Lett. 2010;51:5581–5584. [Google Scholar]
- [9k].Kim M-J, Kim W-H, Han K, Choi YK, Park J. Org. Lett. 2007;9:1157–1159. doi: 10.1021/ol070130d. for an example using Raney metals, see: [DOI] [PubMed] [Google Scholar]
- [9l].Parvulescu AN, Jacobs PA, De Vos DE. Adv. Synth. Catal. 2008;350:113–121. [Google Scholar]
- [10a].Poulhès F, Vanthuyne N, Bertrand MP, Gastaldi S, Gil G. J. Org. Chem. 2011;76:7281–7286. doi: 10.1021/jo201256w. [DOI] [PubMed] [Google Scholar]
- [10b].El Blidi L, Vanthuyne N, Siri D, Gastaldi S, Bertrand MP, Gil G. Org. Biomol. Chem. 2010;8:4165–4168. doi: 10.1039/c0ob00054j. [DOI] [PubMed] [Google Scholar]
- [10c].El Blidi L, Nechab M, Vanthuyne N, Gastaldi S, Bertrand MP, Gil G. J. Org. Chem. 2009;74:2901–2903. doi: 10.1021/jo900074w. [DOI] [PubMed] [Google Scholar]
- [10d].Routaboul L, Vanthuyne N, Gastaldi S, Gil G, Bertrand M. J. Org. Chem. 2008;73:364–368. doi: 10.1021/jo702241y. [DOI] [PubMed] [Google Scholar]
- [10e].Gastaldi S, Escoubet S, Vanthuyne N, Gil G, Bertrand MP. Org. Lett. 2007;9:837–839. doi: 10.1021/ol063062o. [DOI] [PubMed] [Google Scholar]
- [10f].Escoubet S, Gastaldi S, Vanthuyne N, Gil G, Siri D, Bertrand MP. J. Org. Chem. 2006;71:7288–7292. doi: 10.1021/jo061033l. [DOI] [PubMed] [Google Scholar]
- [10g].Escoubet S, Gastaldi S, Vanthuyne N, Gil G, Siri D, Bertrand MP. Eur. J. Org. Chem. 2006:3242–3250. doi: 10.1021/jo061033l. [DOI] [PubMed] [Google Scholar]
- [11].For reviews, see: Warner MC, Casey CP, Bäckvall JE. Top. Organomet. Chem. 2011;37:85–125. [Google Scholar]
- [11b].Karvembu R, Prabhakaran R, Natarajan K. Coord. Chem. Rev. 2005;249:911–918. . For the original report, see: [Google Scholar]
- [11c].Shvo Y, Czarkie D, Rahamim Y, Chodosh DF. J. Am. Chem. Soc. 1986;108:7400–7402. [Google Scholar]
- [12].The transformation of (SR. )-reticuline into the ( )-enantiomer occurs in nature as a strict “one-way” process involving two enzymes, an oxidase and an imine reductase:
- [12a].Hirata K, Poeaknapo C, Schmidt J, Zenk MH. Phytochemistry. 2004;65:1039–1046. doi: 10.1016/j.phytochem.2004.02.015. [DOI] [PubMed] [Google Scholar]
- [12b].De-Eknamkul W, Zenk MH. Phytochemistry. 1992;31:813–821. [Google Scholar]
- [12c].De-Eknamkul W, Zenk MH. Tetrahedron Lett. 1990;31:4855–4858. [Google Scholar]
- [13].Ghislieri D, Green AP, Pontini M, Willies SC, Rowles I, Frank A, Grogan G, Turner NJ. J. Am. Chem. Soc. 2013;135:10863–10869. doi: 10.1021/ja4051235. [DOI] [PubMed] [Google Scholar]
- [14].Steffens P, Nagakura N, Zenk MH. Tetrahedron Lett. 1984;25:951–952. [Google Scholar]
- [15].Apparently, the presence of the borane complex and the natural catalase activity of the E. coli22. cells expressing the MAO were sufficient to degrade any H O formed.
- [16].Substrate 1 c2 c. was transformed too slowly by MAO, resulting in incomplete conversion to.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.