

Abstract
Leiodermatolide is an antimitotic macrolide isolated from the marine sponge Leiodermatium sp. whose potentially novel tubulin-targeting mechanism of action makes it an exciting lead for anticancer drug discovery. In pursuit of a sustainable supply, we report a highly stereocontrolled total synthesis (3.2 % yield) based on a convergent sequence of palladium-mediated fragment assembly and macrolactonization. Boron-mediated aldol reactions were used to configure the three key fragments 2, 5, and 6 by employing the appropriate enantiomer of the lactate-derived ketone 7.
Keywords: aldol reaction, antitumor agents, cross-coupling, macrolides, total synthesis
Tubulin-targeting compounds are perhaps the most validated subset of clinically important anticancer agents, with natural products and analogues representing the mainstay for current chemotherapy,[1–3] recently supplemented by the approval of the antibody–maytansinoid conjugate Kadcyla (trastuzumab emtansine).[2] Leiodermatolide (1; Scheme 1) was isolated (0.001 % wet weight) by the Wright group in 2008 from the lithistid sponge Leiodermatium sp. collected by submersible off the Florida coastline.[4a] Leiodermatolide exhibits potent antiproliferative activity against a panel of human cancer cell lines (e.g. IC50=3.3 nm for A549 lung adenocarcinoma cells, 5.0 nm for PANC-1 pancreatic carcinoma cells), whilst showing reduced toxicity to normal cells. This activity appears to be mediated through the disruption of tubulin dynamics to induce cell-cycle arrest in the G2/M phase and apoptosis. Although the exact mechanism of action of leiodermatolide is currently unknown, it is clearly distinct from that of other tubulin-targeting drugs. Thus, leiodermatolide could serve as a promising lead compound for the development of new anticancer agents, provided a sustainable supply can be generated by chemical synthesis.[5–7]
Scheme 1.
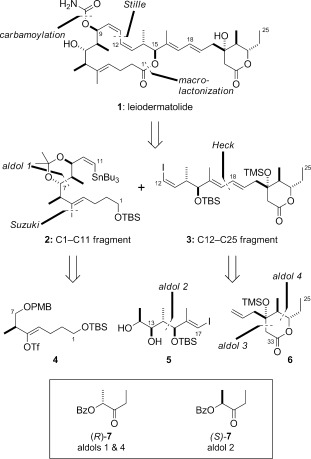
Retrosynthetic analysis and key fragments for the synthesis of leiodermatolide. Bz=benzoyl, PMB=para-methoxybenzyl, TBS=tert-butyldimethylsilyl, Tf=trifluoromethanesulfonyl, TMS=trimethylsilyl.
From a structural perspective, leiodermatolide features a triply unsaturated 16-membered macrolactone appended at C9 with a carbamate group and at C15 with an E,E-dienyl side chain terminating in a δ-lactone ring. This unique structure incorporates a total of nine stereocenters. In association with the Wright research group,[4b] we elucidated the relative configuration of leiodermatolide by using a combination of homo- and heteronuclear NMR spectroscopic analysis, molecular modeling, and computational DP4 NMR prediction.[8] The resulting assignment for the C1–C16 region was further supported by our synthesis of a macrocyclic fragment with a truncated side chain,[5] whereas an alternative stereostructure could be ruled out on the basis of synthetic studies reported earlier.[6a] The full configuration of the isolated C1–C16 and C20–C25 stereoclusters was only recently tied down with the first total synthesis of (−)-leiodermatolide (1) by the Fürstner research group employing an elegant strategy based on ring-closing alkyne metathesis.[7] We now report a highly convergent total synthesis of (−)-leiodermatolide implementing a complementary macrolactonization strategy that also features the extensive application of our versatile lactate aldol chemistry[9] along with a variety of palladium-mediated coupling reactions.[10]
Building on the lessons learned from earlier synthetic efforts directed towards the macrocyclic core,[5] Scheme 1 depicts the main retrosynthetic disconnections and key fragments 2–6 devised for the synthesis of leiodermatolide. The structure was initially simplified by disassembly of the 10Z,12Z-diene region and opening of the macrolactone ring in 1 to reveal the C1–C11 vinyl stannane 2 and the C12–C25 vinyl iodide 3 containing the entire side chain for a planned late-stage Stille coupling. The former fragment was then envisaged to be available by elaboration of vinyl triflate 4 through a Suzuki-type methylation and an anti-selective aldol reaction using (R)-7. The more elaborate fragment 3 would arise in turn through stereocontrolled installation of the 16E,18E diene by a Heck coupling between vinyl iodide 5 and the correctly configured allyl-substituted δ-lactone 6, constructed using (S)- and (R)-7, respectively.
The synthesis of vinyl stannane 2 utilized Roche ester derivative 8 as the source of the C6 methyl-bearing stereocenter (Scheme 2).[11] The required 4E-configured trisubstituted alkene was first introduced via the corresponding stereodefined vinyl triflate 4. In practice, controlled addition of TBSO(CH2)4MgBr to 8 provided the required ketone (88 %), which was converted into 4 with high selectivity (82 %, >20:1 Z/E) by treatment of the kinetically generated lithium enolate (LiHMDS) with the Comins reagent.[12] After screening various methods for methylation, Suzuki coupling of 4 with trimethylboroxine[13] (cat. [Pd(PPh3)4], K2CO3) was found to proceed well to afford the E alkene 9 (96 %, >20:1 E/Z). Following cleavage of the PMB ether (DDQ) and Dess–Martin oxidation (69 %), the resulting aldehyde 10 was treated with the E dicyclohexylboron enolate derived from (R)-7 (c-Hex2BCl, Et3N).[9] This matched aldol addition[14] afforded the anti adduct 11 (96 %, d.r.>20:1) with a high level of control over the C7/C8 stereocenters.
Scheme 2.
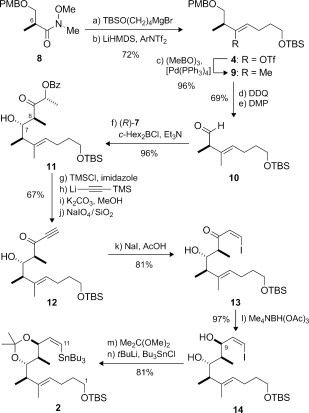
Preparation of vinyl stannane 2. Reagents and conditions: a) TBSO(CH2)4MgBr, THF, −78 °C, 88 %; b) LiHMDS, THF; Comins reagent, −78→−20 °C, 82 %; c) (MeBO)3, [Pd(PPh3)4] (10 mol %), K2CO3, dioxane, 50 °C, 96 % (>20:1 E/Z); d) DDQ, pH 7 buffer, CH2Cl2, 84 %; e) DMP, NaHCO3, CH2Cl2, 82 %; f) (R)-7, c-Hex2BCl, Et3N, Et2O, −78→−20 °C, 96 % (d.r.>20:1); g) TMSCl, imid, CH2Cl2, 96 %; h) LiC=CTMS, THF, −78 °C; i) K2CO3, MeOH; j) NaIO4/SiO2, CH2Cl2, 69 % over 3 steps; k) NaI, AcOH, THF, 81 % (8:1 Z/E); l) Me4NBH(OAc)3, MeCN, AcOH (3:1), −30 °C, 97 % (d.r.>20:1); m) Me2C(OMe)2, PPTS, CH2Cl2, 99 %; n) tBuLi, Bu3SnCl, Et2O, −78 °C, 81 %. Comins reagent=2-(Tf2N)-5-Cl(C5H3N), DDQ=2,3-dichloro-5,6-dicyano-1,4-benzoquinone, DMP=Dess–Martin periodinane, HMDS=hexamethyldisilazide, PPTS=pyridinium para-toluenesulfonate.
Next, 11 was converted into ynone 12 (67 %) by a sequence of silylation, (trimethylsilyl)acetylide addition, basic methanolysis, and oxidative glycol cleavage.[15] The Z iodoenone could then be conveniently accessed through conjugate addition of NaI (AcOH, THF)[16] to 12 to afford 13 (8:1 Z/E, 81 % yield of the isolated Z isomer). To set the C9 configuration, Evans–Saksena reduction[17] (Me4NBH(OAc)3, MeCN, AcOH) of 13 proceeded well to give the 1,3-anti diol 14 (97 %, d.r.>20:1). Although preliminary studies were discouraging,[5] a cyclic C7/C9 protecting group was selected; thus, differentiation of the diol for regioselective carbamate formation at C9 would be required in the final stages of the synthesis. Accordingly, diol 14 was first converted into its acetonide (Me2C(OMe)2, PPTS), and stannylation (tBuLi, Bu3SnCl, 81 %) then provided the C1–C11 fragment 2 (20 % yield over 14 steps) in readiness for installation of the 10Z,12Z diene of leiodermatolide.
The requisite Stille coupling partner 3 was prepared from chiral intermediates 5 and 6 (Scheme 3). Vinyl iodide 5 was readily secured by using a second boron-mediated aldol reaction, this time between (S)-7[9] and aldehyde 15[18] to give the anti adduct 16 (90 %, d.r.>20:1). Silylation (TBSOTf) of 16, selective reduction with LiAlH4 (d.r.>20:1), and methanolysis smoothly afforded diol 5 (91 % over 3 steps). Construction of the δ-lactone fragment 6 required the installation of three contiguous stereocenters, including the axial C21 allyl group. The C22/C23 configuration was set by a third boron-mediated aldol reaction, in this case between (R)-7[9] again and propanal to generate ketone 17 (94 %, d.r.>20:1). Following silylation (TBSOTf),[19] the addition of H2C=CHCH2MgBr gave a 1,2-diol, which underwent oxidative cleavage (NaIO4/SiO2)[15] to afford 18 (83 %). The C21 tertiary alcohol stereocenter could then be configured with good selectivity (d.r. 10:1) through the Mukaiyama aldol addition[20] of silyl ketene acetal 19, mediated by BF3⋅OEt2 at −78 °C. Subsequent acid-mediated cyclization then provided δ-lactone 20 (82 %, 2 steps).[21] In this situation, 1,2-induction by Felkin–Anh control and 1,3-induction based on the Evans polar model are mutually reinforcing.[22] The NMR spectroscopic data for 20 matched well with those for the δ-lactone ring of leiodermatolide, and the stereochemical assignment was further confirmed by single-crystal X-ray crystallography.[4b], [23] Silylation (TMSCl) then afforded 6 (97 %), in readiness for the key Heck reaction. After some experimentation, the exposure of 5 and 6 to Pd(OAc)2 (10 mol %) and Ag2CO3 in DMF at 80 °C[24] generated adduct 21 (73 %) with exclusively the required 16E,18E geometry. Stork–Wittig olefination[25] of the aldehyde arising from oxidative glycol cleavage of 21 (NaIO4/SiO2)[15] provided the Z vinyl iodide 3 (62 %, >20:1 Z/E), thus completing the synthesis of this fragment in 28 % yield over 10 steps.
Scheme 3.
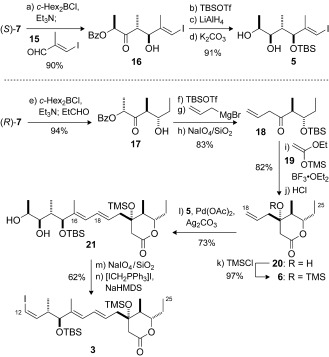
Preparation of vinyl iodide 3. Reagents and conditions: a) (S)-7, c-Hex2BCl, Et3N, Et2O, −78→−20 °C, 90 % (d.r.>20:1); b) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C; c) LiAlH4, THF, −78 °C; d) K2CO3, MeOH, 91 % over 3 steps; e) c-Hex2BCl, Et3N, Et2O; EtCHO, −78→−20 °C, 94 % (d.r.>20:1); f) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 98 %; g) H2C=CHCH2MgBr, THF, −78 °C; h) NaIO4, MeOH, pH 7 buffer, 85 % over 2 steps; i) 19, BF3⋅OEt2, CH2Cl2, −78 °C; j) 3 m HCl, THF, H2O, 82 % over 2 steps (d.r. 10:1); k) TMSCl, imid, CH2Cl2, 97 %; l) 5, Pd(OAc)2 (10 mol %), Ag2CO3, DMF, 80 °C, 73 %; m) NaIO4/SiO2, CH2Cl2; n) [ICH2PPh3]I, NaHMDS, THF, −78 °C, 62 % over 2 steps. DMF=N,N-dimethylformamide.
With an effective means of generating the two key fragments 2 and 3 secured, their controlled linkage to construct the full 25-carbon backbone of leiodermatolide was now executed (Scheme 4). Accordingly, Stille cross-coupling[26] of 2 and 3 under Fürstner conditions[26a] smoothly established the 10Z,12Z diene of 22 (80 %). A sequence of selective desilylation at C1 and C21 (HF⋅py, pyridine), oxidation at C1, and desilylation at C15 (TBAF) then provided the required seco acid 23 (51 % overall). Gratifyingly, Yamaguchi macrolactonization[27] served to efficiently close the 16-membered macrolactone. Acetonide cleavage then gave 24 (73 %), corresponding to the descarbamoyl derivative of leiodermatolide. Notably, the order of steps could be reversed, whereby acetonide cleavage was carried out first on 23 to give the unprotected tetraol, which was then macrolactonized to afford 24 with complete selectivity at C15.[23]
Scheme 4.
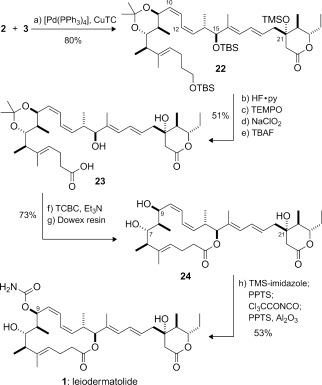
Completion of leiodermatolide (1). Reagents and conditions: a) [Pd(PPh3)4] (10 mol %), CuTC, Bu4NPh2PO2, DMF, 80 %; b) HF⋅py, pyridine, THF; c) TEMPO, PhI(OAc)2, CH2Cl2; d) NaClO2, NaH2PO4, 2-methyl-2-butene, tBuOH, H2O, THF; e) TBAF, THF, 50 °C, 51 % over 4 steps; f) TCBC, Et3N, THF; DMAP, PhMe, 80 %; g) Dowex 50WX8, MeOH, 91 %; h) TMS-imidazole, CH2Cl2; PPTS, MeOH; Cl3CCONCO, CH2Cl2, −78 °C; Al2O3; PPTS, MeOH, 53 %. DMAP=4-dimethylaminopyridine, py=pyridine, TBAF=tetrabutylammonium fluoride, TC=2-thiophenecarboxylate, TCBC=2,4,6-trichlorobenzoyl chloride, TEMPO=2,2,6,6-tetramethylpiperidine 1-oxyl.
Initially, we explored the introduction of the carbamate functionality on triol 24 itself by treatment with Cl3CCONCO (CH2Cl2, −78 °C),[28] but this reaction only afforded a disappointing 4:1 mixture of the C7 and C9 carbamates,[23], [29] a result anticipated from earlier studies with a truncated macrolide core.[5] To solve this problem, an effective sequence of hydroxy-group differentiation was sought to overturn the intrinsic substrate selectivity. Pleasingly, regiocontrolled silylation at C7 (1-(trimethylsilyl)imidazole; PPTS, MeOH) gave the corresponding C9/C21 diol, the treatment of which with Cl3CCONCO and acidic workup exclusively afforded (−)-leiodermatolide (1, 53 %; 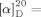 −74.0 (c=0.027, MeOH); lit.:[4]
−74.0 (c=0.027, MeOH); lit.:[4] 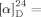 −84.2 (c=0.34, MeOH)). To our satisfaction, all 1H and 13C NMR spectroscopic data for this synthetic material correlated with those recorded for an authentic sample of natural leiodermatolide.
−84.2 (c=0.34, MeOH)). To our satisfaction, all 1H and 13C NMR spectroscopic data for this synthetic material correlated with those recorded for an authentic sample of natural leiodermatolide.
In conclusion, we have achieved a highly convergent total synthesis of the antimitotic marine macrolide (−)-leiodermatolide (1) in 23 steps and 3.2 % yield. This route features a uniformly high level of stereocontrol relying on lactate aldol chemistry,[9] combined with expedient fragment assembly based on a variety of palladium-catalyzed coupling reactions and an efficient macrolactonization step. It should be amenable to the synthesis of useful quantities of this otherwise scarce yet highly promising anticancer agent[30] for further biological evaluation and should also enable structure–activity-relationship studies. Indeed, we have already prepared the first novel leiodermatolide analogues in the form of triol 24 and the regioisomeric C7 carbamate.[31]
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201310164.
References
- [1a].Altmann KH, Gertsch J. Nat. Prod. Rep. 2007;24:327. doi: 10.1039/b515619j. [DOI] [PubMed] [Google Scholar]
- [1b].Cragg GML, Kingston DGI, Newman DJ. Anticancer Agents from Natural Products. Boca Raton: Taylor & Francis Group; 2005. [Google Scholar]
- [1c].Kavallaris M, Verrills NM, Hill BT. Drug Resist. Updates. 2001;4:392. doi: 10.1054/drup.2002.0230. [DOI] [PubMed] [Google Scholar]
- [2].Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh D-Y, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K. N. Engl. J. Med. 2013;367:1783. doi: 10.1056/NEJMoa1209124. Correction: [DOI] [PMC free article] [PubMed] [Google Scholar]; Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh D-Y, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K. N. Engl. J. Med. 2012;368 doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3a].Cragg GM, Newman DJ. Biochim. Biophys. Acta Gen. Subj. 2013;1830:3670. doi: 10.1016/j.bbagen.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3b].Dalby SM, Paterson I. Curr. Opin. Drug Discov. Devel. 2010;13:777. [PubMed] [Google Scholar]
- [3c].Cragg GM, Grothaus PG, Newman DJ. Chem. Rev. 2009;109:3012. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- [3d].Paterson I, Anderson EA. Science. 2005;310:451. doi: 10.1126/science.1116364. [DOI] [PubMed] [Google Scholar]
- [3e].Paterson I, Yeung K-S. Chem. Rev. 2005;105:4237. doi: 10.1021/cr040614c. [DOI] [PubMed] [Google Scholar]
- [4a].Wright AE, Reed JK, Roberts J, Longley RE. 2011. Chem. Abstr2008148, U.S. Pat. Appl. Publ. (USA), US2008033035, 14 pp. [ , 230103];
- [4b].Paterson I, Dalby SM, Roberts JC, Naylor GJ, Guzmán EA, Isbrucker R, Pitts TP, Linley P, Divlianska D, Reed JK, Wright AE. Angew. Chem. 123:3277. [Google Scholar]; Angew. Chem. Int. Ed. 2011;50 [Google Scholar]
- [5].Paterson I, Paquet T, Dalby SM. Org. Lett. 2011;13:4398. doi: 10.1021/ol2017388. [DOI] [PubMed] [Google Scholar]
- [6a].Rink C, Navickas V, Maier ME. Org. Lett. 2011;13:2334. doi: 10.1021/ol200584a. [DOI] [PubMed] [Google Scholar]
- [6b].Navickas V, Rink C, Maier ME. Synlett. 2011:191. [Google Scholar]
- [7].Willwacher J, Kausch-Busies N, Fürstner A. Angew. Chem. 2012;124:12207. doi: 10.1002/anie.201206670. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51 [Google Scholar]
- [8].Smith SG, Goodman JM. J. Am. Chem. Soc. 2010;132:12946. doi: 10.1021/ja105035r. [DOI] [PubMed] [Google Scholar]
- [9a].Paterson I, Wallace DJ, Velázquez SM. Tetrahedron Lett. 1994;35:9083. [Google Scholar]
- [9b].Paterson I, Wallace DJ. Tetrahedron Lett. 1994;35:9087. [Google Scholar]
- [9c].Paterson I, Wallace DJ, Cowden CJ. Synthesis. 1998:639. [Google Scholar]
- [10].Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. 2005;117:4516. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44 [Google Scholar]
- [11a].Paterson I, Florence GJ, Gerlach K, Scott JP. J. Am. Chem. Soc. 2001;123:9535. doi: 10.1021/ja011211m. [DOI] [PubMed] [Google Scholar]
- [11b].Paterson I, Arnott EA. Tetrahedron Lett. 1998;39:7185. [Google Scholar]
- [12].Comins DL, Dehghani A. Tetrahedron Lett. 1992;33:6299. [Google Scholar]
- [13].Gray M, Andrews IP, Hook DF, Kitteringham J, Voyle M. Tetrahedron Lett. 2000;41:6237. [Google Scholar]
- [14].For a review of asymmetric boron-mediated aldol reactions, see: Cowden CJ, Paterson I. Org. React. 1997;51:1. [Google Scholar]
- [15].Zhong Y-L, Shing TKM. J. Org. Chem. 1997;62:2622. doi: 10.1021/jo9621581. [DOI] [PubMed] [Google Scholar]
- [16a].Taniguchi M, Kobayashi S, Nakagawa M, Hino T, Kishi Y. Tetrahedron Lett. 1986;27:4763. [Google Scholar]
- [16b].Shibahara S, Fujino M, Tashiro Y, Okamoto N, Esumi T, Takahashi K, Ishihara J, Hatakeyama S. Synthesis. 2009:2935. [Google Scholar]
- [17].Evans DA, Chapman KT, Carreira EM. J. Am. Chem. Soc. 1988;110:3560. [Google Scholar]
- [18a].Baker R, Castro JL. J. Chem. Soc. Perkin Trans. 1. 1990:47. [Google Scholar]
- [18b].White JD, Blakemore PR, Green NJ, Hauser EB, Holoboski MA, Keown LE, Nylund Kolz CS, Phillips BW. J. Org. Chem. 2002;67:7750. doi: 10.1021/jo020537q. [DOI] [PubMed] [Google Scholar]
- [19].Crossman S, Perkins MV. J. Org. Chem. 2006;71:117. doi: 10.1021/jo051753c. [DOI] [PubMed] [Google Scholar]
- [20a].Mukaiyama T, Banno K, Narasaka K. J. Am. Chem. Soc. 1974;96:7503. [Google Scholar]
- [20b].Kan SBJ, Ng KK-H, Paterson I. Angew. Chem. 2013;125:9267. [Google Scholar]; Angew. Chem. Int. Ed. 2013;52 [Google Scholar]
- [21].Intermediate 20. also featured in the synthesis by the Fürstner group (Ref. [7]), although it was prepared by an entirely different route. They converted this compound into a vinyl boronate by olefin metathesis and then employed a Suzuki cross-coupling to construct the C17–C18 bond.
- [22].Evans DA, Dart MJ, Duffy JL, Young MG. J. Am. Chem. Soc. 1996;118:4322. [Google Scholar]
- [23].See the Supporting Information for full details
- [24].Jeffery T. J. Chem. Soc. Chem. Commun. 1991:324. [Google Scholar]
- [25].Stork G, Zhao K. Tetrahedron Lett. 1989;30:2173. [Google Scholar]
- [26a].Fürstner A, Funel JA, Tremblay M, Bouchez LC, Nevado C, Waser M, Ackerstaff J, Stimson CC. Chem. Commun. 2008:2873. doi: 10.1039/b805299a. [DOI] [PubMed] [Google Scholar]
- [26b].Stille JK, Groh BL. J. Am. Chem. Soc. 1987;109:813. [Google Scholar]
- [27].Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989. [Google Scholar]
- [28a].Kočovský P. Tetrahedron Lett. 1986;27:5521. [Google Scholar]
- [28b].Paterson I, Florence GJ, Gerlach K, Scott JP. Angew. Chem. 2000;112:385. doi: 10.1002/(sici)1521-3773(20000117)39:2<377::aid-anie377>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2000;39 [Google Scholar]
- [29].This reaction was also accompanied by formation of the biscarbamate. In contrast, silylation of 2424. Raspoet G, Nguyen MT, McGarraghy M, Hegarty AF. J. Org. Chem. 1998;63:6878. doi: 10.1021/jo9806411. with chlorotriethylsilane occurred exclusively at the sterically more accessible C9 allylic alcohol. We attribute the contrasting carbamoylation results with the 1,3-diol to a more complex cyclic mechanism; see: [DOI] [PubMed] [Google Scholar]
- [30].For leading references to syntheses of other anticancer macrolides by our research group, see:
- [30a].Dalby SM, Goodwin-Tindall J, Paterson I. Angew. Chem. 2013;125:6645. doi: 10.1002/anie.201301978. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013;52 [Google Scholar]
- [30b].Paterson I, Fink SJ, Lee LYW, Atkinson SJ, Blakey SB. Org. Lett. 2013;15:3118. doi: 10.1021/ol401327r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30c].Paterson I, Maltas P, Dalby SM, Lim JH, Anderson EA. Angew. Chem. 2012;124:2803. doi: 10.1002/anie.201108594. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51 [Google Scholar]
- [30d].Paterson I, Ashton K, Britton R, Cecere G, Chouraqui G, Florence GJ, Knust H, Stafford J. Chem. Asian J. 2008;3:367. doi: 10.1002/asia.200700357. [DOI] [PubMed] [Google Scholar]
- [30e].Paterson I, Britton R, Delgado O, Gardner NM, Meyer A, Naylor GJ, Poullennec KG. Tetrahedron. 2010;66:6534. [Google Scholar]
- [30f].Paterson I, Paquet T. Org. Lett. 2010;12:2158. doi: 10.1021/ol100693c. [DOI] [PubMed] [Google Scholar]
- [31].Detailed biological studies will be reported elsewhere
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.