

Abstract
AIM: To study host gene expression and number of immune cells in liver tissues from patients with fulminant hepatitis E (FH-E).
METHODS: Microarray-based expression profiling was done using Illumina Human WG-6_v3_BeadChip arrays on post-mortem liver tissue from 5 patients with FH-E, and compared with similar tissue from 6 patients with fulminant hepatitis B (FH-B; disease controls) and normal liver tissue from 6 persons. Differential expression was defined as ≥ 2.0-fold change with Benjamini-Hochberg false discovery rate below 0.05 using t-test in liver tissue from FH-B and FH-E, than healthy liver tissue. For some genes that showed differential expression in FH-E, microarray data were validated using quantitative reverse transcription PCR. Differentially expressed gene lists were then subjected to “Gene Ontology” analysis for biological processes, and pathway analysis using BioCarta database on the DAVID server. In addition, tissue sections were stained for CD4+, CD8+ and CD56+ cells using indirect immunohistochemistry; cells staining positive for each of these markers were counted and compared between groups.
RESULTS: Compared to normal livers, those from patients with FH-E and FH-B showed differential expression of 3377 entities (up-regulated 1703, downregulated 1674) and 2572 entities (up 1164, down 1408), respectively. This included 2142 (up 896, down 1246) entities that were common between the two sets; most of these belonged to metabolic, hemostatic and complement pathways, which are active in normal livers. Gene expression data from livers of patients with FH-E but not those of FH-B showed activation of several immune response pathways, particularly those involving cytotoxic T cells. The fold-change values of mRNA for selected genes in livers from FH-E than in normal liver tissue determined using quantitative reverse transcription PCR showed excellent concordance with microarray analysis. At immunohistochemistry, CD8+ T cells showed an increase in liver biopsies from both FH-E [median 53.4 per arbitrary unit area (range 31.2-99.9)] and FH-B [median 49.3 (19.3-51.0); P = 0.005] compared to control liver tissue [median 6.9 (3.1-14.9)].
CONCLUSION: FH-E patients show CD8+ T cell infiltration and increased gene expression of cytotoxic T cell pathways in liver, suggesting a possible pathogenetic role for these cells.
Keywords: Cytotoxic T cells, Gene expression, Hepatitis E, Hepatitis E virus, Immune response, Liver biopsy, Microarray, Natural killer cells, Pathogenesis
Core tip: Data on pathogenesis of hepatitis E virus (HEV) infection, which is a common cause of acute hepatitis in several developing countries, are quite limited. This manuscript reports our data on microarray-based gene expression analysis and immunohistochemistry in liver tissue from patients with HEV infection, as compared to liver tissue from patients with hepatitis B virus infection and normal liver tissue. These data advance the current knowledge about the pathogenesis of HEV infection.
INTRODUCTION
Hepatitis E virus (HEV), a member of genus Hepevirus in family Hepeviridae, consists of 32-34 nm diameter, icosahedral, non-enveloped virions[1]. It has a 7.2-kb single-stranded, positive-sense RNA genome with three open reading frames (ORFs) that code for a viral non-structural polyprotein (ORF1), the major capsid protein (ORF2) and a phosphoprotein with possible regulatory functions (ORF3)[1]. The virus has at least four genotypes, named 1 to 4[2]; however, these all belong to a single serotype.
HEV infection is common in developing countries of Asia and Africa, where it causes an acute disease, known as hepatitis E, either as water-borne outbreaks or as sporadic cases[3,4], caused by infection through the fecal-oral route. The disease occurs predominantly among young adults and resembles acute hepatitis caused by other hepatotropic viruses. It is usually self-limiting, with overall case fatality rate below 0.5%[3]. However, some patients, in particular pregnant women, develop severe liver injury that progresses to fulminant hepatic failure (FH), which is often fatal. In these areas, the virus belongs most often to genotype 1 and sometimes to genotype 2[3,4].
In areas with lower disease endemicity, such as Europe, North America, Japan, etc., occasional cases with locally-acquired HEV infection have been reported[5]. These are mainly elderly men, often with other co-existing diseases, who are believed to acquire infection with genotype 3 or 4 HEV through ingestion of undercooked meat of HEV-infected animals[6]. In immunosuppressed persons, the infection may become persistent[6].
To develop therapeutic measures against hepatitis E, it is important to understand the pathogenesis of liver injury in this disease. In infection with other hepatotropic viruses, such as the hepatitis A, B and C viruses, tissue injury is mediated not by the infectious agent but is related to the host immune response[7-11]. We have shown that HEV infection is associated with activation of specific cellular immune responses in the peripheral blood[12-16]. However, little data are available on the liver tissue from hepatitis E. Therefore, the mechanism of liver injury in this disease remains unclear.
We therefore studied gene expression profile in liver biopsies from patients with FH due to acute hepatitis E (FH-E), in comparison with those from healthy adults and patients with FH due to acute hepatitis B (FH-B; a disease control group).
MATERIALS AND METHODS
Patients and specimens
Post-mortem needle liver biopsies were obtained from patients dying of FH-E (n = 5) or of FH-B (n = 6). The biopsies were obtained within 30 min after death, and separate pieces were collected in RNALater (Qiagen) and stored at -80 °C for gene expression analysis, and in formalin for histology and immunohistochemistry. In addition, a blood specimen was collected from each subject, for biochemical tests and serological markers of viral hepatitis.
Patients dying more than 2 wk after the onset of disease, and those with major sepsis or clinical evidence of pre-existing liver disease were excluded. Diagnosis of acute hepatitis E was based on detection of IgM anti-HEV (Genelabs, Singapore) in the absence of hepatitis B surface antigen (HBsAg), IgM anti-HBc antibody, anti-hepatitis C virus (HCV) antibody and IgM anti-hepatitis A virus (HAV) antibody (all from BioMerieux, Marcy l'Etoile, France), and that of acute hepatitis B on detection of HBsAg and IgM anti-HBc, in the absence of IgM anti-HEV, anti-HCV and IgM anti-HAV.
In addition, normal liver tissue was obtained from six persons undergoing partial hepatic resection for focal diseases, such as hydatid liver disease, gallbladder cancer without biliary obstruction.
The study was reviewed and approved by Sanjay Gandhi Postgraduate Institute of Medical Sciences institutional review board. Written, informed consent was obtained from all patients or their families, as appropriate.
RNA isolation
Total RNA was isolated from tissue biopsies using RNeasy Protect minikit (Qiagen, Carlsbad, CA). RNA concentration was measured using NanoDrop 1000 spectrophotometer (Nanodrop, Wilmington, DE). RNA integrity was assessed using Bioanalyzer 2100 (Agilent, Santa Clara, CA). Specimens with A260/A280 and A260/A230 ratios > 1.9, and RIN > 8.0 were processed further.
Microarray analysis
Gene expression profiling was done using Illumina Human WG-6_v3_BeadChip arrays (Illumina, San Diego, CA), each containing more than 46000 entities (gene probes or probesets) derived from NCBI, RefSeq and UniGene databases. Poly(A)-RNA was reverse transcribed to complementary DNA (cDNA), using an oligo(dT)-primer that contained a phage T7 RNA polymerase promoter sequence at its 5′-end, followed by conversion to double-stranded cDNA. The double-stranded cDNA was transcribed in vitro to yield large quantities of biotin-labelled anti-sense RNA, which was then hybridised to the bead arrays at 55 °C for 16-18 h, and scanned using an Illumina iScan reader.
Bioinformatic analysis
The array intensity data were initially analysed using Illumina Genome Studio Gene Expression Module (v1.1.1) (Illumina, Cambridge, United Kingdom) for visualisation and normalisation. After quantile normalisation and background correction using medians within the BeadStudio software, the data were exported for further analysis in GeneSpring GX software 11.5 (Agilent).
Differential expression between various subject groups was analyzed using Illumina Custom Algorithm. The normalized data were first subjected to quality check using principal component analysis. This was followed by analysis to determine genes showing differential expression (fold-change ≥ 2.0 and Benjamini-Hochberg false discovery rate < 0.05 using t-test) in liver tissue from patients with FH-B and FH-E, as compared to healthy liver tissue. Such gene lists were then subjected to “Gene Ontology” (GO) analysis for biological processes, and pathway analysis using BioCarta database on the DAVID server (http://david.abcc.ncifcrf.gov). Separate analyses were done for genes that were differentially expressed in both FH-E and FH-B, and those differentially expressed in only one of these.
Validation of microarray results
For some genes that showed differential expression in FH-E, microarray data were validated using quantitative reverse transcription PCR (qRT-PCR). In brief, cDNA was prepared using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) from 2 μg of RNA. This was followed by real-time PCR in 20-μL reactions comprising of 50 ng cDNA, primers (Appendix 1) and SYBR Green (Applied Biosystems).
The validation assay included liver tissue for all the subjects used for microarray analysis (a biopsy piece other than that used for microarray analysis) and four additional healthy liver tissues. All assays included RNA from peripheral blood mononuclear cells of a healthy person as a calibrator. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and 18S rRNA was used as housekeeping genes. Relative fold-change was determined for each specimen and gene from cycle threshold (Ct) values as follows, and compared to fold-change in expression in microarray data:
∆∆Ct= [(∆Ctgene of interest - ∆CtGAPDH)patient - [(∆Ctgene of interest - ∆CtGAPDH)calibrator sample] and Fold change = 2-∆∆Ct.
Immunohistochemistry on liver biopsies
Formalin-fixed, paraffin-embedded liver sections were stained with hematoxylin and eosin, and examined to confirm the presence of acute inflammation in patients with FH, and absence of disease in controls.
In addition, immunohistochemistry (IHC) was performed for specific T-cell subsets (CD4+ and CD8+) and NK cells (CD56+). In brief, formalin-fixed, paraffin-embedded 3-μm liver sections on silanized glass slides (Dako) were fixed at 60 °C overnight. The slides were deparaffinised in xylene and rehydrated in graded alcohol. Antigen retrieval was done in 10 mmol/L EDTA (pH 9.0) at 98 °C for 30 min. After washing with distilled water and Tris-buffered saline (TBS; pH 7.4), endogenous peroxidase was blocked using 3% hydrogen peroxide in methanol for 30 min in dark. Sections were then incubated with monoclonal primary antibodies [prediluted mouse anti-human CD8 (clone C8/144B), CD4 (clone 4B12) or CD56 (clone 123C3) (Dako, Denmark)] for 1 h at room temperature, followed by peroxidase enzyme-labelled secondary antibody (Dako) for 30 min. Diaminobenzidine was used as chromogen to detect the bound antibodies. Slides were counterstained with Mayer’s hematoxylin for 1 min, cleared and mounted with DPX (Sigma-Aldrich). Stained cells were counted in five fields, averaged and expressed as number in an area measuring 73000 μm2.
Statistical analysis
Inter-group comparisons were done using t-test with Benjamini-Hochberg correction in case of gene expression data and Mann-Whitney U test for other quantitative data. Relationship of fold-change in the microarray and real-time PCR data was assessed using Pearson’s correlation coefficient. The statistical methods of this study were reviewed by Dr. Rakesh Aggarwal from Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India.
RESULTS
Demographic features and biochemical findings in the three subject groups are shown in Table 1. Alanine aminotransferase levels were higher in FH-B than in FH-E.
Table 1.
Characteristics of study subjects in the three groups
| Characteristic | Healthy controls (n = 6) | Fulminant hepatitis B (n = 6) | Fulminant hepatitis E (n = 5) |
| Age (yr) | 52 (30-58) | 24 (16-60) | 28 (18-32) |
| Gender (Male:Female) | 4:2 | 4:2 | 1:4 |
| Duration of illness before death (d) | - | 9 (6-14) | 10 (7-13) |
| Maximum total serum bilirubin (mg/dL) | 0.7 (0.4-1.4) | 21 (13.1-41.4) | 25 (5.4-32.0) |
| Serum ALTa (IU/L) | 30 (21-58) | 1939 (524-5795) | 770 (240-1302) |
| Serum AST (IU/L) | 28 (18-35) | 859 (250-2345) | 458 (332-1000) |
| Alkaline phosphatase (IU/L) | - | 257 (131-427) | 306 (164-561) |
| Serum albumin (g/dL) | - | 2.8 (2.0-3.3) | 2.8 (2.1-3.2) |
Data are shown as median (range), except for gender distribution; patients with fulminant hepatitis B and fulminant hepatitis E were comparable in all variables except serum ALT levels (aP < 0.05, Mann-Whitney U test). ALT: Alanine aminotransferase.
Principal component analysis
On principal component analysis of gene expression data (Figure 1), all specimens except one (with FH-B) showed clustering with other specimens in the same group.
Figure 1.
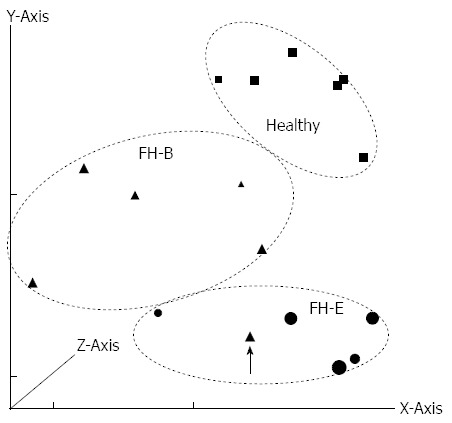
Principal component analysis of gene expression data from various specimens included in the study. The components along X, Y and Z axes were 62.54%, 21.17%, and 8.88%, respectively. Patients with fulminant hepatitis E (FH-E) are represented using circles, those with fulminant hepatitis B (FH-B) using triangles and healthy controls using square symbols. The size of markers varies according to their placement on the Z axis. Patients with FH-E and FH-B clustered separately, except for one patient with FH-B who was an outlier (arrow).
Microarray gene expression
A total of 3377 entities, each representing a discrete gene, showed differential expression of ≥ 2-fold with P-value of < 0.05 in liver tissue from patients with FH-E than that from healthy persons (Figure 2); this included 1703 entities with over-expression and 1674 showing reduced expression in FH-E. In liver tissue from FH-B, 2572 entities showed differential expression than in healthy livers (up-regulation 1164, down-regulation 1408).
Figure 2.
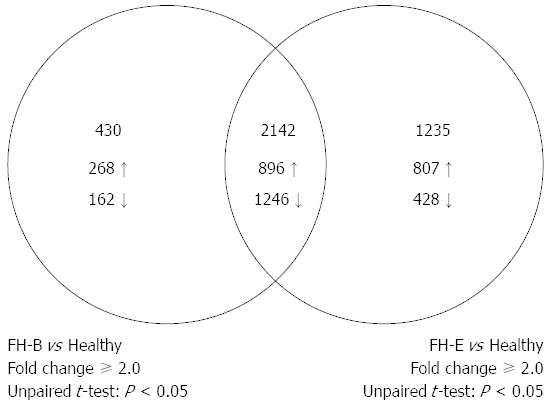
Venn-diagram showing comparison of number of entities differentially expressed in liver tissue from fulminant hepatitis B or E, as compared to healthy liver tissue. The intersection of two circles shows entities that were differentially expressed in both fulminant hepatitis E and fulminant hepatitis B.
Of the entities differentially expressed in FH-E or FH-B compared to normal liver, 2142 were common (up-regulation in both 896, down-regulation in both 1246) (Figure 2, Appendix 2); none of these entities showed discordance in the direction of differential expression between FH-E and FH-B. In contrast, 1235 entities (Appendix 3) were differentially expressed in FH-E but not in FH-B, and 430 entities (Appendix 4) were differentially expressed in FH-B but not in FH-E.
Gene ontology analysis
Genes differentially expressed in FH-E and FH-B livers: Table 2 lists the biological processes that showed differential regulation on GO analysis of the 2142 entities differentially expressed in both FH-E and FH-B. These included several metabolic processes in which the liver plays an active part, such as general body metabolism, or those whose proteins are mainly synthesized in the liver, such as coagulation pathways and complement system. Further, both the disease conditions showed differential regulation of innate immune responses and humoral immune responses mediated by B cells.
Table 2.
Gene Ontology analysis (biological processes) for gene entities differentially expressed in liver tissue from fulminant hepatitis E as compared to normal liver tissue, as well as in fulminant hepatitis B as compared to normal liver tissue
| Gene ontology accession | Gene Ontology term | P value |
| GO:0055114 | Oxidation-reduction process | 2.68E-40 |
| GO:0006082 | Organic acid metabolic process | 4.75E-39 |
| GO:0043436 | Oxoacid metabolic process | 2.17E-38 |
| GO:0019752 | Carboxylic acid metabolic process | 4.09E-38 |
| GO:0006629 | Lipid metabolic process | 3.94E-28 |
| GO:0044281 | Small molecule metabolic process | 9.06E-27 |
| GO:0032787 | Monocarboxylic acid metabolic process | 3.56E-22 |
| GO:0016054 | Organic acid catabolic process | 4.22E-19 |
| GO:0046395 | Carboxylic acid catabolic process | 4.22E-19 |
| GO:0044255 | Cellular lipid metabolic process | 1.35E-17 |
| GO:0044282 | Small molecule catabolic process | 5.16E-17 |
| GO:0044712 | Single-organism catabolic process | 5.16E-17 |
| GO:0006631 | Fatty acid metabolic process | 2.71E-16 |
| GO:0006520 | Cellular amino acid metabolic process | 4.43E-16 |
| GO:0044711 | Single-organism biosynthetic process | 8.25E-14 |
| GO:1901605 | Alpha-amino acid metabolic process | 1.07E-13 |
| GO:0044283 | Small molecule biosynthetic process | 1.60E-13 |
| GO:1901564 | Organonitrogen compound metabolic process | 2.46E-11 |
| GO:0009063 | Cellular amino acid catabolic process | 4.46E-11 |
| GO:0008610 | Lipid biosynthetic process | 8.46E-11 |
| GO:0072376 | Protein activation cascade | 3.87E-10 |
| GO:0008202 | Steroid metabolic process | 4.10E-10 |
| GO:0016053 | Organic acid biosynthetic process | 6.26E-10 |
| GO:0046394 | Carboxylic acid biosynthetic process | 6.26E-10 |
| GO:1901566 | Organonitrogen compound biosynthetic process | 1.94E-08 |
| GO:0008152 | Metabolic process | 4.34E-08 |
| GO:1901606 | Alpha-amino acid catabolic process | 4.58E-08 |
| GO:0072329 | Monocarboxylic acid catabolic process | 1.40E-07 |
| GO:0007596 | Blood coagulation | 1.98E-07 |
| GO:0050817 | Coagulation | 1.98E-07 |
| GO:0007599 | Hemostasis | 2.83E-07 |
| GO:1901615 | Organic hydroxy compound metabolic process | 4.14E-07 |
| GO:0042060 | Wound healing | 4.86E-07 |
| GO:0006956 | Complement activation | 6.77E-07 |
| GO:0009062 | Fatty acid catabolic process | 1.71E-06 |
| GO:0006066 | Alcohol metabolic process | 1.82E-06 |
| GO:0008652 | Cellular amino acid biosynthetic process | 2.42E-06 |
| GO:0050878 | Regulation of body fluid levels | 5.46E-06 |
| GO:0051186 | Cofactor metabolic process | 5.50E-06 |
| GO:0006694 | Steroid biosynthetic process | 8.32E-06 |
| GO:0044242 | Cellular lipid catabolic process | 1.09E-05 |
| GO:0005996 | Monosaccharide metabolic process | 0.000012 |
| GO:0051346 | Negative regulation of hydrolase activity | 1.28E-05 |
| GO:0005975 | Carbohydrate metabolic process | 1.35E-05 |
| GO:0010876 | Lipid localization | 5.30E-05 |
| GO:0044710 | Single-organism metabolic process | 5.82E-05 |
| GO:0006732 | Coenzyme metabolic process | 6.57E-05 |
| GO:0010951 | Negative regulation of endopeptidase activity | 7.22E-05 |
| GO:0019318 | Hexose metabolic process | 7.95E-05 |
| GO:0006869 | Lipid transport | 8.02E-05 |
| GO:0065008 | Regulation of biological quality | 8.96E-05 |
| GO:0010466 | Negative regulation of peptidase activity | 0.0001 |
| GO:0006006 | Glucose metabolic process | 0.0001 |
| GO:0042558 | Pteridine-containing compound metabolic process | 0.0002 |
| GO:0043648 | Dicarboxylic acid metabolic process | 0.0002 |
| GO:0006958 | Complement activation, classical pathway | 0.0004 |
| GO:0006091 | Generation of precursor metabolites and energy | 0.0004 |
| GO:0044262 | Cellular carbohydrate metabolic process | 0.0004 |
| GO:0006575 | Cellular modified amino acid metabolic process | 0.0005 |
| GO:0016051 | Carbohydrate biosynthetic process | 0.0005 |
| GO:0072330 | Monocarboxylic acid biosynthetic process | 0.0006 |
| GO:00024551 | Humoral immune response mediated by circulating immunoglobulin | 0.0010 |
| GO:0044723 | Single-organism carbohydrate metabolic process | 0.0010 |
| GO:0019320 | Hexose catabolic process | 0.0011 |
| GO:0016052 | Carbohydrate catabolic process | 0.0014 |
| GO:0044724 | Single-organism carbohydrate catabolic process | 0.0014 |
| GO:0006820 | Anion transport | 0.0016 |
| GO:0006007 | Glucose catabolic process | 0.0017 |
| GO:0046365 | Monosaccharide catabolic process | 0.0018 |
| GO:0046835 | Carbohydrate phosphorylation | 0.0020 |
| GO:1901617 | Organic hydroxy compound biosynthetic process | 0.0020 |
| GO:00022531 | Activation of immune response | 0.0021 |
| GO:0016125 | Sterol metabolic process | 0.0022 |
| GO:0006096 | Glycolysis | 0.0023 |
| GO:0019395 | Fatty acid oxidation | 0.0037 |
| GO:0034440 | Lipid oxidation | 0.0037 |
| GO:0046165 | Alcohol biosynthetic process | 0.0038 |
| GO:00069591 | Humoral immune response | 0.0042 |
| GO:0009611 | Response to wounding | 0.0044 |
| GO:0006957 | Complement activation, alternative pathway | 0.0044 |
| GO:0006760 | Folic acid-containing compound metabolic process | 0.0044 |
| GO:00450871 | Innate immune response | 0.0053 |
| GO:0052548 | Regulation of endopeptidase activity | 0.0077 |
| GO:0046364 | Monosaccharide biosynthetic process | 0.0077 |
| GO:0006790 | Sulfur compound metabolic process | 0.0078 |
| GO:0051179 | Localization | 0.0079 |
| GO:0052547 | Regulation of peptidase activity | 0.0083 |
| GO:0046942 | Carboxylic acid transport | 0.0083 |
| GO:0015849 | Organic acid transport | 0.0095 |
| GO:0016042 | Lipid catabolic process | 0.0113 |
| GO:0015711 | Organic anion transport | 0.0113 |
| GO:0051234 | Establishment of localization | 0.0113 |
| GO:0006810 | Transport | 0.0119 |
| GO:0044092 | Negative regulation of molecular function | 0.0126 |
| GO:0019319 | Hexose biosynthetic process | 0.0131 |
| GO:0072378 | Blood coagulation, fibrin clot formation | 0.0132 |
| GO:0006558 | L-phenylalanine metabolic process | 0.0146 |
| GO:0006559 | L-phenylalanine catabolic process | 0.0146 |
| GO:0000038 | Very long-chain fatty acid metabolic process | 0.0146 |
| GO:0051289 | Protein homotetramerization | 0.0146 |
| GO:00022521 | Immune effector process | 0.0153 |
| GO:0008203 | Cholesterol metabolic process | 0.0153 |
| GO:0009074 | Aromatic amino acid family catabolic process | 0.0212 |
| GO:0048806 | Genitalia development | 0.0212 |
| GO:00160641 | Immunoglobulin mediated immune response | 0.0214 |
| GO:0050819 | Negative regulation of coagulation | 0.0214 |
| GO:0043086 | Negative regulation of catalytic activity | 0.0224 |
| GO:0034754 | Cellular hormone metabolic process | 0.0243 |
| GO:00197241 | B cell mediated immunity | 0.0263 |
| GO:0051262 | Protein tetramerization | 0.0279 |
| GO:00507781 | Positive regulation of immune response | 0.0292 |
| GO:00507761 | Regulation of immune response | 0.0341 |
| GO:0006633 | Fatty acid biosynthetic process | 0.0372 |
| GO:0003333 | Amino acid transmembrane transport | 0.0389 |
| GO:0016126 | Sterol biosynthetic process | 0.0416 |
| GO:0006811 | Ion transport | 0.0420 |
| GO:0006094 | Gluconeogenesis | 0.0474 |
| GO:0001676 | Long-chain fatty acid metabolic process | 0.0474 |
These pathways are related to immune responses.
Genes differentially expressed in FH-E but not in FH-B: On GO analysis, the 1235 entities that showed differential expression in FH-E but not in FH-B were related mainly to the immune system pathways (Table 3); for each of these immune pathways, several genes showed upregulation.
Table 3.
Results of Gene Ontology analysis (biological processes) for gene entities differentially expressed in liver tissue from fulminant hepatitis E as compared to normal liver, but not in that from fulminant hepatitis B as compared to normal liver
| Gene Ontology term | P value |
| Immune system process1 | 1.67E-11 |
| Immune response1 | 4.81E-09 |
| Regulation of immune system process1 | 5.06E-08 |
| Positive regulation of immune system process1 | 2.72E-06 |
| Defense response | 1.04E-05 |
| Response to wounding | 1.90E-05 |
| Signaling | 7.78E-05 |
| Adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains1 | 2.40E-04 |
| Adaptive immune response1 | 2.40E-04 |
| Response to stimulus | 3.53E-04 |
| Regulation of cell activation | 4.65E-04 |
| Inflammatory response1 | 6.89E-04 |
| Cell adhesion | 0.0010 |
| Biological adhesion | 0.0010 |
| Positive regulation of immune response1 | 0.0015 |
| Cell activation | 0.0018 |
| Signal transduction | 0.0019 |
| Regulation of leukocyte activation1 | 0.0020 |
| Activation of immune response1 | 0.0021 |
| Oxidation reduction | 0.0021 |
| B cell mediated immunity1 | 0.0023 |
| Leukocyte mediated immunity1 | 0.0026 |
| Positive regulation of response to stimulus | 0.0027 |
| Regulation of locomotion | 0.0028 |
| Regulation of immune response1 | 0.0030 |
| Phospholipid efflux | 0.0032 |
| Lymphocyte mediated immunity1 | 0.0032 |
| Regulation of cell migration1 | 0.0048 |
| Regulation of cell motility1 | 0.0048 |
| Response to stress | 0.0050 |
| Regulation of lymphocyte activation1 | 0.0056 |
| Regulation of cellular component movement | 0.0064 |
| Signal transmission | 0.0067 |
| Immunoglobulin mediated immune response1 | 0.0067 |
| Signaling process | 0.0067 |
| Small molecule metabolic process | 0.0083 |
| Cholesterol efflux | 0.0124 |
| Immune effector process1 | 0.0154 |
| Negative regulation of apoptosis | 0.0161 |
| Positive regulation of cell activation | 0.0176 |
| Positive regulation of leukocyte activation1 | 0.0176 |
| Regulation of response to stimulus | 0.0177 |
| Negative regulation of cell death | 0.0179 |
| Negative regulation of programmed cell death | 0.0179 |
| Regulation of lipid metabolic process | 0.0180 |
| Signaling pathway | 0.0206 |
| Acute inflammatory response1 | 0.0241 |
| Phospholipid transport | 0.0257 |
| Regulation of fatty acid metabolic process | 0.0273 |
| Alcohol metabolic process | 0.0282 |
| Cholesterol transport | 0.0310 |
| Sterol transport | 0.0472 |
These pathways are related to immune responses.
Genes differentially expressed in FH-B but not in FH-E: GO analysis of these entities did not reveal significant changes in any specific pathway.
Pathway analysis
Genes differentially expressed in both FH-E and FH-B: Pathway analysis for the 2142 entities differentially expressed in both FH-E and FH-B using the BioCarta database showed downregulation of several pathways related to metabolic processes, hemostasis, and complement system (Table 4). Appendix 5 shows individual genes involved in each of these pathways.
Table 4.
Pathways whose genes were found to be over-represented among entities differentially expressed in both fulminant hepatitis E and fulminant hepatitis B, and in fulminant hepatitis E but not in fulminant hepatitis B (derived using BioCarta through DAVID resource)
| BioCarta pathway | Gene count | P value |
| In both FH-B vs normal, and FH-E vs normal | ||
| Complement pathway | 10 | 0.0002 |
| Intrinsic prothrombin activation pathway | 10 | 0.0002 |
| Nuclear receptors in lipid metabolism and toxicity | 13 | 0.0022 |
| Alternative complement pathway | 6 | 0.0069 |
| Extrinsic prothrombin activation pathway | 6 | 0.0113 |
| Lectin induced complement pathway | 6 | 0.0172 |
| Classical complement pathway | 6 | 0.0172 |
| Acute myocardial infarction | 6 | 0.0248 |
| Vitamin C in the brain | 4 | 0.0385 |
| CBL mediated ligand-induced downregulation of epidermal growth factor receptors | 5 | 0.0543 |
| Catabolic pathways for methionine, isoleucine, threonine and valine | 3 | 0.0955 |
| In FH-E vs normal but not in FH-B vs normal | ||
| T cytotoxic cell surface molecules | 7 | 0.0005 |
| T helper cell surface molecules | 6 | 0.0043 |
| Cytotoxic T lymphocyte mediated immune response against target cells | 7 | 0.0022 |
| Co-stimulatory signal during T-cell activation | 7 | 0.0085 |
| Role of epidermal growth factor receptor transactivation by G protein-coupled receptors in cardiac hypertrophy | 6 | 0.0092 |
| T cell receptor signaling pathway | 10 | 0.0049 |
| Lck and Fyn tyrosine kinases in initiation of T cell receptor activation | 5 | 0.0187 |
| Interleukin-7 signal transduction | 5 | 0.0446 |
| Mitochondrial carnitinepalmitoyltransferase system | 3 | 0.0529 |
| T cell receptor and CD3 complex | 3 | 0.0529 |
| Interleukin-17 signaling pathway | 5 | 0.0562 |
Genes differentially expressed in FH-E but not in FH-B: BioCarta pathways whose genes were significantly over-represented among genes differentially expressed in liver tissue from FH-E compared to normal liver but not in FH-B were mostly related to cellular immune mechanisms (Table 4), e.g., T cytotoxic cell surface molecules, CTL mediated immune response against target cells, T helper cell surface molecules, co-stimulatory signal during T-cell activation, T cell receptor signalling pathway, Lck and Fyn tyrosine kinases in initiation of TCR activation, IL-7 signal transduction, T cell receptor and CD3 complex, IL 17 signalling pathway. Several individual genes involved in these pathways were upregulated in patients with FH-E (Appendix 6).
Genes differentially expressed in FH-B but not in FH-E: Pathway analysis of these 430 entities did not reveal significant change in any pathway.
Validation using qRT-PCR
The fold-change values of mRNA for selected genes (Appendix 1) in livers from FH-E than in normal liver tissue determined using qRT-PCR showed excellent concordance with those at microarray (Figure 3), using either of the two housekeeping genes.
Figure 3.
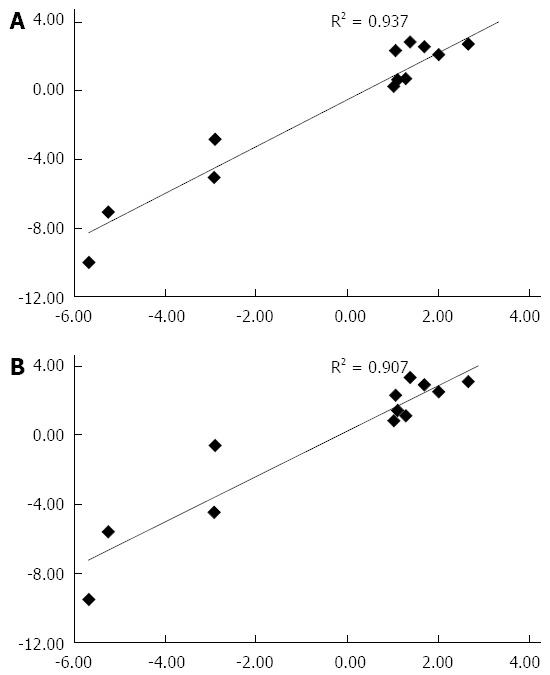
Relationship of fold-change in RNA expression of selected genes in patients with fulminant hepatitis E and fulminant hepatitis B on real-time polymerase chain reaction with fold-change in RNA expression of the same genes on microarray analysis. A and B show the data when reverse transcription polymerase chain reaction results were normalized using the GAPDH gene and 18S rRNA gene as housekeeping gene, respectively.
Immunohistochemistry
IHC showed a few CD4+ positive T-cells and occasional NK cells in all the biopsies. In FH-B as well as FH-E, CD8+ T-cells were found in both the liver parenchyma and the portal areas (Figure 4). The median number of CD8+ T cells was greater in liver tissues from both FH-E [median 53.4 (range 31.2-99.9)]; P = 0.005, 2-sided Mann-Whitney U test) and FH-B [49.3 (19.3-51.0); P = 0.005] than in controls 6.9 (3.1-14.9); however, there was no significant difference between FH-E and FH-B.
Figure 4.
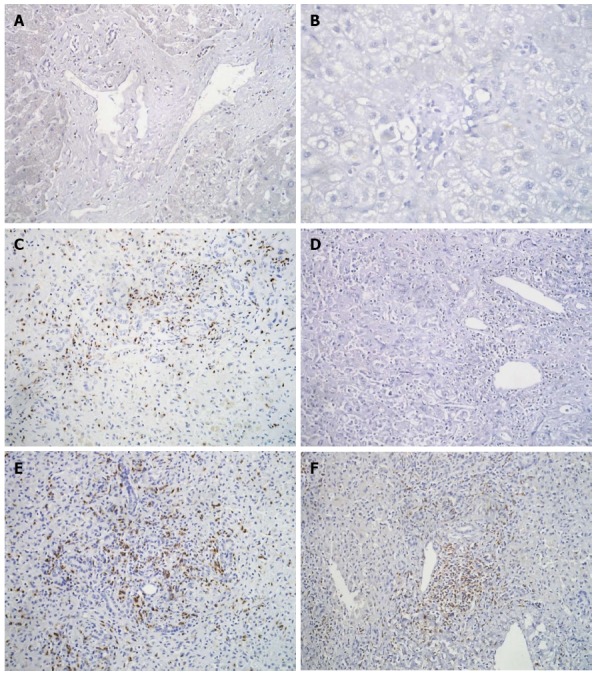
Immunohistochemistry for CD8+ cells in liver biopsies shows occasional cytotoxic T-cells in portal tracts of controls (A), and moderate infiltration in HBV (C) and HEV (E); immunostaining for CD4 shows absence of helper T-cells in portal areas of control (B), an occasional cell in HBV (D) and some small aggregates of helper T-cells in HEV (F). A and B: Diaminobenzidine × 400; C-F: Diaminobenzidine × 200. HBV: Hepatitis B virus; HEV: Hepatitis E virus.
DISCUSSION
Host tissue injury during a viral infection may be caused either by virus-induced cell death, or by immune-mediated killing of infected cells. Understanding the underlying mechanism may not only help understand the pathogenesis of a particular viral disease but also provide a lead to development of strategies for countering the host injury. In infection with hepatitis A, B or C virus, liver injury is mediated primarily by the host immune response[7-11]. However, the mechanisms of liver injury in HEV infection remain unclear.
Initial attempts at delineation of mechanism of liver injury caused by HEV were based on observations during experimental HEV infection in non-human primates. In these studies, liver injury appeared after viremia and fecal viral excretion had started declining and coincided with the appearance of anti-HEV antibodies[17-19]. These findings suggested that host immune response was responsible for the hepatocyte killing. This was followed by attempts to determine whether HEV was capable of producing a cytopathic effect in in vitro models. However, these efforts were largely unsuccessful due to failure of this virus to grow well in cell culture. Recent successful culture[20], though in low titers, of some genotypes 3 and 4 HEV isolates has revealed absence of cytotoxicity[21,22], providing some support to the hypothesis that liver injury in HEV infection is unlikely to be virus-mediated.
Human data on cellular immune responses during HEV infection are limited to studies on peripheral blood mononuclear cells (PBMCs). In these studies, we and others have shown proliferation of HEV-specific CD4+ and CD8+ cells, and increased production of various cytokines by PBMCs during acute and convalescent phases of hepatitis E[14,16]. In a study that looked at patients with different disease severities, HEV-specific T-cell responses in PBMCs were weaker in patients with FH-E than in those with uncomplicated acute hepatitis E[12]. However, observations made on circulating immune cells may not faithfully represent those in the immune cells infiltrating the liver, because of preferential localization of certain cells, including virus-specific T cell subsets, in the inflamed tissue[23,24]. Thus, there is a need to study changes in the liver tissue from patients with hepatitis E.
In the current study, we found changes in expression of genes involved in a variety of immune responses, including the innate, humoral and cellular immune pathways in liver tissue from patients with FH-E. Of these, changes in innate and humoral immune responses were also observed in FH-B, indicating that HEV infection may share some of the pathways causing liver cell injury with HBV infection. In contrast, activation of cellular immune pathways was found only in FH-E, and not in FH-B, indicating a specific role for cellular immune response in inducing liver injury during HEV infection.
In particular, we found several T-cell surface molecules such as CD2, CD3 (γ, δ and ε chains) and CD8-alpha to be up-regulated in liver tissue from patients with FH-E. Furthermore, several T-cell signalling molecules, such as Fyn, phospholipase C gamma-1, NFκB1, protein kinase C and ras-related C3 botulinum toxin substrate (rac) were also overexpressed in these livers. These findings may indicate a role for activated CD8+ T cells in the causation of liver injury in HEV infection. An over-expression of CTLA4, which is expressed only on activated T cells, also supports this hypothesis. An alternative explanation for the overexpression of CD2 in liver tissue could be the presence of this marker on the NK cells. Importantly, the increased expression of CD2 in the liver tissue was also associated with over-expression of perforin, the main cytolytic protein contained in the CD8+ T cells and NK cells. The above findings, taken together, indicate that immune-mediated cytotoxicity of CD8+ or NK cells against virus-infected cells may play a role in producing liver damage in HEV infection.
Some other findings in our study may also point to alterations in CD8+ cells. In viral infections, the number of effector CD8 T cells contracts over time with the formation of a population of protective memory cells, which is maintained by IL-7 and IL-15. Our finding of activation of IL-7-mediated signalling in patients with hepatitis E possibly reflects this phenomenon. In addition, the genes for signal transducer and activator of transcription 5 (STAT5), a key molecule involved in the survival of effector and memory CD8 cells[25], and for BCL2, a key survival molecule that is upregulated by STAT5, were overexpressed in FH-E.
The differential expression in livers from patients with FH-E of several genes which were unaffected in FH-B could have been because of changes in any of the several cell types present in the liver, including hepatocytes, Kupffer cells, cholangiocytes, endothelial cells, stellate cells and a variety of immune cells. However, the demonstration of predominant infiltration with CD8+ T cells in the FH-E livers in the current study as well as in two previous reports[26,27], and the fact that the genes that showed differential expression are not expressed much in other cell types, indicate the CD8+ T cells were the most likely source. It may be pertinent to note that since the immune cells constitute only a minority of all the cells in the liver, the absolute gene expression changes within the intrahepatic immune cells must be more marked than is indicated by the overall gene expression data. It may thus be interesting in future to undertake studies on such immune cells after recovering these using techniques such as laser dissection microscopy.
It may be pertinent to compare our results with those from a recent study of gene expression in serial liver tissues from chimpanzees with experimental HEV infection[28]. In this study, differential expression was limited to a few genes that belonged predominantly to the innate immune response pathways, and was weaker than that observed in chimpanzees with HCV infection. Further, the number of upregulated genes peaked sooner after the onset of viremia in HEV infection than with HCV infection. We did find altered expression of some genes involved in innate immune responses; however, these genes were different from those showing significant changes during HEV infection in the chimpanzee study. In this context, it is important to note that comparison of our data with those from the experimentally-infected chimpanzees may not be valid. Our patients had severe liver disease, whereas experimental HEV infection in primates is milder[19,29]. Further, time-kinetics of different types of immune responses vary, with innate immune responses being prominent during the initial phase after viral infection; thus, in human HEV infection, such responses may occur before an infected person becomes symptomatic and hence not picked up. Also, the experimental animals had been inoculated by the intravenous route, whereas humans acquire infection through the oral route.
Though we studied patients with FH-B primarily as disease controls, it may be interesting to compare liver in this condition with healthy livers. Livers from patients with FH-B showed infiltration with CD8+ cells, but no differential expression of genes belonging to cellular immune response pathways. Previous gene expression data in this disease are available from only two Italian patients[30,31]. These patients too showed prominent CD8+ T cell infiltration with little change in expression of genes associated with T cell activity and a prominent upregulation of B cell response, similar to our observations.
Interestingly, though liver tissues from both FH-E and FH-B in our study showed prominent CD8+ T cell infiltration, only FH-E was associated with a cytotoxic T cell transcriptional signature e.g., increased expression of perforin. This suggests that the infiltrating CD8+ T cells in the two diseases behave quite differently. This may hold the key to pathogenesis of liver injury in HEV infection.
We also found reduced expression of several genes associated with metabolic, hemostatic and complement pathways in liver tissue from FH-E as well as FH-B. Liver is a metabolically active organ which produces several body proteins. Since FH, irrespective of its cause, is characterized by marked destruction of hepatocytes, reduction in the expression of these proteins was thus expected in both FH-E and FH-B, as a consequence of the massive liver injury.
Our data are limited by a small sample size, the use of post-mortem tissue, and of patients with severe liver injury at only one time-point. These limitations are related to the fact that liver biopsy, being invasive, is ethically unacceptable in patients with acute liver disease, forcing us to use tissue collected immediately post-mortem. Though our findings may not be entirely applicable to acute uncomplicated hepatitis E, these should be seen in light of the current absence of any human data on immune events in the liver in acute hepatitis E. Further, we compared our data to those from a control group of post-mortem biopsies from FH-B, which would have been susceptible to similar artefacts as those in FH-E.
In conclusion, liver tissue from our patients with FH-E showed infiltration with CD8+ T cells and overexpression of genes involved in T cell immune responses, especially those related to T cell activation, cytotoxicity and IL-7 signalling. The latter changes were not observed in FH-B, and hence were specific to hepatitis E. These data suggest that the severe liver damage in FH-E is mediated by the host T-cell immune response. Further work on this aspect should help us better understand the pathogenesis of liver injury in hepatitis E.
ACKNOWLEDGMENTS
Microarray data analysis was done at Bio-medical Informatics Centre, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India; this center is supported by the Indian Council of Medical Research (ICMR) and Department of Biotechnology, Government of India during this work. ANS was supported by ICMR during this work. The authors thank Dr. Vivek Chandra for analysis comparing the liver biopsy data with in vitro data from HEV ORF3-expressing cells.
COMMENTS
Background
Infection with hepatitis E virus is the most common cause of acute hepatitis in the world. The mechanism of liver injury in this disease and its pathogenesis are yet fully understood. A better understanding of these may allow attempts at therapeutic intervention in patients with this disease.
Research frontiers
Microarray techniques allow study of expression of several genes in tissues from patients with a particular disease and its comparison with those from healthy controls and persons with similar diseases. These techniques have been applied for the study of pathogenesis of several diseases.
Innovations and breakthroughs
The authors used microarray techniques to determine the expression of various genes in the liver tissues from patients dying of hepatitis E and compared this to that in healthy liver tissue (control tissue) and liver tissues from patients dying of a disease with similar morphologic changes but caused by infection with another virus (hepatitis B; disease controls). The data showed several differences in gene expression between these groups. In particular, livers of patients with fulminant hepatitis E, but not those of hepatitis B, showed activation of several immune response pathways, particularly those involving cytotoxic T cells. This difference was observed even though tissues from both hepatitis B and E showed infiltration with cytotoxic T cells.
Applications
All data suggest that immune cells may play a role in the pathogenesis of hepatitis E, though further work is required in this regard.
Peer-review
Minor improvements, such as addition of statistical methods to the manuscript.
Footnotes
Supported by National Institutes of Health, Bethesda, United States, No. 5R01AI076192.
Institutional review board statement: The study was reviewed and approved by Sanjay Gandhi Postgraduate Institute of Medical Sciences institutional review board. Written, informed consent was obtained from and all patient or their families, as appropriate.
Institutional animal care and use committee statement: Exempt, since this study did not include the use of any animals.
Conflict-of-interest statement: None of the authors listed on the above manuscript had any conflict of interest, either financial or non-financial in relation to this paper.
Data sharing statement: Participants gave informed consent for data sharing. The corresponding author would be willing to share the data as required for the above manuscript with the editors.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: January 30, 2015
First decision: March 10, 2015
Article in press: May 4, 2015
P- Reviewer: Ambrosioni J, Li WG, Ma L S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.Aggarwal R, Jameel S. Hepatitis E. Hepatology. 2011;54:2218–2226. doi: 10.1002/hep.24674. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal R, Naik S. Epidemiology of hepatitis E: current status. J Gastroenterol Hepatol. 2009;24:1484–1493. doi: 10.1111/j.1440-1746.2009.05933.x. [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal R, Shukla R, Jameel S, Agrawal S, Puri P, Gupta VK, Patil AP, Naik S. T-cell epitope mapping of ORF2 and ORF3 proteins of human hepatitis E virus. J Viral Hepat. 2007;14:283–292. doi: 10.1111/j.1365-2893.2006.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agrawal V, Goel A, Rawat A, Naik S, Aggarwal R. Histological and immunohistochemical features in fatal acute fulminant hepatitis E. Indian J Pathol Microbiol. 2012;55:22–27. doi: 10.4103/0377-4929.94849. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad I, Holla RP, Jameel S. Molecular virology of hepatitis E virus. Virus Res. 2011;161:47–58. doi: 10.1016/j.virusres.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Exley MA, Koziel MJ. To be or not to be NKT: natural killer T cells in the liver. Hepatology. 2004;40:1033–1040. doi: 10.1002/hep.20433. [DOI] [PubMed] [Google Scholar]
- 7.Farci P, Diaz G, Chen Z, Govindarajan S, Tice A, Agulto L, Pittaluga S, Boon D, Yu C, Engle RE, et al. B cell gene signature with massive intrahepatic production of antibodies to hepatitis B core antigen in hepatitis B virus-associated acute liver failure. Proc Natl Acad Sci USA. 2010;107:8766–8771. doi: 10.1073/pnas.1003854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 9.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 10.Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, Liu Y, Kaech SM. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci USA. 2010;107:16601–16606. doi: 10.1073/pnas.1003457107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Husain MM, Aggarwal R, Kumar D, Jameel S, Naik S. Effector T cells immune reactivity among patients with acute hepatitis E. J Viral Hepat. 2011;18:e603–e608. doi: 10.1111/j.1365-2893.2011.01489.x. [DOI] [PubMed] [Google Scholar]
- 12.Kamar N, Bendall R, Legrand-Abravanel F, Xia NS, Ijaz S, Izopet J, Dalton HR. Hepatitis E. Lancet. 2012;379:2477–2488. doi: 10.1016/S0140-6736(11)61849-7. [DOI] [PubMed] [Google Scholar]
- 13.Kamar N, Selves J, Mansuy JM, Ouezzani L, Péron JM, Guitard J, Cointault O, Esposito L, Abravanel F, Danjoux M, et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med. 2008;358:811–817. doi: 10.1056/NEJMoa0706992. [DOI] [PubMed] [Google Scholar]
- 14.Klenerman P, Thimme R. T cell responses in hepatitis C: the good, the bad and the unconventional. Gut. 2012;61:1226–1234. doi: 10.1136/gutjnl-2011-300620. [DOI] [PubMed] [Google Scholar]
- 15.Krawczynski K, Bradley DW. Enterically transmitted non-A, non-B hepatitis: identification of virus-associated antigen in experimentally infected cynomolgus macaques. J Infect Dis. 1989;159:1042–1049. doi: 10.1093/infdis/159.6.1042. [DOI] [PubMed] [Google Scholar]
- 16.Krawczynski K, Meng XJ, Rybczynska J. Pathogenetic elements of hepatitis E and animal models of HEV infection. Virus Res. 2011;161:78–83. doi: 10.1016/j.virusres.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Lu L, Li C, Hagedorn CH. Phylogenetic analysis of global hepatitis E virus sequences: genetic diversity, subtypes and zoonosis. Rev Med Virol. 2006;16:5–36. doi: 10.1002/rmv.482. [DOI] [PubMed] [Google Scholar]
- 18.McCaustland KA, Krawczynski K, Ebert JW, Balayan MS, Andjaparidze AG, Spelbring JE, Cook EH, Humphrey C, Yarbough PO, Favorov MO, et al. Hepatitis E virus infection in chimpanzees: a retrospective analysis. Arch Virol. 2000;145:1909–1918. doi: 10.1007/s007050070065. [DOI] [PubMed] [Google Scholar]
- 19.Nissim O, Melis M, Diaz G, Kleiner DE, Tice A, Fantola G, Zamboni F, Mishra L, Farci P. Liver regeneration signature in hepatitis B virus (HBV)-associated acute liver failure identified by gene expression profiling. PLoS One. 2012;7:e49611. doi: 10.1371/journal.pone.0049611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okamoto H. Hepatitis E virus cell culture models. Virus Res. 2011;161:65–77. doi: 10.1016/j.virusres.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Pal R, Aggarwal R, Naik SR, Das V, Das S, Naik S. Immunological alterations in pregnant women with acute hepatitis E. J Gastroenterol Hepatol. 2005;20:1094–1101. doi: 10.1111/j.1440-1746.2005.03875.x. [DOI] [PubMed] [Google Scholar]
- 22.Prabhu SB, Gupta P, Durgapal H, Rath S, Gupta SD, Acharya SK, Panda SK. Study of cellular immune response against Hepatitis E virus (HEV) J Viral Hepat. 2011;18:587–594. doi: 10.1111/j.1365-2893.2010.01338.x. [DOI] [PubMed] [Google Scholar]
- 23.Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 24.Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest. 2009;119:1745–1754. doi: 10.1172/JCI39133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srivastava R, Aggarwal R, Jameel S, Puri P, Gupta VK, Ramesh VS, Bhatia S, Naik S. Cellular immune responses in acute hepatitis E virus infection to the viral open reading frame 2 protein. Viral Immunol. 2007;20:56–65. doi: 10.1089/vim.2006.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srivastava R, Aggarwal R, Sachdeva S, Alam MI, Jameel S, Naik S. Adaptive immune responses during acute uncomplicated and fulminant hepatitis E. J Gastroenterol Hepatol. 2011;26:306–311. doi: 10.1111/j.1440-1746.2010.06356.x. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi M, Hoshino Y, Tanaka T, Takahashi H, Nishizawa T, Okamoto H. Production of monoclonal antibodies against hepatitis E virus capsid protein and evaluation of their neutralizing activity in a cell culture system. Arch Virol. 2008;153:657–666. doi: 10.1007/s00705-008-0045-6. [DOI] [PubMed] [Google Scholar]
- 28.Ticehurst J, Rhodes LL, Krawczynski K, Asher LV, Engler WF, Mensing TL, Caudill JD, Sjogren MH, Hoke CH, LeDuc JW. Infection of owl monkeys (Aotus trivirgatus) and cynomolgus monkeys (Macaca fascicularis) with hepatitis E virus from Mexico. J Infect Dis. 1992;165:835–845. doi: 10.1093/infdis/165.5.835. [DOI] [PubMed] [Google Scholar]
- 29.Yu C, Boon D, McDonald SL, Myers TG, Tomioka K, Nguyen H, Engle RE, Govindarajan S, Emerson SU, Purcell RH. Pathogenesis of hepatitis E virus and hepatitis C virus in chimpanzees: similarities and differences. J Virol. 2010;84:11264–11278. doi: 10.1128/JVI.01205-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang HY, Chen DS, Wu YQ, He QG, Chen HC, Liu ZF. Both swine and human cells are capable to support the replication of swine hepatitis E virus type 4 in vitro. Virus Res. 2011;158:289–293. doi: 10.1016/j.virusres.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y, Callendret B, Xu D, Brasky KM, Feng Z, Hensley LL, Guedj J, Perelson AS, Lemon SM, Lanford RE, et al. Dominance of the CD4(+) T helper cell response during acute resolving hepatitis A virus infection. J Exp Med. 2012;209:1481–1492. doi: 10.1084/jem.20111906. [DOI] [PMC free article] [PubMed] [Google Scholar]