

Abstract
Purpose
Albinism is a heterogeneous genetic disorder of melanin synthesis that results in hypopigmented eyes (in patients with ocular albinism) or hair, skin, and eyes (in individuals with oculocutaneous albinism). It is associated with decreased visual acuity, nystagmus, strabismus, and photophobia. The tyrosinase gene is known to be involved in both oculocutaneous albinism and autosomal recessive ocular albinism. In this study, we aimed to screen the mutations in the TYR gene in the nonsyndromic OCA and autosomal recessive ocular albinism patients from Iran.
Methods
The tyrosinase gene was examined in 23 unrelated patients with autosomal recessive ocular albinism or nonsyndromic OCA using DNA sequencing and bioinformatics analysis.
Results
TYR gene mutations were identified in 14 (app. 60%) albinism patients.
Conclusions
We found 10 mutations, 3 of which were novel. No mutation was found in our ocular albinism patients, but one of them was heterozygous for the p.R402Q polymorphism.
Introduction
Albinism is a heterogeneous group of genetic disorders that affect 1 in 20,000 individuals worldwide, although the prevalence of the different subtypes of albinism varies considerably among the different ethnic backgrounds. It is caused by deficiencies in pigmentation, and clinically is divided into ocular and oculocutaneous albinism [1-3].
Oculocutaneous albinism (OCA) is a heterogeneous and autosomal recessive disorder that involves a lack of pigment in the skin, hair, and eyes, and is accompanied by optic defects such as photophobia, strabismus, poor vision, and nystagmus [2,4,5]. It is identified as nonsyndromic OCA (if the mutations occur in the TYR, OCA2, TYRP1, MATP (SLC45A2), SLC24A5, or C10ORF11 genes) or syndromic OCA (if the mutations occur in the HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, PLDN, LYST, MYO5A, RAB27A, or MLPH genes) [1,6]. OCA-1 is the most common subtype found in Caucasians, and accounts for about 50% of cases worldwide. It results from mutations in the TYR gene [7].
TYR null mutations producing inactive or incomplete polypeptides result in OCA1A (tyrosinase-negative OCA), in which melanin formation never occurs throughout the patient’s life. In contrast, mutations producing a partially active or hypomorphic tyrosinase enzymes result in the OCA1B (yellow mutant OCA), in which the patients completely lack detectable pigment at birth but rapidly develop yellow hair pigment in the first few years of life and then continue to slowly accumulate pigment in the hair, eyes, and skin with time [3,8].
Ocular albinism (OA) has similar eye findings to those of OCA but does not affect the hair and skin [7]. Both X-linked recessive (OA1) and autosomal recessive forms of ocular albinism exist [9]. Previous studies have shown that autosomal recessive ocular albinism in some cases constitutes a clinically mild phenotype of OCA, due to mutations in either the TYR or OCA2 (P) genes [10].
Clinical diagnosis of albinism type is difficult, due to the observed range of phenotypic variation. Thus, genetic analysis may be helpful with respect to a more accurate diagnosis and genetic counseling [2,11]. In this study, we screened mutations in the TYR gene in nonsyndromic OCA and autosomal recessive ocular albinism patients from Iran.
Methods
Twenty three Iranian albinism patients, including 2 autosomal recessive ocular albinism and 21 oculocutaneous albininsm patients from unrelated families, were recruited for this study. Features such as hypopigmentation of the hair, skin, and iris; nystagmus; strabismus; poor vision; photophobia; and foveal hypoplasia were observed in our patients. The affected individuals were examined by ophthalmologists, and syndromic forms of OCA were excluded. Informed written consent was obtained from all patients and their parents for carrying out research on their specimens. The study was approved by the local ethics committee. Genomic DNA was extracted from peripheral whole blood of the patients and their parents using the salting out method.
The primers were designed for all five exons and intron-exon boundaries of the TYR gene (NG_008748.1) using gene runner software and also the primer blast website (NCBI) to prevent amplifying the TYRL pseudogene, which is similar to the 3′ half of this gene (Table1). These primers were used for PCR amplification and cycle sequencing.
Table 1. Primers designed for amplifying the TYR exons and intron-exon boundaries.
| Primer name | Sequence | Amplicon size | Annealing temperature (°C) |
|---|---|---|---|
| TYR-1F |
TTAACTGGGTTTGCTTAGGTC |
1230 bp |
57 |
| TYR-1R |
TATACCCTGCCTGAAGAAGTG |
||
| TYR-2F |
CTCCTCAGGAGAAGTCTAAC |
429 bp |
58 |
| TYR-2R |
AACTCAGAAAATTCTGAATTC |
||
| TYR-3F |
ACACACTGGGTATCCAGAATG |
430 bp |
57 |
| TYR-3R |
ACAATAGACTACCATAACTTCTTAGC |
||
| TYR-4F |
TCAAGGCCTGAAAGAATAAACTA |
570 bp |
60 |
| TYR-4R |
GCCTATGTTAAGCAAAATGACC |
||
| TYR-5F |
TGTCTACTCCAAAGGACTGT |
918 bp | 58 |
| TYR-5R | ACTTAGCTGGATGTGTTATAGA |
The PCR condition was: 1x PCR buffer, 2 mM MgCl2, 0.2 mM of each dNTPs, 0.25 µM of each primers, 0.25 unit/25 µl CinnaGen Taq DNA polymerase, and 50–100 ng template DNA in 25 µl final volume. PCR was performed as: 95 °C for 5 min (pre-denaturation), 94 °C for 40 s, annealing temperature (according to Table 1) for 40 s, 72 °C for 40 s for 26 cycles, and 72 °C for 10 min (final extension). Cycle sequencing was performed by Macrogen (Seoul, Korea). New missense mutations were analyzed using SIFT, PolyPhen, I Mutant 2, and Mutation Taster tools, and were interpreted based on American College of Medical Genetics standards and guidelines [12].
Results
TYR sequences were studied in 23 albinism (2 autosomal recessive ocular albinism and 21 nonsyndromic OCA) patients. Mutations were found in 14 OCA individuals; no mutations were found in OA patients. The heterozygosity of the patients parents for the identified mutations was confirmed. Eleven patients were homozygous for TYR mutations, two were compound heterozygous, and one had only one detected mutation (Table 2).
Table 2. TYR mutations and polymorphisms (p.S192Y and p.R402Q) in 23 OCA / OA Iranian patients.
| #P | Clinical diagnosis | Female/Male | Proband |
Type of OCA | Consanguinity | |||
|---|---|---|---|---|---|---|---|---|
| Mutation 1 | Mutation 2 | Polymorphisms |
||||||
| p.S192Y | p.R402Q | |||||||
| 1 |
OCA |
F |
c.286dupA |
c.286dupA |
- |
- |
OCA1A |
+ |
| 2 |
OCA |
F |
p.P406L (Maternal) |
- |
- |
- |
ND |
+ |
| 3 |
OA |
F |
- |
- |
- |
Hetero (Maternal) |
ND |
+ |
| 4 |
OCA |
F |
- |
- |
Hetero (Maternal) |
Hetero (Paternal) |
ND |
+ |
| 5 |
OCA |
M |
p.M332I |
p.M332I |
- |
- |
OCA1B |
+ |
| 6 |
OCA |
M |
- |
- |
Hetero |
- |
ND |
+ |
| 7 |
OCA |
F |
p.M332I |
p.M332I |
- |
- |
OCA1B |
+ |
| 8 |
OCA |
F |
- |
- |
- |
Homo |
ND |
+ |
| 9 |
OCA |
M |
p.R239W (Paternal) |
p.M332I (Maternal) |
- |
- |
ND |
ND |
| 10 |
OCA |
F |
- |
- |
- |
- |
ND |
+ |
| 11 |
OCA |
M |
c.286dupA |
c.286dupA |
- |
- |
OCA1A |
+ |
| 12 |
OCA |
F |
- |
- |
- |
- |
ND |
+ |
| 13 |
OCA |
F |
c.286dupA |
c.286dupA |
- |
- |
OCA1A |
+ |
| 14 |
OCA |
M |
p.R77Q |
p.R77Q |
- |
Homo |
OCA1A |
+ |
| 15 |
OCA |
F |
p.G47S (Paternal) |
c.del1276–82 (Maternal) |
Hetero |
Hetero (Paternal) |
OCA1A |
+ |
| 16 |
OCA |
F |
p.P21S |
p.P21S |
- |
- |
OCA1A |
+ |
| 17 |
OCA |
M |
p.R77Q |
p.R77Q |
- |
Homo |
OCA1A |
+ |
| 18 |
OCA |
M |
p.G419R |
p.G419R |
- |
- |
OCA1A |
+ |
| 19 |
OCA |
F |
p.P301L |
p.P301L |
- |
- |
OCA1A |
+ |
| 20 |
OCA |
M |
- |
- |
Hetero |
- |
ND |
ND |
| 21 |
OCA |
F |
c.286dupA |
c.286dupA |
- |
- |
OCA1A |
+ |
| 22 |
OCA |
F |
- |
- |
Homo |
- |
ND |
+ |
| 23 | OA | F | - | - | - | - | ND | + |
ND: not determined. The type of OCA1 is delineated based on the hair color with increasing age (white in OCA1A and yellow/blonde in OCA1B).
One of the patients (patient 15) was heterozygous for two novel mutations that included one missense (p.G47S) and one frameshift (c.del1276–82) mutation, as well as two polymorphisms of p.S192Y and p.R402Q. One patient was homozygous for another novel mutation (p.P301L).
The p.R402Q and p.S192Y polymorphisms were detected in six and five cases, respectively.
Discussion
In this study, we performed mutation analysis of the TYR gene on 23 unrelated albinism patients, including 2 autosomal recessive ocular albinism and 21 nonsyndromic OCA patients. We found 10 different mutations in 14 out of 23 (app. 60%) albinism patients. No mutations were found in OA patients, although one of them was heterozygous for the p.R402Q polymorphism.
Eight missense (p.M332I, p.R77Q, p.P21S, p.G419R, p.P301L, p.P406L, p.R239W, and p.G47S) and two frameshift (c.286dupA and c.del1276–82) mutations were found in this study, and no mutation was found in exons 3 and 5. The frequency percentage of the detected mutations and polymorphisms (p.S192Y and p.R402Q) are shown in Table 3.
Table 3. The frequency of the 10 mutations and the p.R402Q and p.S192Y polymorphisms in our 23 albinism patients.
| Nucleotide change | Amino acid change | Exon No. | Status (Number of the patients) | Frequency percentage |
|---|---|---|---|---|
| c.286dupA |
Frameshift |
Ex 1 |
Homo (4) |
17.39 |
| c.61C>T |
p.P21S |
Ex 1 |
Homo (1) |
4.34 |
| c.139G>A |
p.G47S |
Ex 1 |
Hetero (1) |
2.17 |
| c.230G>A |
p.R77Q |
Ex 1 |
Homo (2) |
8.69 |
| c.715C>T |
p.R239W |
Ex 1 |
Hetero (1) |
2.17 |
| c.902C>T |
p.P301L |
Ex 2 |
Homo (1) |
4.34 |
| c.0996G>A |
p.M332I |
Ex 2 |
Homo (2), Hetero (1) |
10.86 |
| c.1217C>T |
p.P406L |
Ex 4 |
Hetero (1) |
2.17 |
| c.1255G>A |
p.G419R |
Ex 4 |
Homo (1) |
4.34 |
| c.del1276–82 |
Frameshift |
Ex 4 |
Hetero (1) |
2.17 |
| c.575C>A |
p.S192Y |
Ex 1 |
Homo (1), Hetero (4) |
13.04 |
| c.1205G>A | p.R402Q | Ex 4 | Homo (3), Hetero (3) | 19.56 |
Patient 15 had two nonreported mutations (p.G47S and c.del1276–82), as well as both p.R402Q and p.S192Y polymorphisms, all in heterozygous forms. To our knowledge, the p.G47S (c.139G>A) has not been previously reported, although pathogenic mutations have been previously reported for codon 47 (p.G47D and p.G47V) [11,13-15]. Tools such as SIFT, Polyphen-2, I Mutant 2, and Mutation Tasters predict p.G47S as a disease-causing mutation (Table 4). According to the ACMG standards and guidelines for the interpretation of sequence variants, this novel mutation is likely pathogenic. Our second novel mutation, c.del1276–82, is a frameshifting mutation and causes a termination signal in the 57th codon. According to the ACMG standards and guidelines, this frameshift mutation is interpreted to be pathogenic.
Table 4. Results of the analysis of the novel missense mutations with the bioinformatics tools.
| Novel missense mutations | Polyphen-2 |
SIFT |
I Mutant 2 |
Mutation Taster | ||
|---|---|---|---|---|---|---|
| Prediction | Score | Prediction | Score | Prediction (sign of DDG) | ||
| p.G47S |
Probably damaging |
1.000 |
damaging |
0 |
Decrease stability |
disease causing |
| p.P301L | Probably damaging | 1.000 | damaging | 0 | Decrease stability | disease causing |
Patient 19 was homozygous for another novel mutation (p.P301L). No mutation has been reported in this codon before. Mentioned bioinformatics tools predicted p.P301L to be a disease-causing mutation (Table 4). According to the ACMG standards and guidelines, this novel mutation is likely pathogenic.
In this study, nine patients did not show any causative mutations in the TYR gene. This may be due to the involvement of other OCA genes, variants in the promoter or other regulatory elements that were not covered in this study, or deletions/duplications of the complete gene or single exons that are not detectable by the cycle sequencing procedure.
The contribution of the p.R402Q variant to the albinism phenotype has been heavily disputed in the literature [7,16]. According to our study, it doesn’t seem to be pathogenic. In Figure 1, Figure 2, and Figure 3, the pedigrees of our three OCA patients (patients 4, 9, and 15) are in support of this phenomenon. Nonpathogenicity of p.S192Y can also be inferred from these pedigrees. Since the parents of patient 4 are both homozygous for p.R402Q and p.S192Y, respectively, and do not show the albinism phenotype, none of these variants could be considered pathogenic in the homozygous state (Figure 1). Also, p.S192Y is not pathogenic in heterozygous form in combination with other mutations such as p.R239W or p.M332I in the parents of patient 9 (Figure 2). Moreover, according to the pedigree of patient 15, both p.S192Y and p.R402Q, together with p.G47S, are not pathogenic (Figure 3).
Figure 1.
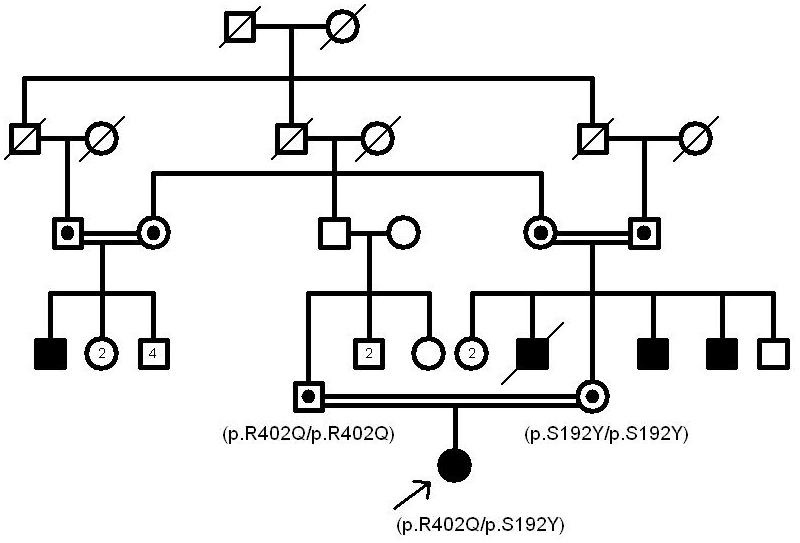
The pedigree of patient 4. The nonpathogenic nature of p.R402Q and p.S192Y can be inferred from the above pedigree in which the patient’s parents do not show any albinism features.
Figure 2.
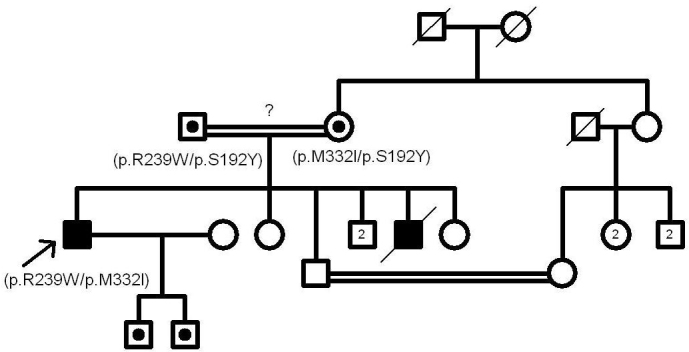
The pedigree of patient 9. p.S192Y is not pathogenic in heterozygous form in combination with p.R239W or p.M332I in the patient’s parents.
Figure 3.
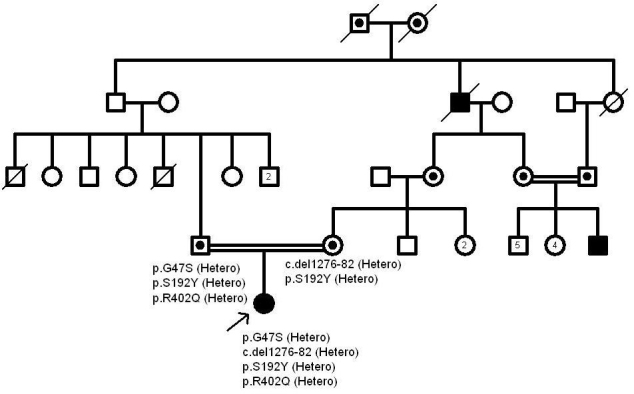
The pedigree of patient 15. Both p.S192Y and p.R402Q together with the p.G47S are not pathogenic in the patient’s father according to the above pedigree.
In Table 2, the patients with identified mutations in the TYR gene are classified in two types of OCA1A and OCA1B based on their hair color with increasing age. Patients with permanent white hair are diagnosed to have OCA1A, and patients with white hair at birth and yellow/blonde coloration later in their life are classified as OCA1B. In patient 2, the hair color was white at birth and yellow afterwards, but because we did not find her second mutation and digenic OCA is reported [4], we couldn’t determine her type of OCA.
Here we found homozygous TYR mutations in 11 patients whose parents had consanguineous marriage (Table 2). This suggests that the disease-causing mutations in these patients are identical by descent. As reported previously, the overall rate of consanguineous marriage is about 38.6% in Iran, and this can increase the probability of incidence of genetic disorders due to common ancestors [17,18].
Acknowledgments
The authors thank the albinism families for their collaboration. The personnel of Tehran Medical Genetics Laboratory are acknowledged for their help and support. This project was financially supported by Tehran Medical Genetics Laboratory, grant number 93,003.
References
- 1.Kamaraj B, Purohit R. Mutational analysis of oculocutaneous albinism: a compact review. Biomed Res Int. 2014;2014:905472. doi: 10.1155/2014/905472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Passmore LA, Kaesmann-Kellner B, Weber BH. Novel and recurrent mutations in the tyrosinase gene and the P gene in the German albino population. Hum Genet. 1999;105:200–10. doi: 10.1007/s004390051090. [DOI] [PubMed] [Google Scholar]
- 3.Gargiulo A, Testa F, Rossi S, Di Iorio V, Fecarotta S, de Berardinis T, Iovine A, Magli A, Signorini S, Fazzi E, Galantuomo MS, Fossarello M, Montefusco S, Ciccodicola A, Neri A, Macaluso C, Simonelli F, Surace EM. Molecular and clinical characterization of albinism in a large cohort of Italian patients. Invest Ophthalmol Vis Sci. 2011;52:1281–9. doi: 10.1167/iovs.10-6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei AH, Yang XM, Lian S, Li W. Genetic analyses of Chinese patients with digenic oculocutaneous albinism. Chin Med J (Engl) 2013;126:226–30. [PubMed] [Google Scholar]
- 5.Wilk MA, McAllister JT, Cooper RF, Dubis AM, Patitucci TN, Summerfelt P, Anderson JL, Stepien KE, Costakos DM, Connor TB, Jr, Wirostko WJ, Chiang PW, Dubra A, Curcio CA, Brilliant MH, Summers CG, Carroll J. Relationship between foveal cone specialization and pit morphology in albinism. Invest Ophthalmol Vis Sci. 2014;55:4186–98. doi: 10.1167/iovs.13-13217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Visser M, Kayser M, Grosveld F, Palstra RJ. Genetic variation in regulatory DNA elements: the case of OCA2 transcriptional regulation. Pigment Cell Melanoma Res. 2014;27:169–77. doi: 10.1111/pcmr.12210. [DOI] [PubMed] [Google Scholar]
- 7.Simeonov DR, Wang X, Wang C, Sergeev Y, Dolinska M, Bower M, Fischer R, Winer D, Dubrovsky G, Balog JZ, Huizing M, Hart R, Zein WM, Gahl WA, Brooks BP, Adams DR. DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Hum Mutat. 2013;34:827–35. doi: 10.1002/humu.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomita Y, Miyamura Y. Oculocutaneous albinism and analysis of tyrosinase gene in Japanese patients. Nagoya J Med Sci. 1998;61:97–102. [PubMed] [Google Scholar]
- 9.Fukai K, Holmes SA, Lucchese NJ, Siu VM, Weleber RG, Schnur RE, Spritz RA. Autosomal recessive ocular albinism associated with a functionally significant tyrosinase gene polymorphism. Nat Genet. 1995;9:92–5. doi: 10.1038/ng0195-92. [DOI] [PubMed] [Google Scholar]
- 10.Hutton SM, Spritz RA. A comprehensive genetic study of autosomal recessive ocular albinism in Caucasian patients. Invest Ophthalmol Vis Sci. 2008;49:868–72. doi: 10.1167/iovs.07-0791. [DOI] [PubMed] [Google Scholar]
- 11.Hutton SM, Spritz RA. Comprehensive analysis of oculocutaneous albinism among non-Hispanic caucasians shows that OCA1 is the most prevalent OCA type. J Invest Dermatol. 2008;128:2442–50. doi: 10.1038/jid.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oetting WS, Witkop CJ, Jr, Brown SA, Colomer R, Fryer JP, Bloom KE, King RA. A frequent tyrosinase gene mutation associated with type I-A (tyrosinase-negative) oculocutaneous albinism in Puerto Rico. Am J Hum Genet. 1993;52:17–23. [PMC free article] [PubMed] [Google Scholar]
- 14.Oetting WS, Handoko HY, Mentink MM, Paller AS, White JG, King RA. Molecular analysis of an extended family with type IA (tyrosinase-negative) oculocutaneous albinism. J Invest Dermatol. 1991;97:15–9. doi: 10.1111/1523-1747.ep12477808. [DOI] [PubMed] [Google Scholar]
- 15.Opitz S, Kasmann-Kellner B, Kaufmann M, Schwinger E, Zuhlke C. Detection of 53 novel DNA variations within the tyrosinase gene and accumulation of mutations in 17 patients with albinism. Hum Mutat. 2004;23:630–1. doi: 10.1002/humu.9248. [DOI] [PubMed] [Google Scholar]
- 16.Oetting WS, Pietsch J, Brott MJ, Savage S, Fryer JP, Summers CG, King RA. The R402Q tyrosinase variant does not cause autosomal recessive ocular albinism. Am J Med Genet A. 2009;149A:466–9. doi: 10.1002/ajmg.a.32654. [DOI] [PubMed] [Google Scholar]
- 17.Saadat M, Ansari-Lari M, Farhud DD. Consanguineous marriage in Iran. Ann Hum Biol. 2004;31:263–9. doi: 10.1080/03014460310001652211. [DOI] [PubMed] [Google Scholar]
- 18.Akrami SM, Montazeri V, Shomali SR, Heshmat R, Larijani B. Is there a significant trend in prevalence of consanguineous marriage in Tehran? A review of three generations. J Genet Couns. 2009;18:82–6. doi: 10.1007/s10897-008-9191-y. [DOI] [PubMed] [Google Scholar]