

Abstract
Objective
Pathology in both cortex and deep gray matter contribute to disability in multiple sclerosis (MS). We used the increased signal-to-noise ratio of 7-tesla (7T) MRI to visualize small lesions within the thalamus and to relate this to clinical information and cortical lesions.
Methods
7T MRI scans were obtained on 34 MS cases and 15 healthy volunteers. Thalamic lesion number and volume were related to demographic data, clinical disability measures, and lesions in cortical gray matter.
Results
Thalamic lesions were found in 24/34 of MS cases. Two lesion subtypes were noted: discrete, ovoid lesions, and more diffuse lesional areas lining the periventricular surface. The number of thalamic lesions was greater in progressive MS compared to relapsing remitting (mean ± SD, 10.7 ± 0.7 vs. 3.0 ± 0.7, respectively, p < 0.001). Thalamic lesion burden (count and volume) correlated with EDSS score and measures of cortical lesion burden, but not with white matter lesion burden or white matter volume.
Conclusions
7T MRI allows identification of thalamic lesions in MS, which are associated with disability, progressive disease, and cortical lesions. Thalamic lesion analysis may be a simpler, more rapid estimate of overall gray matter lesion burden in MS.
Introduction
Demyelination and neurodegeneration in gray matter (GM) are critical aspects of multiple sclerosis (MS) pathology.1 Autopsy studies have demonstrated demyelination and axonal loss in the cerebral cortex,2, 3 spinal cord,4 hippocampus,5 and deep GM structures.6 Pathologic alterations of GM structures have been found in early and late disease, with greater changes noted in those with a progressive clinical phenotype.7 GM pathology is clearly clinically relevant, as it is associated with cognitive and physical disability.8, 9
Similar to what is seen in white matter (WM), GM pathology can manifest as subtle changes to normal appearing GM in addition to distinct lesions. In recent years, it has become possible to quantify GM pathology with advances in magnetic resonance imaging (MRI) protocols. Techniques such as double inversion recovery, phase sensitive inversion recovery, and ultrahigh-field MRI have been used to characterize the extent of cortical GM lesions in MS.10–12 However, neuroimaging studies of deep GM structures, such as the thalamus, have concentrated on non-lesion measures of pathology, such as alterations in the concentrations of metabolites, atrophy, and iron deposition.13–16 To date, in vivo characterization of the extent and clinical impact of deep GM lesions, particularly those in the thalamus, has been limited.
The objective of this study was to take advantage of the improved signal-to-noise ratio (SNR) and resultant spatial resolution of 7-tesla (7T) MRI to identify and characterize thalamic MS lesions. The extent to which thalamic lesions are a marker for cortical lesions and their relationship with disability was also explored.
Methods
Standard protocol approvals and patient consents
Protocols were approved by the Institutional Review Boards at the Johns Hopkins University School of Medicine and the Kennedy Krieger Institute. Written, informed consent was obtained from all participants.
Participants
MS participants were recruited from the Johns Hopkins MS Center. Individuals with diagnoses of relapsing remitting (RRMS), secondary progressive (SPMS), and primary progressive (PPMS) MS were enrolled. Participants were excluded if they had experienced an MS relapse in the prior 30 days or if they were experiencing symptoms of a major depressive episode. A cohort of age-matched healthy volunteers was also recruited.
MRI protocol and image analysis
MRI was performed with a 7T Philips Achieva scanner with a volume transmit/32-channel receive head coil (Novamedical). Dielectric padding was used to improve image homogeneity. Whole brain, 3D, T1-weighted MPRAGE (magnetization prepared rapid acquisition of gradient echoes) images were acquired with 0.5mm isotropic resolution (repetition time 5.2ms, delay time 4500ms, echo time 2.3ms, flip angle 7 degrees, parallel imaging factor 2.5 (AP) x 2 (RL), 13 minutes, 12 seconds). Whole brain, 3D, T2-weighted MPFLAIR (magnetization prepared fluid attenuated inversion recovery) images were acquired with 1.0mm isotropic resolution (repetition time 8107ms, inversion time 2175ms, echo time 293ms, flip angle 90 degrees, TSE factor 115, parallel imaging factor 2 (AP) x 3 (RL), 8 minutes, 14 seconds).
Images were transferred to an offline workstation and processed with the MIPAV software package (version 5.3, http://mipav.cit.nih.gov). Using MIPAV’s built-in algorithms, the MPRAGE images were smoothed with an anisotropic diffusion filter and the MPFLAIR was rigidly registered to the MPRAGE. Linked MPRAGE and MPFLAIR image slices were viewed at four times magnification and lesions were manually demarcated by a neurologist (DH) who was blinded to subject identity and diagnostic category. Thalamic lesions were identified as hyperintense on MPFLAIR and hypointense on MPRAGE. Cortical lesions were required to be hypointense on MPRAGE; the MPFLAIR was used for visual guidance, but hyperintensity was not required as 7T MPFLAIR images typically have artifacts along the cortical ribbon. Cortical lesions were required to be a minimum of 15% hypointense relative to adjacent normal appearing cortex, at least 1mm wide in at least one image plane, and distinctly different from cortical blood vessels.
Thalamic lesion subtypes were defined in accord with prior pathological analysis.6 Two lesion subtypes were noted (Figure 1): periventricular lesions, which were diffuse, confluent areas of signal change lining the periventricular surface of the thalamus; and ovoid lesions, which were smaller, discrete, ovoid areas of signal change. The cortical lesion subtypes were also defined in accord with pathology.3 Three lesion subtypes were noted (Figure 2): Type 1 (leukocortical) – lesion borders traversing both white and GM, Type 2 (intracortical) – lesions located exclusively in GM, and Type 3 (subpial) – widespread areas of demyelination extending inward from the pial surface, usually located in deep sulci.
Figure 1.
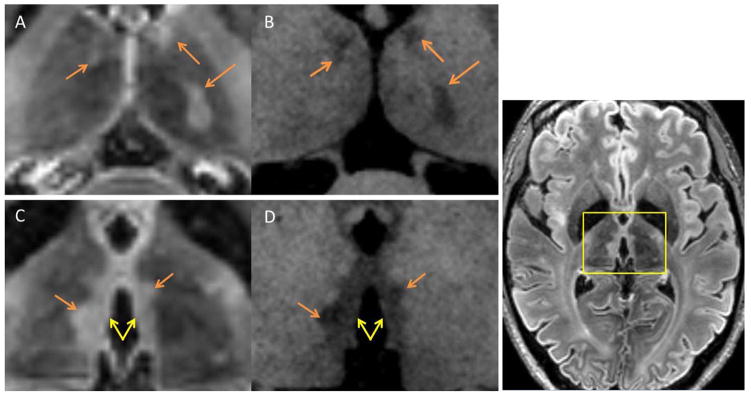
Thalamic Lesion Subtypes
Examples of thalamic lesions from axial MPRAGE and MPFLAIR are seen above. The yellow box shown in MPFLAIR image to the far right shows the approximate localization of panels A–D. Two subtypes of lesions were noted on inspection of 7T MRI images. Panels A (MPFLAIR) and B (MPRAGE) show examples of ovoid lesions, which were discrete ovoid-shaped lesions (orange arrows). Panels C (MPFLAIR) and D (MPRAGE) show examples of periventricular lesions, which were widespread areas of abnormality lining the periventricular surface of the thalamus (yellow double arrow). Note: ovoid lesions are also present in panels C and D, highlighted in yellow.
Figure 2.

Cortical Lesion Subtypes
Examples of cortical lesion subtypes are seen above (yellow arrows). Saggital MPRAGE images are seen in the top row, and saggital MPFLAIR images are seen in the bottom row.
In order to obtain brain structure volumes and WM lesion volumes, the Lesion-TOADS segmentation algorithm17 was modified for use with 7T images. Co-registered MPRAGE and MPFLAIR images underwent N4 inhomogeneity correction, skull and dura stripping, and Lesion-TOADS segmentation. The segmented images were then reviewed for segmentation errors, with manual correction of the WM lesion masks. WM lesion masks were then used for in painting of MPRAGE and MPFLAIR images and the TOADS algorithm was repeated. The subsequent thalamus masks were then manually corrected for segmentation errors and all corrected masks were used to create a final segmentation mask. The intracranial volume (ICV) was calculated from the skull-stripped brain mask. Raw volumes were normalized to ICV and brain parenchymal fraction (BPF) was calculated as a sum of brain structure volumes divided by ICV.
Disability Measures
Neurologic examinations were performed to determine the Expanded Disability Status Scale (EDSS) score. The MS severity score (MSSS) was also calculated based on disease duration and the EDSS score. The timed 25-foot walk, 9-hole peg test, and Paced Auditory Serial Addition Test were administered to determine the MS Functional Composite (MSFC) score. Z-scores for individual test results and the MSFC total score were calculated according to the recommendations of the National MS Society Clinical Outcomes Assessment Task Force, using the Task Force dataset for normalization.18, 19
Statistical analysis
Statistical analysis was performed in Stata 10.1 IC (StataCorp, College Station, Texas). Group differences were tested with Student’s t-test for continuous variables and Wilcoxon Rank-Sum test for discrete variables. Group differences found in univariate analysis were re-tested in multivariate analysis in a logistic regression model for prediction of group assignment by the variable of interest, adjusted for potential confounds, such as age, disease duration, and sex. Univariate correlation testing was performed with Spearman’s rank correlation testing. Significant correlations were evaluated in multivariate analysis by Pearson correlation adjusted for potential confounds. All tests were two-tailed, and statistical significance was set at p < 0.05 and was not adjusted for multiple comparisons given the small sample size and exploratory nature of this study.
Results
Thirty-four MS cases were imaged and analyzed, along with 15 healthy volunteers. There were no differences in age or sex between the MS cases and healthy volunteers (Table 1). Twenty-eight (83%) of the MS cases had RRMS and 6 (17%) had SPMS (3) or PPMS (3). The relapsing and progressive cohorts did not differ in mean age or sex ratio, but there was a higher proportion of RRMS on disease modifying treatment (79% vs. 33%) and SPMS/PPMS cases had significant worse disability as measured by EDSS and MSFC.
Table 1.
Demographic and Clinical Characteristics.
| HV (n= 15) | All MS (n = 34) | RRMS (n = 28) | SPMS/PPMS (n = 6) | |
|---|---|---|---|---|
| Mean Age (sd) | 41 (10) | 43 (10) | 42 (10) | 46 (9) |
| % Female | 67 % | 53 % | 57 % | 33 % |
| Mean Years of Education (sd) | 18 (3) | 17 (2) | 17 (2) | 18 (3) |
| Mean Disease Duration in Years (sd) | n/a | 10 (7) | 9 (7) | 13 (10) |
| % on MS Treatment | n/a | 79 % | 89 % | 33 %† |
| Median EDSS (Range) | n/a | 3.0 (1–6.5) | 2.5 (1–6) | 5.5 (2–6.5)† |
| Mean MSSS (sd) | n/a | 4.6 (2.3) | 4.27 (2.4) | 6.1 (1.2) |
| Mean MSFC z-score (sd) | 0.76 (0.40) | 0.16 (0.50)* | 0.24 (0.40) | −0.29 (0.79)† |
| 9-Hole Peg Test z-score (sd) | 1.3 (0.68) | 0.01 (0.96)* | 0.16 (0.89) | −0.68 (1.1) |
| Timed 25 Foot Walk z-score (sd) | 0.50 (0.41) | 0.26 (0.44)* | 0.36 (0.24) | −0.17 (0.85)† |
| PASAT z-score (sd) | 0.51 (0.65) | 0.18 (0.82) | 0.22 (0.78) | −0.06 (1.1) |
Demographic and clinical characteristics of the study cohort. HV = healthy volunteers. RRMS = relapsing remitting multiple sclerosis. SPMS = secondary progressive multiple sclerosis. PPMS = primary progressive multiple sclerosis. MSSS = Multiple Sclerosis Severity Score.
p < 0.05 for difference from healthy volunteers.
p < 0.05 for difference from RRMS.
Most MS cases had thalamic lesions (24, 71%). Thalamic lesions were either small, discrete, ovoid lesions distributed throughout the body of the thalamus, or wider areas of signal abnormality lining the third ventricle (Figure 1). Although 5 healthy volunteers were found to have very small areas of signal abnormality in the thalamus, normalized volume was very low (mean 3.79 × 10−6 vs. 7.65 × 10−5 in MS cases, p < 0.01), and none was periventricular. Progressive MS cases had a greater thalamic lesion burden than RRMS cases as measured by lesion count and burden (Figure 3). Ovoid thalamic lesion count was significantly greater in SPMS/PPMS (9.0 (SD 1.9)) than in RRMS (1.9 (SD 0.5)), as was ovoid thalamic lesion volume (1.29 × 10−4 (SD 9.71 × 10−5 vs. 2.04 × 10−5 (SD 4.82 × 10−5)). There were no differences between subgroups in periventricular thalamic lesion burden. Total thalamic lesion volume (but not lesion count) correlated with disease duration (ρ = 0.41, p = 0.02), but not age. Multivariate logistic regression for differences in thalamic lesion burden adjusted for disease duration, sex, and white matter lesion volume did not alter the RRMS vs. SPMS/PPMS comparisons noted above.
Figure 3.

Thalamic Lesion Count and Volume
Mean thalamic lesion count and volumes (normalized to intracranial volume) for all MS subjects and per subtype. SPMS and PPMS combined due to low number in both groups. The total thalamic lesion burden and ovoid lesion burden were increased in SPMS/PPMS subjects compared to RRMS. There was no difference between subgroups for the extent of periventricular lesion burden. * = p < 0.05 for comparison to healthy volunteers. ** = p < 0.05 for comparison to RRMS. Error bars represent the 95% confidence interval for estimate of the mean.
Thalamic lesion burden correlated with disability in univariate analysis (Figure 4 and Table 2), with correlations found between the total and ovoid thalamic lesion burden and the EDSS score and 9-hole peg test time. However, adjustment for age, disease duration and sex in multivariate analysis removed these correlations, and there was no association between MSSS and thalamic lesion burden. The correlations between total and thalamic lesion burden and EDSS were of similar magnitude as seen for WM lesion volume and EDSS, but weaker than for BPF. In contrast, correlations between thalamic volume and disability did not yield significant results. Periventricular thalamic lesions did not correlate with any disability measure.
Figure 4.

Correlation of thalamic lesions with disability.
Scatter plots of total and subtype lesion count and volume (normalized to intracranial volume) against EDSS score. Line of best fit is shown, along with results of Spearman rank correlation analysis. Total thalamic lesion count and ovoid lesion count and normalized volume significantly correlated with EDSS.
Table 2.
Correlation of Thalamic Lesion Burden and Disability.
| Disability Measure |
Thalamic Lesions | Conventional MRI Measures | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total Lesion Count |
Total Lesion Volume |
Periventricular Lesion Count |
Periventricular Lesion Volume |
Ovoid Lesion Count |
Ovoid Lesion Volume |
WM Lesion Volume |
BPF | Cortical GM Volume |
WM Volume |
Thalamic Volume |
|
| EDSS | 0.36* | 0.34* | 0.13 | 0.16 | 0.40* | 0.36* | 0.36* | −0.66* | −0.14 | −0.51* | −0.28 |
| MSSS | 0.14 | 0.03 | −0.15 | −0.18 | 0.31 | 0.26 | −0.05 | −0.26 | 0.04 | −0.32 | −0.05 |
| 9-HPT | −0.35* | −0.36* | −0.17 | −0.17 | −0.34* | −0.28 | −0.27 | 0.52* | 0.20 | 0.38* | 0.30 |
| T25-FW | −0.27 | −0.21 | −0.04 | −0.04 | −0.34 | −0.32 | −0.15 | 0.42* | 0.05 | 0.40* | 0.09 |
| PASAT-3 | 0.00 | 0.12 | −0.19 | −0.17 | 0.12 | 0.19 | 0.02 | 0.31 | −0.21 | 0.54* | −0.16 |
| MSFC | −0.17 | −0.12 | −0.21 | −0.19 | −0.09 | −0.04 | −0.12 | 0.55* | 0.01 | 0.56* | 0.16 |
Spearman rank correlation coefficient for relationship between thalamic lesion burden parameter is shown above. For comparison, conventional MRI measures and their correlations with disability measures are also shown. Volumes are normalized to intracranial volume prior to correlation analysis. Significant correlations are shown in bold and marked with “*”. EDSS = Kurtzke Expanded Disability Status Scale. MSSS = MS Severity Scale. 9HPT = 9 Hold Peg Test. T25-FW = Timed 25 Foot Walk. PASAT = Paced Auditory Serial Addition Test-3. MSFC = Multiple Sclerosis Functional Composite. WM = White Matter. BPF = Brain Parenchymal Fraction. GM = Gray Matter.
Total cortical lesion count was increased in subjects with SPMS/PPMS (40.7, SD 30.4) compared to RRMS (20.1, SD 16.3) (Table 3). Total and ovoid thalamic lesion count correlated with total cortical lesion count and all three cortical lesion subtypes (Table 4). Total and ovoid thalamic lesion volumes correlated well with cortical lesion counts and total, type 1, and type 2 cortical lesion volumes, with the strongest correlations being found with type 2 cortical lesion burden. Total and ovoid thalamic lesion burden did not correlate with WM lesion volume, measures of brain atrophy, or thalamic volume. However, correlations were found between thalamic volume and periventricular lesion count (ρ = −0.35, p = 0.04) and volume (ρ = −0.41, p = 0.02).
Table 3.
Cortical Lesions
| HV (n=15) | All MS (n = 34) | RRMS (n = 28) | SPMS/PPMS (n = 6) | |
|---|---|---|---|---|
| Total Lesion Count | 1.9 (3.3) | 23.8 (20.6)* | 20.1 (16.3) | 40.7 (30.4)† |
| Total Lesion Volume | 1.26 × 10−5 (2.05 × 10−5) | 2.90 × 10−4 (2.55 × 10−4)* | 2.70 × 10−4 (2.52 × 10−4) | 3.89 × 10−4 (2.68 × 10−4) |
| Type 1 Lesion Count | 1.2 (1.9) | 12.2 (9.6)* | 11.0 (9.6) | 17.7 (7.7) |
| Type 1 Lesion Volume | 8.81 × 10−6 (1.65 × 10−5) | 1.53 × 10−4 (1.59 × 10−4)* | 1.46 × 10−4 (1.62 × 10−4) | 1.88 × 10−4 (1.47 × 10−4) |
| Type 2 Lesion Count | 0.6 (1.5) | 5.6 (7.8)* | 4.1 (4.9) | 12.0 (14.1)† |
| Type 2 Lesion Volume | 1.68 × 10−6 (3.77 × 10−6) | 1.44 × 10−5 (2.01 × 10−5)* | 1.19 × 10−5 (1.64 × 10−5) | 2.65 × 10−5 (3.20 × 10−5) |
| Type 3 Lesion Count | 0.1 (0.3) | 6.0 (7.6)* | 4.9 (6.3) | 11.0 (11.3) |
| Type 3 Lesion Volume | 2.08 × 10−6 (7.51 × 10−6) | 1.22 × 10−4 (1.42 × 10−4)* | 1.12 × 10−4 (1.36 × 10−4) | 1.73 × 10−4 (1.74 × 10−4) |
Mean (sd) cortical lesion count and volume (normalized to intracranial volume) for healthy volunteers and MS in total and by subtype. Type 1 lesions = leukocortical lesions. Type 2 lesions = intracortical lesions. Type 3 lesions = subpial lesions.
p < 0.05 for comparison to healthy volunteers.
p < 0.05 for comparison to RRMS.
Table 4.
Correlation of thalamic lesion burden with cortical and white matter lesion burden and atrophy.
| Cortical Lesions | WM Lesion Volume |
BPF | Cortical GM Volume |
WM Volume |
Thalamic Volume |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lesion Count | Lesion Volume | ||||||||||||||
| Total | Type 1 | Type 2 | Type 3 | Total | Type 1 | Type 2 | Type 3 | ||||||||
| Thalamic Lesions | Lesion Count | Total | 0.53* | 0.54* | 0.60* | 0.47* | 0.36* | 0.34 | 0.58* | 0.31 | 0.14 | −0.06 | <0.01 | −0.09 | −0.13 |
| Periventricular | 0.21 | 0.27 | 0.32 | 0.21 | 0.09 | 0.11 | 0.34 | 0.04 | 0.06 | −0.17 | −0.18 | −0.09 | −0.35* | ||
| Ovoid | 0.55* | 0.54* | 0.57* | 0.47* | 0.40* | 0.34* | 0.55* | 0.36* | 0.11 | −0.01 | 0.14 | −0.08 | −0.07 | ||
| Lesion Volume | Total | 0.51* | 0.45* | 0.62* | 0.44* | 0.39* | 0.34 | 0.59* | 0.27 | 0.14 | −0.28 | 0.06 | 0.00 | −0.18 | |
| Periventricular | 0.25 | 0.35* | 0.34 | 0.19 | 0.19 | 0.23 | 0.38* | 0.02 | 0.19 | −0.21 | 0.07 | 0.03 | −0.34* | ||
| Ovoid | 0.50* | 0.40* | 0.60* | 0.42* | 0.33 | 0.24 | 0.57* | 0.31 | 0.09 | −0.28 | 0.06 | −0.06 | −0.06 | ||
Spearman rank correlation coefficients shown for relationship between cortical lesion and thalamic lesion burden. Raw volumes were used in correlations with other volumes in the same subject. Normalized volumes were used in correlations with non-volumetric measures. Values with p < 0.05 are bold and marked with “*”. BPF = brain parenchymal fraction, GM = gray matter, WM = white matter.
Discussion
In this investigation of the thalamus in MS at 7T, we found thalamic lesions in the majority of MS cases (>70%). Two distinct types of thalamic lesions were noted: ovoid (discrete, ovoid shaped lesions) and periventricular (diffuse areas of confluent signal abnormality lining the ventricular surface of the thalamus). Thalamic lesion burden was greatest in those with a progressive clinical phenotype- independent of age, disease duration, sex, or white matter lesion volume. The thalamic lesion burden was also greater in those with more physical disability in univariate analysis, although this relationship was not present when adjusted for confounding demographic variables. The overall thalamic lesion burden was also related to the extent of cortical lesion burden, mostly driven by associations between ovoid thalamic lesions and cortical lesions.
The differing properties of the two thalamic lesion subtypes may provide interesting insights into their causation. Our findings are consistent with those found on autopsy by Vercellino et al.,6 a comprehensive pathologic study of MS lesions in deep gray matter structures. In that study, ovoid and periventricular lesions were found mostly in the caudate and thalamus. Ovoid thalamic lesions were noted to develop around a central blood vessel, with inflammation, demyelination, and neuronal loss radiating outward. The inflammatory response around these lesions is described as being less robust, however, than WM lesions, and perhaps more consistent with that occurring in type 2 cortical lesions. In our study, ovoid thalamic lesions showed the strongest correlations with type 2 cortical lesions, possibly cluing towards manifestations of a common pathology.
Periventricular thalamic lesions likely evolve from differing pathologic processes than ovoid thalamic lesions. Vercillino et al. described this lesion type as extensive areas of demyelination lining the thalamic surface of the third ventricle, with no clear relationship to a central vessel. We found no correlations between periventricular thalamic lesion burden and cortical lesion burden, WM lesion burden, or overall brain atrophy, perhaps indicating an independent pathologic source. Their visual similarity to subpial cortical lesions, which are thought to arise from the cytotoxic effects of chemokines from local meningeal follicles,20–22 may possibly indicate a similar pathologic origin. Despite their similarities, we did not find a specific correlation between periventricular thalamic lesion burden and subpial cortical lesion burden in this study. Additionally, the thalamic-ventricular surface lacks a pial meningeal layer, so the source of cytotoxic chemokines would have to be unrelated, but could be freely diffusing in cerebrospinal fluid (CSF). Of note, thalamic periventricular lesion burden did correlate with thalamic atrophy, cluing towards a joint pathologic process leading to both hyperintense signal around the third ventricle and neurodegeneration of thalamic nuclei.
Of the two lesion subtypes, ovoid thalamic lesions were most clinically relevant, as they significantly correlated with disability in the univariate analysis and were more prominent in progressive patients. This may indicate that periventricular inflammation in deep GM results in less neuronal damage than perivascular inflammation. Alternately, the differences in correlation with disability between the thalamic lesion subtypes may signify that standard measures of disability (such as EDSS) are not sensitive to the particular consequences of dysfunction in the medial thalamic nuclei.
Pathologic analysis of deep GM lesions demonstrates that most deep GM lesions involve both GM and WM.6 Deep GM lesion burden in the Vercellino et al. study did not correlate with overall WM lesion burden, however, but did correlate with the extent of cortical demyelination (ρ = 0.77). We similarly observed a correlation between thalamic and cortical lesion burden in absence of a correlation between thalamic lesion burden and WM lesions or measures of brain atrophy. Similar to deep gray matter lesions, cortical lesion burden also has previously been shown to lack strong pathologic correlation to WM lesion burden.23 These observations may be indicative of a mechanistic link between the formation of deep and cortical GM lesions that is distinct from WM lesions. The unique characteristics of the blood-brain barrier seen in cortical MS lesions,24 other aspects of the microvasculature, or specific tissue antigens may share enough similarity between deep and cortical GM as to allow for the same pathological processes to affect both. Additionally, neuronal-immunologic cell interactions can inhibit microglia/macrophage activation and induce regulatory T-cell responses,25,26 explaining why signs of inflammation are less common in cortical GM lesions.27 Given the presence of neuronal cell bodies in thalamic nuclei, the immunologic milieu of the thalamus may thus be more similar to cortical GM than WM.
Although our study does not have pathologic and neuroimaging correlation in the same subjects, we believe that the consistency between our results and those seen in the Vercellino et al. study indicate that our imaging approach is sensitive to the same pathologic processes in vivo. This confirms he ability of 7T MRI to facilitate in vivo detection of small-scale pathology.28, 29 Further, the correlation between lesions in the cortex and thalamus without a similar link to WM lesions suggests that measurement of lesions in deep GM structures may be a suitable estimate of overall GM lesion burden. Given that there are no automated methods for identification and quantification of cortical lesions, having such a marker would be valuable. If thalamic lesion burden is an adequate marker of overall GM lesion burden, we propose that quantification of thalamic and/or overall deep GM lesion burden may be a quicker, more practical measure of GM lesion burden for future observational studies and clinical trials.
Despite limitations of a small sample size, our study supports a link between the development of gray matter disease and a progressive MS phenotype. Importantly, the increase in thalamic lesion burden seen in progressive MS subjects in this study was found to be independent of the confounds of age, disease duration, sex, and white matter lesion volume. The independent relationship of thalamic lesion burden and clinical phenotype we observed, along with the increased cortical lesion burden in SPMS/PPMS vs. RRMS seen in this and other studies,7, 30 and data linking relative cortical sparing with a benign clinical course31 may all indicate that a transition to the development of gray matter lesions may be a contributor to a more progressive phenotype of MS. If this is the case, having a rapid measure of gray matter pathology (such as thalamic lesion burden), without the need for intensive cortical lesion quantification, would have utility in clinical care and future studies of progressive MS.
The demonstration of a relationship between thalamic pathology and clinical disability in this study is not a unique finding. Previous studies at lower field strengths have related thalamic damage to disability in MS.14, 15, 32–34 However, all previous studies chose to evaluate more global measures of thalamic damage, such as structural atrophy, iron deposition, or spectroscopic changes, without characterizing lesions. Descriptions of focal demyelinating lesions in deep GM structures, and in the thalamus specifically, are rare in the MS imaging literature- making this study unique. The paucity of attention to the imaging of thalamic lesions in MS may be due to the inherent signal characteristics of deep GM lesions on standard clinical imaging. Despite the presence of demyelination and neuroglial cell loss, deep GM lesions tend to have less overall tissue destruction than WM lesions,6 which could affect their intensity on MRI. Deep GM lesions contain less inflammation and minimal gliosis compared to WM lesions,6 also resulting in a less hyperintense T2 signal. Finally, deep GM lesions tend to be small, making them less apparent at standard resolution. The increased SNR and concomitant improved resolution possible with 7T MRI35 may help overcome these limitations.
Our study does have some shortcomings. Given the subtle nature of some lesions, manual lesion identification of the type performed in this study could be limited by poor reproducibility. Also, the sample size is small and thus our conclusions, especially for the differences noted between relapsing and progressive MS cases, should be taken as preliminary. Future work should confirm these findings in a larger sample, compare these results directly to scans at lower field strengths, and measure longitudinal changes. Despite limitations, the data presented here suggest 7T MRI may represent an important tool for quantification of deep GM tissue damage in MS. In doing so, we have demonstrated that thalamic lesions in MS have clinical relevance, and that their measurement may provide a useful estimate of overall gray matter lesion burden and may be indicative of processes linked with the clinical phenotype of progressive MS.
Acknowledgments
This study was supported in part by NIH Supplemental Grants 5P41 RR15241-09S1 and 5P41EB15909 (van Zijl) and Bayer Schering Pharma (Calabresi). Drs. Reich and Ms. Wood are supported by the Intramural Research Program of NINDS. Time for data analysis was supported by NIH Mentored Grant K23NS072366 (Harrison).
Special thanks to Janelle Aquino and Stephanie Syc, who both had helped with participant recruitment and study visit logistics. In addition, special thanks to the dedicated Kirby Center staff, who make studies of this type possible.
References
- 1.Geurts JJ, Barkhof F. Grey matter pathology in multiple sclerosis. Lancet Neurol. 2008;7:841–851. doi: 10.1016/S1474-4422(08)70191-1. [DOI] [PubMed] [Google Scholar]
- 2.Kidd D, Barkhof F, McConnell R, Algra PR, et al. Cortical lesions in multiple sclerosis. Brain. 1999;122 (Pt 1):17–26. doi: 10.1093/brain/122.1.17. [DOI] [PubMed] [Google Scholar]
- 3.Peterson JW, Bo L, Mork S, Chang A, et al. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 4.Gilmore CP, Geurts JJ, Evangelou N, et al. Spinal cord grey matter lesions in multiple sclerosis detected by post-mortem high field MR imaging. Mult Scler. 2009;15:180–188. doi: 10.1177/1352458508096876. [DOI] [PubMed] [Google Scholar]
- 5.Geurts JJ, Bo L, Roosendaal SD, et al. Extensive hippocampal demyelination in multiple sclerosis. J Neuropathol Exp Neurol. 2007;66:819–827. doi: 10.1097/nen.0b013e3181461f54. [DOI] [PubMed] [Google Scholar]
- 6.Vercellino M, Masera S, Lorenzatti M, et al. Demyelination, inflammation, and neurodegeneration in multiple sclerosis deep gray matter. J Neuropathol Exp Neurol. 2009;68:489–502. doi: 10.1097/NEN.0b013e3181a19a5a. [DOI] [PubMed] [Google Scholar]
- 7.Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–2712. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- 8.Benedict RH, Zivadinov R, Carone DA, et al. Regional lobar atrophy predicts memory impairment in multiple sclerosis. AJNR Am J Neuroradiol. 2005;26:1824–1831. [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher E, Rudick RA, Cutter G, et al. Relationship between brain atrophy and disability: an 8-year follow-up study of multiple sclerosis patients. Mult Scler. 2000;6:373–377. doi: 10.1177/135245850000600602. [DOI] [PubMed] [Google Scholar]
- 10.Geurts JJ, Pouwels PJ, Uitdehaag BM, Polman CH, et al. Intracortical lesions in multiple sclerosis: improved detection with 3D double inversion-recovery MR imaging. Radiology. 2005;236:254–260. doi: 10.1148/radiol.2361040450. [DOI] [PubMed] [Google Scholar]
- 11.Nelson F, Poonawalla AH, Hou P, Huang F, et al. Improved identification of intracortical lesions in multiple sclerosis with phase-sensitive inversion recovery in combination with fast double inversion recovery MR imaging. AJNR Am J Neuroradiol. 2007;28:1645–1649. doi: 10.3174/ajnr.A0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kilsdonk ID, de Graaf WL, Lopez Soriano A, et al. Multicontrast MR Imaging at 7T in Multiple Sclerosis: Highest Lesion Detection in Cortical Gray Matter with 3D-FLAIR. AJNR Am J Neuroradiol. 2012 doi: 10.3174/ajnr.A3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geurts JJ, Reuling IE, Vrenken H, et al. MR spectroscopic evidence for thalamic and hippocampal, but not cortical, damage in multiple sclerosis. Magn Reson Med. 2006;55:478–483. doi: 10.1002/mrm.20792. [DOI] [PubMed] [Google Scholar]
- 14.Houtchens MK, Benedict RH, Killiany R, et al. Thalamic atrophy and cognition in multiple sclerosis. Neurology. 2007;69:1213–1223. doi: 10.1212/01.wnl.0000276992.17011.b5. [DOI] [PubMed] [Google Scholar]
- 15.Ge Y, Jensen JH, Lu H, et al. Quantitative assessment of iron accumulation in the deep gray matter of multiple sclerosis by magnetic field correlation imaging. AJNR Am J Neuroradiol. 2007;28:1639–1644. doi: 10.3174/ajnr.A0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammond KE, Metcalf M, Carvajal L, et al. Quantitative in vivo magnetic resonance imaging of multiple sclerosis at 7 Tesla with sensitivity to iron. Ann Neurol. 2008;64:707–713. doi: 10.1002/ana.21582. [DOI] [PubMed] [Google Scholar]
- 17.Shiee N, Bazin P-L, Ozturk A, et al. A topology-preserving approach to the segmentation of brain images with multiple sclerosis lesions. Neuroimage. 2010;49(2):1524–35. doi: 10.1016/j.neuroimage.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischer JS, Rudick RA, Cutter GR, Reingold SC. The Multiple Sclerosis Functional Composite Measure (MSFC): an integrated approach to MS clinical outcome assessment. National MS Society Clinical Outcomes Assessment Task Force. Mult Scler. 1999;5:244–250. doi: 10.1177/135245859900500409. [DOI] [PubMed] [Google Scholar]
- 19.Rudick R, Antel J, Confavreux C, et al. Recommendations from the National Multiple Sclerosis Society Clinical Outcomes Assessment Task Force. Ann Neurol. 1997;42:379–382. doi: 10.1002/ana.410420318. [DOI] [PubMed] [Google Scholar]
- 20.Villar LM, Sadaba MC, Roldan E, et al. Intrathecal synthesis of oligoclonal IgM against myelin lipids predicts an aggressive disease course in MS. J Clin Invest. 2005;115:187–194. doi: 10.1172/JCI22833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asgari N, Khorooshi R, Lillevang ST, Owens T. Complement-dependent pathogenicity of brain-specific antibodies in cerebrospinal fluid. J Neuroimmunol. 2013;254:76–82. doi: 10.1016/j.jneuroim.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Magliozzi R, Howell OW, Reeves C, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68:477–493. doi: 10.1002/ana.22230. [DOI] [PubMed] [Google Scholar]
- 23.Bo L, Geurts JJ, van der Valk P, Polman C, et al. Lack of correlation between cortical demyelination and white matter pathologic changes in multiple sclerosis. Arch Neurol. 2007;64:76–80. doi: 10.1001/archneur.64.1.76. [DOI] [PubMed] [Google Scholar]
- 24.van Horssen J, Brink BP, de Vries HE, van der Valk P, et al. The blood-brain barrier in cortical multiple sclerosis lesions. J Neuropathol Exp Neurol. 2007;66:321–328. doi: 10.1097/nen.0b013e318040b2de. [DOI] [PubMed] [Google Scholar]
- 25.Chitnis T, Imitola J, Wang Y, et al. Elevated neuronal expression of CD200 protects Wlds mice from inflammation-mediated neurodegeneration. Am J Pathol. 2007;170:1695–1712. doi: 10.2353/ajpath.2007.060677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518–525. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- 27.Bo L, Vedeler CA, Nyland H, Trapp BD, et al. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler. 2003;9:323–331. doi: 10.1191/1352458503ms917oa. [DOI] [PubMed] [Google Scholar]
- 28.Mainero C, Benner T, Radding A, et al. In vivo imaging of cortical pathology in multiple sclerosis using ultra-high field MRI. Neurology. 2009;73:941–948. doi: 10.1212/WNL.0b013e3181b64bf7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tallantyre EC, Brookes MJ, Dixon JE, Morgan PS, et al. Demonstrating the perivascular distribution of MS lesions in vivo with 7-Tesla MRI. Neurology. 2008;70:2076–2078. doi: 10.1212/01.wnl.0000313377.49555.2e. [DOI] [PubMed] [Google Scholar]
- 30.Calabrese M, Filippi M, Rovaris M, et al. Morphology and evolution of cortical lesions in multiple sclerosis. A longitudinal MRI study. Neuroimage. 2008;42:1324–1328. doi: 10.1016/j.neuroimage.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 31.Calabrese M, Filippi M, Rovaris M, et al. Evidence for relative cortical sparing in benign multiple sclerosis: a longitudinal magnetic resonance imaging study. Mult Scler. 2009;15:36–41. doi: 10.1177/1352458508096686. [DOI] [PubMed] [Google Scholar]
- 32.Neema M, Arora A, Healy BC, et al. Deep gray matter involvement on brain MRI scans is associated with clinical progression in multiple sclerosis. J Neuroimaging. 2009;19:3–8. doi: 10.1111/j.1552-6569.2008.00296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bakshi R, Benedict RH, Bermel RA, et al. T2 hypointensity in the deep gray matter of patients with multiple sclerosis: a quantitative magnetic resonance imaging study. Arch Neurol. 2002;59:62–68. doi: 10.1001/archneur.59.1.62. [DOI] [PubMed] [Google Scholar]
- 34.Cifelli A, Arridge M, Jezzard P, Esiri MM, et al. Thalamic neurodegeneration in multiple sclerosis. Ann Neurol. 2002;52:650–653. doi: 10.1002/ana.10326. [DOI] [PubMed] [Google Scholar]
- 35.Vaughan JT, Garwood M, Collins CM, et al. 7T vs. 4T: RF power, homogeneity, and signal-to-noise comparison in head images. Magn Reson Med. 2001;46:24–30. doi: 10.1002/mrm.1156. [DOI] [PubMed] [Google Scholar]