

Abstract
Nontypeable Haemophilus influenzae (NTHi) colonize the human pharynx asymptomatically, and are also an important cause of otitis media (OM). Previous studies have demonstrated that some genes are more prevalent in OM-causing NTHi strains than in commensal strains, suggesting a role in virulence. These studies, however, are unable to investigate the possible associations between gene polymorphisms and disease. This study examined amino acid polymorphisms and sequence diversity in a potential virulence gene, the hemin receptor hemR, from a previously characterized NTHi strain collection containing both commensal and OM organisms to identify possible associations between the polymorphisms and otitis media. The full open reading frame of hemR was sequenced from a total of 146 NTHi isolates, yielding a total of 47 unique HemR amino acid sequences. The predicted structure of HemR showed substantial similarity to a class of monomeric TonB dependent, ligand-gated channels involved in iron acquisition in other gram negative bacteria. Fifteen amino acid polymorphisms were significantly more prevalent at the 90% confidence level among commensal compared to OM isolates. Upon controlling for the confounding effect of population structure, over half of the polymorphism-otitis media relationships lost statistical significance, emphasizing the importance of assessing the effect of population structure in association studies. The seven polymorphisms that retained significance were dispersed throughout the protein in various functional and structural domains, including the signal peptide, N-terminal plug domain, and intra- and extracellular loops. The alternate amino acid of only one of these seven polymorphisms was more common among OM isolates, demonstrating a strong trend toward the consensus sequence among disease causing NTHi. We hypothesize that variability at these positions in HemR may result in a reduced ability to acquire iron, rendering NTHi with such versions of the gene less fit for survival in the middle ear environment.
Keywords: Nontypeable Haemophilus influenzae, NTHi, Population structure, Otitis media, Polymorphism, Iron acquisition
1. Introduction
Nontypeable Haemophilus influenzae (NTHi), which lack a polysaccharide capsule, frequently colonize the human nasopharynx, particularly in young children in whom the carriage rate is up to 80% (Bou et al., 2000; Farjo et al., 2004; Kilian, 2005; Schumacher et al., 2012; St Sauver et al., 2000). Colonization is typically a dynamic process, marked by simultaneous colonization with multiple strains and apparent rapid turnover (Dhooge et al., 2000; LaCross et al., 2008; Murphy et al., 1999). NTHi also has the potential to be pathogenic, causing a variety of respiratory infections, including otitis media (OM), sinusitis, pneumonia, and chronic bronchitis (Casey et al., 2013; van Wessel et al., 2011; Zhang et al., 2012). A number of genes and genetic islands have been associated with OM, including those encoding adhesins, pili, lipooligosaccharide biosynthesis enzymes, and the histidine biosynthesis operon (Ecevit et al., 2004; Juliao et al., 2007; Pettigrew et al., 2002; Xie et al., 2006).
Genes involved in the acquisition of iron and iron containing molecules have also been implicated in NTHi virulence. Given the importance of iron for growth in nearly all bacteria, and the absolute requirement of heme for Haemophilus influenzae (Hi) aerobic growth, it is not surprising that Hi have several partially redundant systems to acquire iron from a variety of sources, including heme, hemoglobin, transferrin, hemoglobin:haptoglobin complexes, and heme:hemopexin complexes. A study by Morton et al. demonstrated that a mutant NTHi strain lacking the hemoglobin binding proteins (hgp's) had significantly reduced virulence in an animal model of otitis media compared to the isogenic wild type strain (Morton et al., 2004). In a later study, Morton and colleagues showed that infant rats infected with a mutant H. influenzae type b (Hib) strain lacking the hxuCBA operon (responsible for the utilization of heme:hemopexin complexes) had significantly lower bacteremic titers and improved survival rates as compared to those infected with the wild type strain (Morton et al., 2007a).
The hemin receptor of NTHi, hemR, has been the focus of considerably less research. Thomas et al. found that TdhA of Haemophilus ducreyi, a distant relative of H. influenzae, has significant sequence homology to HemR, as well as other heme receptors from gram negative bacteria, including HxuC from Hi, HmuR from Yersinia pestis, and ChuA from E. coli (Thomas et al., 1998). An E. coli mutant unable to synthesize heme and lacking native heme and hemoglobin receptors but expressing H. ducreyi tdhA grew on low levels of heme only when an intact H. ducreyi Ton system plasmid was present, demonstrating functional TonB dependence. Leduc et al. found no statistically significant difference in pustule formation or number of bacteria recovered from the pustules in six human volunteers experimentally inoculated with both wild type H. ducreyi and an isogenic tdhA mutant (Leduc et al., 2008). These data led the authors to suggest that the HemR homologue TdhA is not necessary for virulence in H. ducreyi.
The presence of hemR in NTHi, however, has been associated with otitis media strains. Xie and colleagues, using dot blot hybridization, found hemR to be significantly more common among OM NTHi isolates (99.2%) as compared to commensal NTHi isolates (86.9%) (prevalence ratio of 1.14, p=0.0002) (Xie et al., 2006). Similarly, hemR was more common among invasive Hib isolates (97.4%) than among commensal NTHi isolates (86.9%), with a prevalence ratio of 1.15 (p=0.0169). Whitby et al. used microarray and qPCR analyses to demonstrate that expression of hemR was increased under iron/heme limiting conditions in OM NTHi strain R2866, capsule deficient type d strain Rd, and invasive Hib strain 10810 (Whitby et al., 2009).
However, many iron acquisition genes are present in the majority of NTHi strains assayed and demonstrate only modest prevalence differences between disease-causing and commensal isolates, if a significant difference exists at all. Such genes may still play an important role in pathogenesis, however, and certain alleles may be able to better provide NTHi with iron and heme in normally privileged environments such as the middle ear, thus enhancing virulence.
As hemR has been previously shown to have significantly increased prevalence in OM isolates, it was chosen to investigate the hypothesis that polymorphisms may also be associated with NTHi disease. The full coding sequence for hemR was obtained from 85 OM and 61 commensal NTHi isolates. Amino acid polymorphisms that differed in prevalence between the OM and commensal isolates were identified from the translated hemR nucleotide sequences and related to the theoretical three dimensional structure of the protein. Most genetic association studies in bacteria, including those described in the preceding paragraphs, do not attempt to control for the potentially confounding effect of bacterial population structure, despite numerous studies demonstrating the presence of structure in bacterial populations (den Bakker et al., 2008; Erwin et al., 2008; Falush and Bowden, 2006; Falush et al., 2003; LaCross et al., 2013; Musser et al., 1986; Musser et al., 1990; Musser et al., 1988; Sheppard et al., 2010; Smith et al., 2000).
The research presented in this study identified amino acid polymorphisms in the NTHi hemin receptor HemR associated with otitis media while adjusting for the population structure of this set of strains as identified previously in our laboratory (LaCross et al., 2013). In addition, the theoretical three dimensional protein structure of HemR was assessed, allowing otitis media-associated HemR amino acid polymorphisms to be mapped onto potential functional and structural domains.
2. Materials and Methods
2.1. DNA Amplification and Sequencing
The 170 NTHi previously characterized by phylogenetic clustering as Haemophilus influenza and isolated from three distinct geographic regions (Finland, Israel, and the US) were utilized in this study; genomic DNA extraction and PCR protocols used to analyze hemR were performed as described therein (LaCross et al., 2013). Since some primer pairs did not successfully amplify some isolates, alternate primers covering the same region of the gene were designed, yielding a total of seven primer pairs covering the entire coding region of hemR (Supplemental Table 1). Sequences for the full coding region of hemR for all isolates used in this study are available from GenBank under accession numbers JN229266 through JN229411.
2.2. Data and Statistical Analysis
SeqMan Pro 8.1.5 (DNASTAR, Madison, WI) was used to align and trim the sequenced PCR products into contigs covering the hemR open reading frame. Potential signal peptides were identified using the hidden Markov model implemented by the SignalP online server (Bendtsen et al., 2004; Nielsen and Krogh, 1998). The predicted three dimensional structures of the HemR protein from selected isolates were determined from their amino acid sequences by the I-TASSER server (Roy et al., 2010; Zhang, 2007, 2008). Yasara View 11.6.16 was used to visualize the resulting theoretical protein structures (Krieger et al., 2002).
All hemR sequences were translated and aligned in CLC Sequence Viewer 6.5.2 (CLC bio, Aarhus, Denmark); this amino acid sequence alignment was then examined visually to identify amino acid polymorphisms associated with otitis media. Unadjusted prevalence ratios (PRs), odds ratios (ORs), and associated confidence intervals (CIs) were calculated to describe the association between each potentially significant polymorphism and otitis media using OpenEpi 2.3.1 (Dean et al.). All ratios compare OM isolates to commensal isolates. For all statistical analyses, isolates with the consensus amino acid at a polymorphic position based on the above alignment were coded with a ‘1’, and isolates with any other amino acid at that position were coded with a ‘0’. Thus, a PR or OR greater than one for a given polymorphism indicates that OM isolates are more likely to have the consensus amino acid at that position, while ratios less than one indicate a greater likelihood of having an alternative amino acid there.
To adjust the association between each HemR polymorphism and otitis media for the presence of population structure, a series of logistic regression models were constructed in R version 2.13.0 (R_Development_Core_Team., 2011). Population structure information based on multilocus sequence typing (MLST) data from the all isolates dataset (as described extensively by LaCross et al. (LaCross et al., 2013)) was used, and the estimated individual membership proportions (Q) were coded as eight continuous variables (one for each population) with ranges between 0, indicating no ancestry from that population, and 100, indicating all ancestry was from that population. To obtain p-values for the ORs adjusted for population structure, permutation tests were constructed by randomly rearranging the polymorphism data labels and recalculating the OR 10,000 times. The proportion of permuted ORs as extreme or more extreme than the observed OR was the p-value for that permutation test.
The p-values were not adjusted for the number of comparisons, as recommended by some statisticians (Feise, 2002; Rothman, 1990). Common methods to adjust for multiple comparisons attempt to reduce the frequency of type I errors (incorrectly rejecting a true null hypothesis), which has the side effect of increasing type II errors (erroneously not rejecting a false null hypothesis). In this study, all comparisons are reported, and the effect of multiple comparisons was informally assessed by contrasting the expected number of type I errors with the empirically derived number of p-values below the specified significance level (α=0.10).
3. Results
3.1. HemR Characteristics
The nucleotide sequences of the full hemR protein coding regions of the 170 NTHi isolates were sought using the primers listed in Supplemental Table 1. In ten isolates (one OM and nine commensal) no amplification was observed with any of the hemR specific primers; these isolates were judged to be missing the gene entirely, which was not an unexpected result since a previous study from this laboratory found hemR to be absent by dot blot DNA hybridization in 0.8% of NTHi OM isolates and in 13.1% of commensal isolates. The increased prevalence of HemR among OM isolates compared to commensal isolates (99.2% versus 86.9%, PR = 1.14, p=0.0002) reported in the earlier study (Xie et al., 2006) is confirmed in the present study, (98.9% versus 88.0%, PR = 1.12, p=0.0010). Intriguingly, all nine hemR negative commensal isolates were also fucK negative, suggesting that divergent NTHi strains, such as those that are fucK negative, are less likely to have potential virulence loci like hemR. However, the remaining five fucK negative commensal isolates, as well as the sole OM fucK negative isolate, were positive for hemR, so the association is by no means absolute. A further 14 isolates (nine OM and 5 commensal) had consistently poor or absent amplification for at least one of the primer pairs that could not be resolved. As the full open reading frame of the gene could not be determined, these isolates were excluded from further analyses. The full protein coding sequence of hemR was obtained for a total of 146 NTHi isolates. A summary of these results appears in Table 1.
Table 1.
Summary of the NTHi isolates used for hemR amplification.
| Initiala | Gene Missingb | Poor Amplificationc | Total Removedd | Final | |
|---|---|---|---|---|---|
| Finland OM | 30 | 0 | 3 | 3 | 27 |
| Finland Commensal | 26 | 3 | 3 | 23 | |
| Israel OM | 28 | 0 | 5 | 5 | 23 |
| Israel Commensal | 22 | 1 | 2 | 3 | 19 |
| US OM | 37 | 1 | 1 | 2 | 35 |
| US Commensal | 27 | 8 | 0 | 8 | 19 |
| Total OM | 95 | 1 | 9 | 10 | 5 |
| Total Commensal | 75 | 9 | 5 | 14 | 61 |
Initial number of NTHi isolates characterized.
Isolates in which hemR is not present.
Isolates in which full sequencing of hemR was not possible due to poor PCR amplification.
Isolates removed from hemR sequence analyses.
The length of the HemR protein was variable between isolates, with an average and median of 745 residues, ranging from 724 to 751 residues. The isolate with the shortest HemR at 724 amino acids (K35LE2.1, a US OM isolate) was an outlier, however, as the next shortest HemR contained 740 residues. The majority of the insertions and deletions responsible for the variable length occurred in two locations, approximately 510 and 580 residues past the N-terminus.
Most bacterial outer membrane proteins contain a short peptide chain, often near the N-terminus, that assists in directing the transport of the protein through the periplasm. As HemR is an outer membrane protein, the presence of such a signal peptide was assessed. Based on the HemR alignment, four NTHi isolates (F162-7, F433-3, I207, and C09.2.3) that adequately covered the small amount of variation observed near the N-terminus were chosen for further analysis. A high probability for the presence a signal peptide, as assessed by the SignalP algorithm, was observed in all four isolates (mean ± SD = 96.5% ± 1.8%), with the cleavage site between residues 24 and 25 (96.1% ± 1.9%). Similarly high probabilities were estimated for the presence of a canonical n-region (a positively charged stretch of amino acids), h-region (the hydrophobic core area), and c-region (a polar amino acid stretch that includes the cleavage site). A representative plot of the SignalP results is shown in Figure 1.
Figure 1.
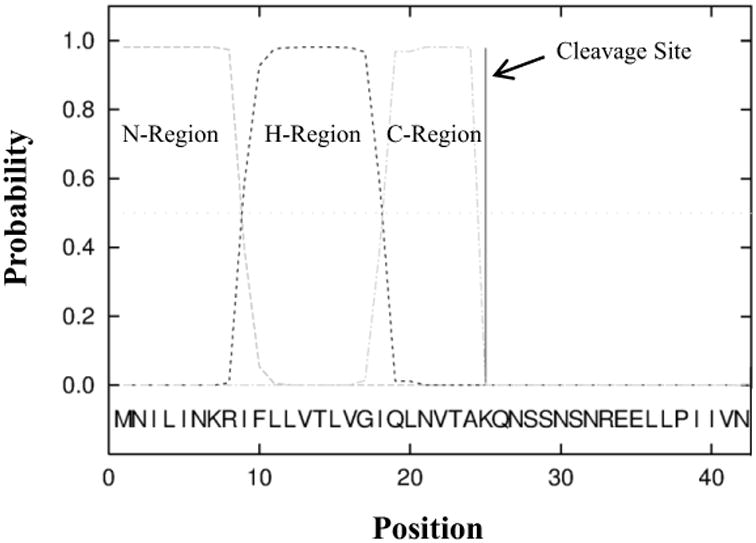
Signal peptide probabilities estimated by SignalP. HemR from isolate F433-3 was used, and position zero is the N-terminus. The n-, h-, and c-regions are canonical in most signal peptides and are a stretch of positively charged amino acids, a hydrophobic area, and a polar region, respectively.
The overall level of amino acid sequence conservation was quite high, with an average identity of 94.5%. However, the level of conservation fluctuated considerably across the length of the protein, with some regions identical in all 146 isolates and others exhibiting substantial variation (Figure 2). The N-terminal 250 amino acids, approximately 1/3 of the protein, shows considerable conservation, with only one position at lower than 80% identity. The picture is different after residue 250, where regions of substantial diversity (40 – 75% identity) are interspersed with regions of high conservation. This pattern is directly related to the different functional domains and motifs of the protein, which are color coded to match the domains elucidated in Figure 3 and discussed further therein.
Figure 2.
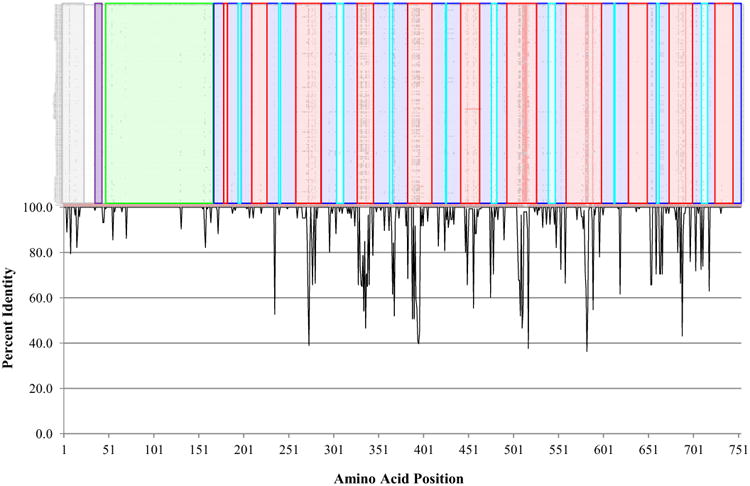
HemR amino acid sequence conservation by structural domain. A. Alignment of the 146 NTHi HemR amino acid sequences. Identical residues are coded with a dot. Structural domains match those in Figure 3 and are colored as follows: gray – signal peptide; purple – putative TonB box; green – N-terminal plug domain; dark blue – transmembrane β-strands; red – extracellular loops; light blue – intracellular loops. B. Plot of the percent identity by amino acid position.
Figure 3.
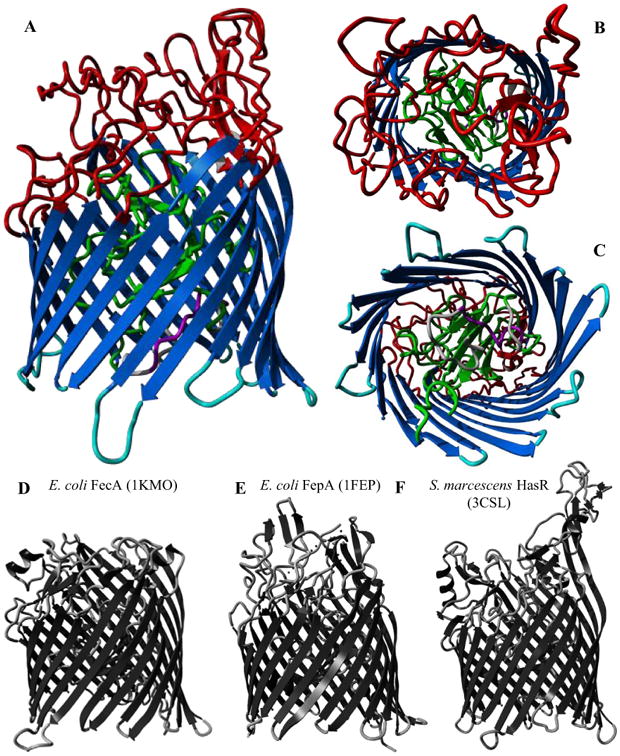
A-C: Predicted structure of HemR from Finnish OM isolate F433-3 (C-score = 0.74). Structural domains are colored as follows: gray – signal peptide; purple – putative TonB box; green – N-terminal plug domain; dark blue – transmembrane β-strands; red – extracellular loops; light blue – intracellular loops. A. Side view. B. Extracellular side. C. Periplasmic side. D-F: Experimentally derived structures for three iron acquisition receptors in other gram negative species, with PDB IDs in parentheses. D. E. coli ferric citrate uptake transporter FecA. E. E. coli ferric enterobactin receptor FepA. F. S. marcescens hemophore receptor HasR.
In all but four cases, multiple isolates with the same MLST genotype also shared identical HemR sequences. Isolates of STs 13 and 156 had single non-synonymous SNPs at positions 171 and 235, respectively. Of the two isolates comprising ST11, one was missing the entire gene. Finally, two of the three isolates of ST3 had identical HemR sequences, while the third showed only 94.5% sequence identity to the other two. As the residue differences were spread throughout the sequence and not concentrated within one PCR amplicon, it seems likely that this isolate's HemR sequence is legitimately different from other members of its MLST genotype, probably due to a recent recombination event.
Based on amino acid identity, 47 unique HemR protein sequences (differing by at least one amino acid, and henceforth referred to as HemR ‘types’) were identified among the 146 NTHi isolates. While the majority of HemR types consisted of only a single isolate, six types were comprised of strains from at least five different STs. The largest, HemR type 4 (arbitrarily named), contains 19 isolates of 11 different STs and consists of a mix of OM and commensal isolates from all three geographic regions, a theme that is repeated amongst most of the other multi-ST HemR types. This pattern derived from the HemR types is reminiscent of the population structure inferred by the structure program based on MLST data described from the all isolates dataset previously by our laboratory (LaCross et al., 2013). Supplemental Table 2 sorts the HemR types, and the isolates they contain, by the individual membership proportion (Q) for each isolate (i.e. the proportion of an isolate's ancestry from a given population). It is apparent that for the most part, when HemR types include isolates of multiple STs, those isolates belong to the same structure population. For example, HemR type 15 in population 5 and HemR type 2 in population 6 are composed of five and nine STs, respectively, but belong to single structure populations.
3.2. Otitis Media Associated HemR Polymorphisms
Visual assessment of the HemR alignment identified 14 amino acid polymorphisms significantly associated with otitis media at the 95% confidence level, with a further four polymorphisms significant at the 90% confidence level (Table 2). The name of each polymorphism references its position within the HemR multiple sequence alignment. Four of these polymorphisms (I659V, N663Y, F664L, and A666V) always co-occurred and are treated as a single entity for the statistical analyses, leaving a total of 15 polymorphisms significant at the 90% confidence level. If all null hypotheses are true (i.e. no polymorphism is truly associated with otitis media), we would expect 10% of these comparisons to yield p-values below the confidence level by chance.
Table 2.
Distribution of HemR polymorphisms among OM and commensal NTHi isolates.
| Polymorphism | OM | Commensal | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1° aaa | Position | 2° aab | N1°/Ntotalc | % | N1°/Ntotalc | % | PR (95% CI) | p-value | OR (95% CI) | p-value | Perm. p-valued |
|
|
|
|
|
|
|||||||
| L | 4 | I/F 11/5 | 80/85 | 94.1% | 50/61 | 82.0% | 1.15 (1.01 - 1.31) | 0.02 | 3.52 (1.15 - 10.73) | 0.03 | 0.02 |
| R | 8 | H | 63/85 | 74.1% | 53/61 | 86.9% | 0.85 (0.73 - 1.00) | 0.06 | 0.43 (0.18 - 1.05) | 0.06 | 0.04 |
| L | 15 | F | 77/85 | 90.6% | 43/61 | 70.5% | 1.29 (1.08 - 1.53) | <0.01 | 4.03 (1.62 - 10.04) | <0.01 | <0.01 |
| V | 16 | I | 80/85 | 94.1% | 51/61 | 83.6% | 1.13 (1.00 - 1.27) | 0.04 | 3.14 (1.01 - 9.71) | 0.05 | 0.03 |
| A | 70 | V | 67/85 | 78.8% | 59/61 | 96.7% | 0.82 (0.72 - 0.92) | <0.01 | 0.13 (0.03 - 0.57) | 0.01 | <0.01 |
| R | 131 | K | 81/85 | 95.3% | 51/61 | 83.6% | 1.14 (1.01 - 1.29) | 0.02 | 3.97 (1.18 - 13.33) | 0.03 | 0.02 |
| K | 157 | R | 80/85 | 94.1% | 51/61 | 83.6% | 1.13 (1.00 - 1.27) | 0.04 | 3.14 (1.01 - 9.71) | 0.05 | 0.03 |
| Q | 164 | K | 82/85 | 96.5% | 54/61 | 88.5% | 1.09 (0.99 - 1.20) | 0.06 | 3.54 (0.88 - 14.30) | 0.08 | 0.06 |
| N | 303 | H/R 13/4 | 79/85 | 92.9% | 50/61 | 82.0% | 1.13 (0.99 - 1.29) | 0.04 | 2.90 (1.01 - 8.33) | 0.05 | 0.03 |
| T | 324 | I/E 8/4 | 81/85 | 95.3% | 53/61 | 86.9% | 1.10 (0.98 - 1.22) | 0.07 | 3.06 (0.88 - 10.66) | 0.08 | 0.07 |
| Y | 405 | F | 83/85 | 97.6% | 54/61 | 88.5% | 1.10 (1.00 - 1.21) | 0.02 | 5.38 (1.08 - 26.87) | 0.04 | 0.02 |
| D | 578 | N | 50/85 | 94.1% | 52/61 | 85.2% | 1.10 (0.98 - 1.24) | 0.07 | 2.77 (0.88 - 8.73) | 0.08 | 0.07 |
| P | 589 | I | 53/85 | 62.4% | 27/61 | 44.3% | 1.41 (1.02 - 1.95) | 0.03 | 2.09 (1.07 - 4.07) | 0.03 | 0.02 |
| I* | 659* | V* | |||||||||
| N* | 663* | Y* | 67/85 | 78.8% | 36/61 | 59.0% | 1.34 (1.05 - 1.69) | 0.01 | 2.58 (1.25 - 5.36) | 0.01 | 0.01 |
| F* | 664* | L* | |||||||||
| A* | 666* | V* | |||||||||
| I | 718 | V | 60/85 | 70.6% | 32/61 | 52.5% | 1.35 (1.02 - 1.77) | 0.03 | 2.18 (1.10 - 4.32) | 0.03 | 0.02 |
Consensus amino acid at that position.
Alternative amino acid at that position. Polymorphisms at positions 4, 303, and 324 have multiple alternative amino acids.
Prevalence of the consensus (or 1°) amino acid at a given position within OM or commensal isolates.
P-values calculated by a permutation test.
The I659V, N663Y, F664L, and A666V polymorphisms always co-occur, and are thus treated as a single entity in the statistical analyses.
While prevalence ratios are perhaps the most intuitive and straightforward measure of the crude association between otitis media and these polymorphisms, the equivalent ORs are also reported in Table 2 for comparison purposes, as later analyses rely on logistic regression models that output ORs, and there is unfortunately no straightforward comparison between the two measures of association in most situations. Consequently, it is important to note the difference in interpretation between PRs and ORs, given the large differences in value for the same relationship. As an example, consider the first polymorphism in Table 2, L4I/F. The PR is 1.15, indicating that the prevalence of the consensus amino acid leucine at position four of HemR is 1.15 times (or 15%) greater among OM isolates than among commensal isolates. The OR for that same relationship is much greater at 3.52, and indicates that the odds of an OM isolate having a leucine at position four are 3.52 times higher than the odds for a commensal isolate. When the outcome (a unique polymorphism, in this study) is rare, the OR will usually approximate the PR, but this is clearly not the case here. Both the regular p-value and a p-value calculated by a permutation test are reported for the ORs, again for comparison purposes. The permutation tests were performed by randomly rearranging the polymorphism data labels and recalculating the OR 10,000 times. The p-values were calculated as the proportion of permuted ORs as extreme or more extreme than the observed OR.
All of the significant PRs presented in Table 2 are modest in magnitude, with largest at 1.41 for the P589I polymorphism. This is not unexpected, as both truly commensal NTHi and NTHi capable of causing disease are found in the naso- and oropharynges, which can have a diluting effect on ratio measures of association. Of the 15 identified polymorphisms, 13 have PRs greater than the null value of one, signifying that OM isolates are significantly more likely to have the consensus amino acid at those positions within HemR. This suggests a constraint on the diversity among these isolates, consistent with the idea that NTHi isolates able to cause otitis media differ from those in the pharynx that typically do not. It is possible that alterations at certain loci like HemR can modify the virulence potential of an isolate, and that most alterations either provide no benefit or are deleterious.
3.3. HemR Protein Structure
To assist in identifying the potential functional effects of these polymorphisms, the three dimensional tertiary structure of the full HemR protein was predicted using the I-TASSER protein structure and function server (Roy et al., 2010; Zhang, 2007, 2008). The confidence score, or C-score, is a statistic calculated by I-TASSER for estimating the quality of the predicted models. It typically has a range of -5 to 2, such that a higher value signifies a model with high confidence. Yasara View 11.6.16 was used to visualize the resulting theoretical protein structures (Krieger et al., 2002).
Figure 3 shows the predicted structure for HemR from an NTHi isolate in panels A – C. The C-score for this model was 0.74, indicating high confidence for its quality. Predicted models for other HemR types had similarly high C-scores (not shown). Panels D – F show the experimentally derived (via X-ray diffraction) structures of three iron acquisition receptors from other gram negative species. There is marked similarity between the predicted structures for NTHi HemR and the known structures from other bacteria, despite very low amino acid sequence identity (approximately 18%, 15%, and 22% between HemR and FecA, FepA, and HasR, respectively).
Knowing the theoretical three dimensional structure of HemR allows a more detailed assessment of the variable amino acid conservation observed in this sample. Panels A-C of Figure 3 show a structural model of HemR with various domains color coded as in Figure 2, including extra- and intracellular loops and the N-terminal plug domain. By mapping these structural domains onto the alignment of all 146 NTHi HemR sequences, it becomes readily apparent that much of the variation in sequence conservation corresponds to these different domains (Figure 2). The majority of the most variable regions map to extracellular loops, as one would expect for an outer membrane protein exposed to the immune system. Interestingly, four of the extracellular loops appear to be quite conserved; they may be shielded from the immune system and thus under little selective pressure to vary, or they may be involved in ligand binding and be functionally constrained. Likewise, most of the intracellular loops are fairly conserved, but several show relatively high levels of variation.
Prediction of the three dimensional structure of HemR also allows more informative hypotheses of the potential functional effect of the otitis media-associated polymorphisms. Figure 4A highlights the A70V, R131K, K157R, Q164K, N303H/R, and T324I/E polymorphisms, while Figure 4B depicts the Y405F, D578N, P598I, and I718V polymorphisms, as well as the I659V, N663Y, F664L, and A666V quartet of polymorphisms that are treated as a single entity. All figures are modeled on the HemR structure from Finland OM isolate F199-3 of ST57 and population 7. The HemR sequences from the ST57 isolates are useful models in that they have the OM-associated amino acid at all the polymorphic positions identified in Table 2 (i.e. if the PR > 1 they have the consensus amino acid, and if the PR < 1 the alternate amino acid is present).
Figure 4.
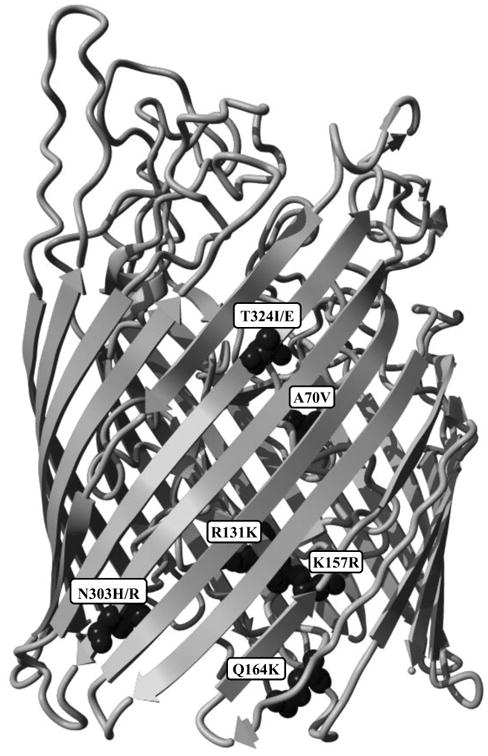
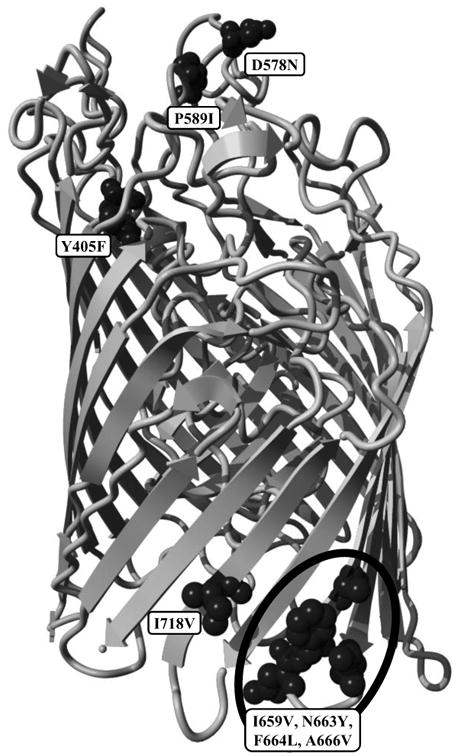
A. Side view of the predicted structure of HemR from ST57 isolate F199-3 showing the locations of polymorphisms Y405F through I718V. The N-terminal signal peptide has been removed in this figure. The six polymorphisms have been depicted using a space-filling model for visibility. B. Side view of the predicted structure of HemR from ST57 isolate F199-3 showing the locations of polymorphisms Y405F through I718V. The N-terminal signal peptide has been removed in this figure. The five polymorphisms have been depicted using a space-filling model for visibility.
It should be noted that the N-terminal 24 residue signal peptide identified in Figure 1 has been removed in Figure 4. This region contains the first four polymorphisms (L4I/F, R8H, L15F, and V16I) in Table 2. It is unknown what effect, if any, alteration in this region may produce. Studies of human membrane and secreted proteins have identified mutations within signal peptide regions that are implicated in various diseases, including Wolman disease, aspartylglucosaminuria, and Bernard–Soulier syndrome (Jarjanazi et al., 2008). It is thought that alterations within signal peptide domains impair the proper translocation and/or secretion of the proteins associated with these outcomes. The polymorphisms identified within the HemR signal peptide domain may also have a similar adverse effect on the proper localization of the protein.
It is apparent from Figure 4 that the amino acid polymorphisms listed in Table 2 occupy a variety of structural, and potentially functional, domains. Aside from the four polymorphisms within the signal peptide region listed above, four further polymorphisms, A70V, R131K, K157R, and Q164K, are located in the plug domain that occludes the interior channel (Figures 4A). In other, more widely studied TonB dependent receptors (primarily in E. coli), the plug domain is thought to either undergo a conformational change or exit the protein channel entirely upon interaction with TonB, thus allowing transit of the ligand (Krewulak and Vogel, 2011). The four HemR polymorphisms identified within its plug domain could potentially alter these functions. Furthermore, in at least some TonB dependent receptors, regions on the extracellular side of the plug domain are involved in ligand recognition and binding (Krewulak and Vogel, 2008). The A70V polymorphism is in close proximity to the extracellular apex of the plug domain, and could influence ligand interaction.
Four other HemR polymorphisms, N303H/R and T324I/E (Figures 4A) and I718V and the quartet of polymorphisms beginning at I659V (Figure 4B), are located within β-strands that form the transmembrane domain. All but T324I/E are on the periplasmic side of the protein close to, but not within, intracellular loops. Interestingly, the I659V quartet surrounds one of the intracellular loops and includes both residues immediately adjacent, while the loop itself (consisting of three residues) is identical in all 146 NTHi isolates examined. The final three polymorphisms, Y405F, D578N, and P589I (Figure 4B), are all positioned within extracellular loops, with D578N and P589I located within the same loop. The size of this loop is variable due to several small insertions and deletions, and D578N and P589I are separated by seven to ten amino acids. In E. coli and other bacteria, the extracellular loops of TonB dependent receptors are involved in ligand recognition and binding (Braun and Endriss, 2007; Buchanan et al., 1999; Chakraborty et al., 2003; Krewulak and Vogel, 2008, 2011; Pawelek et al., 2006; Tong and Guo, 2009). The extracellular loops of HemR in NTHi likely have a similar role, and alterations within them could affect this function.
3.4. Adjustment for Population Structure
To adjust for the possible confounding effect of population structure on the association between HemR polymorphisms and otitis media, multiple logistic regression models were constructed in R version 2.13.0 (R_Development_Core_Team., 2011) with the individual membership proportions for each population coded as continuous variables (eight in total). This population structure was previously inferred by structure based on MLST data (LaCross et al., 2013), and an individual membership proportion is the fraction of an isolate's ancestry from a given population. A separate set of models was run for each polymorphism, and permutation tests were used to calculate p-values. In addition to the crude (i.e. unadjusted) models, models controlling for all eight populations simultaneously as well as models adjusting for those populations that minimized the Akaike information criterion (AIC) values were created. The AIC is a measure of the relative goodness of fit of a statistical model, and offers a measure of the information lost when a given model is used to describe reality. Additionally, it includes a penalty that increases with the number of estimated parameters, thus discouraging overfitting. For a set of candidate models, the model with the minimum AIC value is preferred. Typically, models with AIC values within one or two of the minimum have substantial support, models with values within four to seven of the minimum have considerably less support, and models with values greater than ten above the minimum have essentially no support (Burnham and Anderson, 2002).
Table 3 presents the OR, permutation test p-value, and AIC value for all three sets of models run for each polymorphism. The unadjusted results are repeated from Table 2 for comparison purposes. The mean AIC for the unadjusted models was 197.13, with a high of 199.26 for D578N and a low of 191.17 for A70V. Upon adjustment for all eight populations identified previously (LaCross et al., 2013), the majority (ten of fifteen) of the HemR polymorphism – otitis media associations lost statistical significance. Four of those remaining (R8H, A70V, Y405F and the I659V quartet) retained significance at the 95% confidence level, and D578N was significant at the 90% confidence level. The average AIC for these models was considerably lower (188.78) than the AIC values for the unadjusted models, indicating that the models adjusted for all populations were a better fit. Every polymorphism – disease association adjusted for all populations had a lower AIC than its unadjusted counterpart, albeit only slightly so for A70V.
Table 3.
Association between HemR polymorphisms and otitis media, adjusted for population structure.
| Unadjusted | All Populatinsa | AIC Populationsb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| OR | p-value | AIC | OR | p-value | AIC | OR | p-value | AIC | |
|
|
|
|
|||||||
| L4I/F | 3.52 | 0.02 | 197.11 | 1.50 | 0.26 | 189.54 | 1.35 | 0.31 | 183.30 |
| R8H | 0.43 | 0.04 | 198.74 | 4.50 | <0.01 | 187.36 | 5.01 | <0.01 | 180.47 |
| L15F | 4.03 | 0.00 | 192.69 | 1.74 | 0.14 | 188.99 | 1.93 | 0.09 | 181.79 |
| V16I | 3.14 | 0.03 | 198.23 | 2.00 | 0.14 | 188.79 | 2.25 | 0.10 | 181.54 |
| A70V | 0.13 | <0.01 | 191.17 | 0.37 | 0.05 | 189.31 | 0.11 | <0.01 | 180.95 |
| R131K | 3.97 | 0.02 | 196.86 | 1.42 | 0.30 | 189.62 | 1.43 | 0.29 | 183.25 |
| K157R | 3.14 | 0.03 | 198.23 | 1.01 | 0.50 | 189.80 | 1.11 | 0.43 | 183.49 |
| Q164K | 3.54 | 0.06 | 198.95 | 0.50 | 0.21 | 189.34 | 0.74 | 0.36 | 183.41 |
| N303H/R | 2.90 | 0.03 | 198.33 | 1.15 | 0.41 | 189.76 | 1.10 | 0.43 | 183.49 |
| T324I/E | 3.06 | 0.07 | 199.15 | 1.19 | 0.41 | 189.76 | 0.89 | 0.44 | 183.49 |
| Y405F | 5.38 | 0.02 | 197.27 | 5.34 | 0.03 | 186.28 | 3.92 | 0.05 | 180.29 |
| D578N | 2.77 | 0.07 | 199.26| | 2.42 | 0.09 | 187.67 | 2.37 | 0.09 | 181.34 |
| P589I | 2.09 | 0.02 | 197.73 | 1.60 | 0.12 | 189.04 | 1.02 | 0.49 | 183.51 |
| I659V | |||||||||
| N663Y | 2.58 | 0.01 | 195.78 | 2.27 | 0.03 | 187.07 | 1.75 | 0.08 | 181.52 |
| F664L | |||||||||
| A666V | |||||||||
| I718V | 2.18 | 0.02 | 197.44 | 1.40 | 0.21 | 189.32 | 1.46 | 0.16 | 182.60 |
|
| |||||||||
| Average AIC | 197.13 | 188.78 | 182.30 | ||||||
All p-values were calculated by permutation tests, permuting the polymorphism data labels.
Adjustment for all eight populations. Associations significant at the 90% confidence level are shaded.
Adjustment for the populations that minimized the AIC. These were populations 2 and 8 for A70V, and populations 2, 7, and 8 for all other polymorphisms. Associations significant at the 90% confidence level are shaded.
Models with various combinations of populations as covariates were run until the model with the minimum AIC was found. With only a single exception, adjusting for populations 2, 7, and 8 resulted in the lowest AIC value. For A70V, the model that minimized the AIC was adjusted for populations 2 and 8. The strong collinearity between that polymorphism and population 7 may explain the difference (18 of 20 isolates with a valine at position 70 have greater than 99% of their ancestry from population 7, and one further claims a third of its ancestry from population 7). The five polymorphisms that were statistically significant at the 90% confidence level remained so after adjusting for all populations, and a further two (L15F and V16I) joined their ranks. If all null hypotheses are true (i.e. no polymorphism is truly associated with otitis media), we would expect 10% of all comparisons (or 1.5) to yield p-values below the α=0.10 level by chance, considerably fewer than the number empirically determined. The average AIC for this set of models was 182.30, which was 6.48 lower than the mean AIC when adjusting for all populations and 14.83 lower than the mean unadjusted AIC. This suggests that controlling for all eight populations resulted in moderate overfitting, and that the lowest amount of information loss and the best fit was typically obtained by adjusting only for populations 2, 7, and 8.
The seven polymorphisms that remained statistically significant after adjustment for those populations that minimized the AIC were not limited to particular structural or functional domains. R8H, L15F, and V16I are all located within the putative signal peptide, the A70V polymorphism is situated near the extracellular side of the plug domain, Y405F and D578N are positioned within different extracellular loops, and the four polymorphisms beginning with I659V are immediately adjacent to a conserved intracellular loop. These results demonstrate the importance of assessing and controlling for the confounding effects of population structure in association studies, as well as again implicating HemR, and various alleles thereof, in NTHi virulence.
4. Discussion
Iron is an essential element for life in virtually all organisms, including H. influenzae. Hi has the additional absolute requirement of heme for aerobic growth, as it is unable to synthesize the heme precursor protoporphyrin IX (White and Granick, 1963). However, due to the instability of ferrous iron (the biologically relevant form) in aerobic environments and to protect against infection by iron-dependent pathogenic pathogens, nearly all iron in the human body is sequestered by high affinity iron binding proteins, such as transferrin, lactoferrin, and haptoglobin, or by incorporation within molecules such as heme (Krewulak and Vogel, 2008, 2011; Tong and Guo, 2009). In response, bacteria have developed numerous, often redundant mechanisms for acquiring iron that has been sequestered by their human hosts.
Nontypeable H. influenzae are no exception, and have a variable number of systems to scavenge iron and heme from many different host sources, including transferrin, hemoglobin, heme, heme:hemopexin, heme:albumin, hemoglobin:haptoglobin complexes, and siderophores produced by other microorganisms (Morton et al., 2010; Whitby et al., 2009). A number of these systems have been implicated in Hi virulence. Using a chinchilla model of otitis media and 5-and 30-day-old rat models of bacteremia, isogenic strains lacking certain iron acquisition genes have lower pathogenicity. These studies have found significant differences in virulence using the hemoglobin:haptoglobin binding proteins (hgpA-C) (Morton et al., 2004), the heme binding lipoprotein (hbpA) (Morton et al., 2009), lipoprotein e (P4) (hel) (Morton et al., 2007b), and the heme:hemopexin utilization proteins (hxuCBA). Some iron acquisition systems have also been found at different frequencies in OM and commensal NTHi isolates. In a 2006 study, Xie et al. found by dot blot hybridization that hgpB was 1.36 times more prevalent among OM NTHi isolates than among commensal NTHi isolates (Xie et al., 2006). Together, these data present a convincing argument that the presence of genes for the acquisition of iron and heme from a variety of sources is important not just for growth in H. influenzae, but pathogenesis as well.
HemR in NTHi appears to be a member of a common class of TonB dependent, ligand-gated channels formed by a monomeric, 22 strand, anti-parallel beta-barrel. Many highly specific, high affinity outer membrane receptors fall into this category, including many iron acquisition proteins such as the heme:hemopexin binding protein C (HxuC) and the transferrin binding protein 1 (Tbp1) in Hi (predicted structures; data not shown). Typically, the N-terminal 150 – 200 residues form a plug domain in the periplasmic end of the beta-barrel, blocking diffusion through the receptor. Ligand bound to the plug's extracellular domains combined with TonB bound to the periplasmic side induces a conformational alteration of the channel, allowing passage of the ligand. Given the predicted similarity between the structures of HemR and known TonB dependent receptors, it is likely that HemR functions in the same manner.
TonB dependent receptors also have two relatively conserved domains, one at the N-terminus and the other at the C-terminus. The N-terminal domain, known as the TonB box, is involved in the interaction with TonB. PROSITE (http://prosite.expasy.org) lists the TonB box pattern as <x(10,115)-[DENF]- [ST]- [LIVMF]- [LIVSTEQ]-V-[AGPN]- [AGP]-[STANEQPK]; a key feature appears to be the invariant valine in the midst of the domain (PROSITE accession number PS00430). There is a short sequence in HemR beginning 37 residues from the N-terminus (LPIIVNTN) that is a partial match with the TonB box pattern (five of eight residues, including the invariant valine). This may be the equivalent domain for this protein in NTHi. The C-terminus domain (PROSITE accession number PS01156) is marked by two invariant residues; the HemR sequence in this region matches this pattern exactly.
A total of 47 unique HemR amino acid sequences (or ‘types’) were identified among the 146 NTHi isolates used in the study. Most isolates with identical STs also had identical HemR sequences (89.5%), and the majority of STs that shared a HemR sequence had a large proportion of their ancestry from the same population, based on population structure analyses of MLST sequences. This is suggestive of two scenarios. In the simplest, the isolates within a population are more related and thus are more likely to have identical sequences at other loci. However, this fails to explain why isolates within HemR type 4, for example, have a wide range of ancestry proportions from population 2, from over 99% to just 50% (Supplemental Table 2). Some of these isolates are, in fact, not very closely related (on an intraspecific scale, at any rate) despite having identical HemR sequences. Furthermore, five isolates of two STs that share HemR type 4 have the majority of their ancestry from another population altogether. Similar patterns are also evident in HemR type 8 of population 3, HemR type 12 of population 5, and HemR type 1 of population 8.
In the other scenario, there are a number of HemR sequences present within the NTHi populations, and recombination could lead to both the admixture observed in the population structure as well as different STs sharing the same HemR type. Recombination may be more frequent among members of the same population, so most STs that share a HemR type have the majority of their ancestry from the same population. This hypothesis would also explain why multiple isolates of the same ST occasionally have different HemR types. Further support for this hypothesis can be found in those STs that have a different primary population from the other STs with that HemR type (shaded in brown in Supplemental Table 2). With only a single exception, these STs acquired the second highest proportion of their ancestry from the most common primary population for that HemR type. For example, in HemR type 4 the majority of isolates have the highest membership proportion in population 2. There are five isolates in that same HemR type whose primary population is 5, but they also have significant membership in population 2. This could be the result of one or more recombination events that brought in sizeable amounts of genetic material from population 2, including hemR. The truth, most likely, is a combination of the two hypotheses presented above.
The predicted structure of the NTHi HemR protein was remarkably similar to the experimentally derived structures of TonB dependent receptors involved in iron acquisition from other gram negative bacteria, despite low amino acid sequence identity. A beta-barrel consisting of 22 anti-parallel β-strands composes a transmembrane channel that is occluded by an approximately 150 residue N-terminal plug domain, preventing the free diffusion of small molecules through the channel. In other systems, TonB interacts preferentially with ligand-bound receptors, inducing conformational changes in the plug domain that allow passage of the ligand through the receptor channel and into the periplasm. Even in these well studied systems, exactly how the interaction between TonB and ligand-bound receptors results in translocation of the ligand across the outer membrane is unclear (Krewulak and Vogel, 2011). It is likely that NTHi HemR operates in a similar fashion, with TonB harnessing the proton motive force of the cytoplasmic membrane to provide the energy necessary for hemin translocation.
Prior to adjusting for population structure, 18 amino acid polymorphisms were found to have statistically significant prevalence differences between the OM and commensal isolates. Four of these polymorphisms (I659V, N663Y, F664L, and A666V) were in close proximity to each other and always co-occurred. As such, they were treated as a single entity, leaving a total of 15 polymorphisms significant at the 90% confidence level. Most methods of adjusting for multiple comparisons reduce the frequency of type I errors (incorrectly rejecting a true null hypothesis) at the expense of increasing the risk of type II errors (erroneously not rejecting a false null hypothesis). Rather than adjust for the number of comparisons, their effect was assessed by reporting all comparisons and contrasting the expected number of type I errors with the experimentally derived number of p-values below the α=0.10 level. If all null hypotheses are true (i.e. no polymorphisms are truly associated with otitis media), 10% of all comparisons would be expected to yield p-values below the α=0.10 level by chance alone (i.e., 10% of all comparisons would result in type I errors). As this was an exploratory and descriptive study, it was felt that identifying potential associations between polymorphisms and disease was more important than minimizing the potential for type I errors.
The HemR polymorphisms were not limited to a particular structural or functional domain, but were instead dispersed throughout the HemR sequence in the putative signal peptide, the N-terminal plug domain, transmembrane β-strands, and extracellular loops. After the confounding effect of population structure was adjusted for, over half of the HemR polymorphism – otitis media associations identified during the crude analysis lost statistical significance. This is clear evidence that the assessment and control of population structure is just as important for association studies in bacteria as it is in human studies.
As mentioned previously, four of the polymorphisms (I659V, N663Y, F664L, and A666V) are very closely spaced and always co-occur. While this may be the result of a common lineage between the isolates that contain the polymorphisms, these isolates derive a wide range of their ancestry (from greater than 90% to less than 50%) from at least five of the eight identified populations, suggesting little commonality among them. It seems more likely that this group of polymorphisms has spread via recombination (alternatively, the consensus amino acids could be the relative newcomers that have proliferated). It is also possible that the co-occurrence of these polymorphisms is required for the proper folding or function of HemR (i.e. if one changes, all must change for the receptor to operate adequately). If this is the case, it is conceivable that isolates with only a subset of the four polymorphisms have a low or nonfunctioning HemR and are more likely to lose the gene entirely as the cost of producing the protein would not be offset by a beneficial gain in hemin acquisition. This could leave primarily isolates with either all four polymorphisms or none of them, as was observed in this study.
There is only one polymorphism in which the alternate amino acid is more common among OM isolates than among commensal isolates, A70V, exposing a strong trend toward the consensus amino acids among NTHi isolates associated with otitis media. Interestingly, this polymorphism seems to be associated with population 7, which is composed solely of OM isolates of ST57. Twenty isolates have a valine at position 70, of which 18 are the ST57 isolates and one further has a third of its ancestry from population 7. It is possible that the statistical significance of the association between otitis media and this polymorphism is the result of having multiple genotypically identical isolates within the study, analogous to the identification of the ST57 isolates as a distinct subpopulation by LaCross et al. (LaCross et al., 2013). Even if that is the case, however, the A70V polymorphism may still be involved in virulence. All 18 ST57 isolates were collected from the middle ears of children with otitis media from all three geographic regions in roughly equal proportions. Despite being by far the most commonly identified ST among the 170 NTHi isolates, it was completely unrepresented in the commensal isolates. This would appear to be strong evidence for heightened virulence in this genotype, and it is possible that the A70V HemR polymorphism plays a role, possibly by leading to enhanced acquisition of heme by the bacteria while in the middle ear.
While a number of HemR amino acid polymorphisms were found to be significantly associated with otitis media after adjusting for population structure, no host or environmental factors were considered in the analyses, an important limitation of this study. A number of host and environmental factors have been implicated in NTHi pathogenesis, including Eustachian tube dysfunction, preceding viral respiratory infection, allergies, exposure to cigarette smoke, and attending a daycare center (Berman, 1995; Bhetwal and McConaghy, 2007; Hardy et al., 2003). These factors, and others as yet undescribed, could influence the way bacterial virulence factors such as HemR affect pathogenesis. Most association studies on bacterial virulence factors do not consider host or environmental factors, frequently because such information is simply not available, as is the case here. The bacterial strains utilized in such analyses are often originally isolated for other purposes and host and environment data are not collected. In other cases, privacy concerns or the cost associated with collecting such information can be significant barriers. Nevertheless, association studies linking bacterial factors to disease have yielded a great deal of invaluable knowledge, and provide an effective starting point for future studies to incorporate the effects of bacterial, host, and environmental factors into a cohesive whole.
Supplementary Material
Acknowledgments
This work was supported with funding from the Molecular Mechanisms of Microbial Pathogenesis Training Program (NCL) (AI007528), and the National Institutes of Health (JRG) (R01-DC05840 and R01-AI125630).
We wish to thank Drs. Noah Rosenberg and Paul Verdu for their valuable advice, and Mayuri Patel for her excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Berman S. Otitis media in children. N Engl J Med. 1995;332:1560–1565. doi: 10.1056/NEJM199506083322307. [DOI] [PubMed] [Google Scholar]
- Bhetwal N, McConaghy JR. The evaluation and treatment of children with acute otitis media. Prim Care. 2007;34:59–70. doi: 10.1016/j.pop.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Bou R, Dominguez A, Fontanals D, Sanfeliu I, Pons I, Renau J, Pineda V, Lobera E, Latorre C, Majo M, Salleras L. Prevalence of Haemophilus influenzae pharyngeal carriers in the school population of Catalonia. Working Group on invasive disease caused by Haemophilus influenzae. Eur J Epidemiol. 2000;16:521–526. doi: 10.1023/a:1007632122570. [DOI] [PubMed] [Google Scholar]
- Braun V, Endriss F. Energy-coupled outer membrane transport proteins and regulatory proteins. Biometals. 2007;20:219–231. doi: 10.1007/s10534-006-9072-5. [DOI] [PubMed] [Google Scholar]
- Buchanan SK, Smith BS, Venkatramani L, Xia D, Esser L, Palnitkar M, Chakraborty R, van der Helm D, Deisenhofer J. Crystal structure of the outer membrane active transporter FepA from Escherichia coli. Nat Struct Biol. 1999;6:56–63. doi: 10.1038/4931. [DOI] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR. Model selection and multimodel inference: a practical information-theoretic approach. 2nd. Springer; New York: 2002. [Google Scholar]
- Casey JR, Kaur R, Friedel VC, Pichichero ME. Acute otitis media otopathogens during 2008 to 2010 in Rochester, new york. Pediatr Infect Dis J. 2013;32:805–809. doi: 10.1097/INF.0b013e31828d9acc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R, Lemke EA, Cao Z, Klebba PE, van der Helm D. Identification and mutational studies of conserved amino acids in the outer membrane receptor protein, FepA, which affect transport but not binding of ferric-enterobactin in Escherichia coli. Biometals. 2003;16:507–518. doi: 10.1023/a:1023485632520. [DOI] [PubMed] [Google Scholar]
- Dean AG, Sullivan KM, Soe MM. OpenEpi: open source epidemiologic statistics for public health, version 2.3.1 [Google Scholar]
- den Bakker HC, Didelot X, Fortes ED, Nightingale KK, Wiedmann M. Lineage specific recombination rates and microevolution in Listeria monocytogenes. BMC evolutionary biology. 2008;8:277. doi: 10.1186/1471-2148-8-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhooge I, Vaneechoutte M, Claeys G, Verschraegen G, Van Cauwenberge P. Turnover of Haemophilus influenzae isolates in otitis-prone children. Int J Pediatr Otorhinolaryngol. 2000;54:7–12. doi: 10.1016/s0165-5876(00)00321-9. [DOI] [PubMed] [Google Scholar]
- Ecevit IZ, McCrea KW, Pettigrew MM, Sen A, Marrs CF, Gilsdorf JR. Prevalence of the hifBC, hmw1A, hmw2A, hmwC, and hia Genes in Haemophilus influenzae Isolates. J Clin Microbiol. 2004;42:3065–3072. doi: 10.1128/JCM.42.7.3065-3072.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin AL, Sandstedt SA, Bonthuis PJ, Geelhood JL, Nelson KL, Unrath WC, Diggle MA, Theodore MJ, Pleatman CR, Mothershed EA, Sacchi CT, Mayer LW, Gilsdorf JR, Smith AL. Analysis of genetic relatedness of Haemophilus influenzae isolates by multilocus sequence typing. J Bacteriol. 2008;190:1473–1483. doi: 10.1128/JB.01207-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Bowden R. Genome-wide association mapping in bacteria? Trends Microbiol. 2006;14:353–355. doi: 10.1016/j.tim.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, Yamaoka Y, Megraud F, Otto K, Reichard U, Katzowitsch E, Wang X, Achtman M, Suerbaum S. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–1585. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- Farjo RS, Foxman B, Patel MJ, Zhang L, Pettigrew MM, McCoy SI, Marrs CF, Gilsdorf JR. Diversity and sharing of Haemophilus influenzae strains colonizing healthy children attending day-care centers. Pediatr Infect Dis J. 2004;23:41–46. doi: 10.1097/01.inf.0000106981.89572.d1. [DOI] [PubMed] [Google Scholar]
- Feise RJ. Do multiple outcome measures require p-value adjustment? BMC Med Res Methodol. 2002;2:8. doi: 10.1186/1471-2288-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy GG, Tudor SM, St Geme JW., 3rd . The pathogenesis of disease due to nontypeable Haemophilus influenzae. In: Herbert MA, Hood DW, Moxon ER, editors. Haemophilus influenzae Protocols. Humana Press; Totowa, NJ: 2003. pp. 1–28. [DOI] [PubMed] [Google Scholar]
- Jarjanazi H, Savas S, Pabalan N, Dennis JW, Ozcelik H. Biological implications of SNPs in signal peptide domains of human proteins. Proteins. 2008;70:394–403. doi: 10.1002/prot.21548. [DOI] [PubMed] [Google Scholar]
- Juliao PC, Marrs CF, Xie J, Gilsdorf JR. Histidine auxotrophy in commensal and disease-causing nontypeable Haemophilus influenzae. J Bacteriol. 2007;189:4994–5001. doi: 10.1128/JB.00146-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian M. Genus Haemophilus. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. 2. Springer-Verlag; New York: 2005. pp. 883–904. [Google Scholar]
- Krewulak KD, Vogel HJ. Structural biology of bacterial iron uptake. Biochim Biophys Acta. 2008;1778:1781–1804. doi: 10.1016/j.bbamem.2007.07.026. [DOI] [PubMed] [Google Scholar]
- Krewulak KD, Vogel HJ. TonB or not TonB: is that the question? Biochem Cell Biol. 2011;89:87–97. doi: 10.1139/o10-141. [DOI] [PubMed] [Google Scholar]
- Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- LaCross NC, Marrs CF, Gilsdorf JR. Population structure in nontypeable Haemophilus influenzae. Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases. 2013;14:125–136. doi: 10.1016/j.meegid.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCross NC, Marrs CF, Patel M, Sandstedt SA, Gilsdorf JR. High Genetic Diversity of Nontypeable Haemophilus influenzae Among Two Children Attending a Daycare Center. J Clin Microbiol. 2008;46:3817–3821. doi: 10.1128/JCM.00940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc I, Banks KE, Fortney KR, Patterson KB, Billings SD, Katz BP, Spinola SM, Elkins C. Evaluation of the repertoire of the TonB-dependent Receptors of Haemophilus ducreyi for their role in virulence in humans. J Infect Dis. 2008;197:1103–1109. doi: 10.1086/586901. [DOI] [PubMed] [Google Scholar]
- Morton DJ, Bakaletz LO, Jurcisek JA, VanWagoner TM, Seale TW, Whitby PW, Stull TL. Reduced severity of middle ear infection caused by nontypeable Haemophilus influenzae lacking the hemoglobin/hemoglobin-haptoglobin binding proteins (Hgp) in a chinchilla model of otitis media. Microb Pathog. 2004;36:25–33. doi: 10.1016/j.micpath.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Morton DJ, Seale TW, Bakaletz LO, Jurcisek JA, Smith A, Vanwagoner TM, Whitby PW, Stull TL. The heme-binding protein (HbpA) of Haemophilus influenzae as a virulence determinant. Int J Med Microbiol. 2009 doi: 10.1016/j.ijmm.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton DJ, Seale TW, Madore LL, VanWagoner TM, Whitby PW, Stull TL. The haem-haemopexin utilization gene cluster (hxuCBA) as a virulence factor of Haemophilus influenzae. Microbiology. 2007a;153:215–224. doi: 10.1099/mic.0.2006/000190-0. [DOI] [PubMed] [Google Scholar]
- Morton DJ, Smith A, VanWagoner TM, Seale TW, Whitby PW, Stull TL. Lipoprotein e (P4) of Haemophilus influenzae: role in heme utilization and pathogenesis. Microbes Infect. 2007b;9:932–939. doi: 10.1016/j.micinf.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton DJ, Turman EJ, Hensley PD, VanWagoner TM, Seale TW, Whitby PW, Stull TL. Identification of a siderophore utilization locus in nontypeable Haemophilus influenzae. BMC Microbiol. 2010;10:113. doi: 10.1186/1471-2180-10-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TF, Sethi S, Klingman KL, Brueggemann AB, Doern GV. Simultaneous respiratory tract colonization by multiple strains of nontypeable Haemophilus influenzae in chronic obstructive pulmonary disease: implications for antibiotic therapy. J Infect Dis. 1999;180:404–409. doi: 10.1086/314870. [DOI] [PubMed] [Google Scholar]
- Musser JM, Barenkamp SJ, Granoff DM, Selander RK. Genetic relationships of serologically nontypable and serotype b strains of Haemophilus influenzae. Infect Immun. 1986;52:183–191. doi: 10.1128/iai.52.1.183-191.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musser JM, Kroll JS, Granoff DM, Moxon ER, Brodeur BR, Campos J, Dabernat H, Frederiksen W, Hamel J, Hammond G, Hoiby EA, Jonsdottir KE, Kabeer M, Kallings I, Koornhof HJ, Law B, Li KI, Montgomery J, Pattison PE, Piffaretti J, Takala AK, Thong ML, Wall RA, Ward JI, Selander RK. Global genetic structure and molecular epidemiology of encapsulated Haemophilus influenzae. Rev Infect Dis. 1990;12:75–111. doi: 10.1093/clinids/12.1.75. [DOI] [PubMed] [Google Scholar]
- Musser JM, Kroll JS, Moxon ER, Selander RK. Clonal population structure of encapsulated Haemophilus influenzae. Infect Immun. 1988;56:1837–1845. doi: 10.1128/iai.56.8.1837-1845.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H, Krogh A. Prediction of signal peptides and signal anchors by a hidden Markov model. Proc Int Conf Intell Syst Mol Biol. 1998;6:122–130. [PubMed] [Google Scholar]
- Pawelek PD, Croteau N, Ng-Thow-Hing C, Khursigara CM, Moiseeva N, Allaire M, Coulton JW. Structure of TonB in complex with FhuA, E. coli outer membrane receptor. Science. 2006;312:1399–1402. doi: 10.1126/science.1128057. [DOI] [PubMed] [Google Scholar]
- Pettigrew MM, Foxman B, Marrs CF, Gilsdorf JR. Identification of the lipooligosaccharide biosynthesis gene lic2B as a putative virulence factor in strains of nontypeable Haemophilus influenzae that cause otitis media. Infect Immun. 2002;70:3551–3556. doi: 10.1128/IAI.70.7.3551-3556.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R_Development_Core_Team. R: a language and environment for statistical computing. 2.13.0. R Foundation for Statistical Computing; Vienna: 2011. [Google Scholar]
- Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology. 1990;1:43–46. [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher SK, Marchant CD, Loughlin AM, Bouchet V, Stevenson A, Pelton SI. Prevalence and genetic diversity of nontypeable haemophilus influenzae in the respiratory tract of infants and primary caregivers. Pediatr Infect Dis J. 2012;31:145–149. doi: 10.1097/INF.0b013e31823aaeb3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard SK, Colles F, Richardson J, Cody AJ, Elson R, Lawson A, Brick G, Meldrum R, Little CL, Owen RJ, Maiden MC, McCarthy ND. Host association of Campylobacter genotypes transcends geographic variation. Appl Environ Microbiol. 2010;76:5269–5277. doi: 10.1128/AEM.00124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM, Feil EJ, Smith NH. Population structure and evolutionary dynamics of pathogenic bacteria. Bioessays. 2000;22:1115–1122. doi: 10.1002/1521-1878(200012)22:12<1115::AID-BIES9>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- St Sauver J, Marrs CF, Foxman B, Somsel P, Madera R, Gilsdorf JR. Risk factors for otitis media and carriage of multiple strains of Haemophilus influenzae and Streptococcus pneumoniae. Emerg Infect Dis. 2000;6:622–630. doi: 10.3201/eid0606.000611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CE, Olsen B, Elkins C. Cloning and characterization of tdhA, a locus encoding a TonB-dependent heme receptor from Haemophilus ducreyi. Infect Immun. 1998;66:4254–4262. doi: 10.1128/iai.66.9.4254-4262.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Guo M. Bacterial heme-transport proteins and their heme-coordination modes. Arch Biochem Biophys. 2009;481:1–15. doi: 10.1016/j.abb.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wessel K, Rodenburg GD, Veenhoven RH, Spanjaard L, van der Ende A, Sanders EA. Nontypeable Haemophilus influenzae invasive disease in The Netherlands: a retrospective surveillance study 2001-2008. Clin Infect Dis. 2011;53:e1–7. doi: 10.1093/cid/cir268. [DOI] [PubMed] [Google Scholar]
- Whitby PW, Seale TW, Vanwagoner TM, Morton DJ, Stull TL. The iron/heme regulated genes of Haemophilus influenzae: Comparative transcriptional profiling as a tool to define the species core modulon. BMC Genomics. 2009;10:6. doi: 10.1186/1471-2164-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DC, Granick S. Hemin biosynthesis in Haemophilus. J Bacteriol. 1963;85:842–850. doi: 10.1128/jb.85.4.842-850.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Juliao PC, Gilsdorf JR, Ghosh D, Patel M, Marrs CF. Identification of new genetic regions more prevalent in nontypeable Haemophilus influenzae otitis media strains than in throat strains. J Clin Microbiol. 2006;44:4316–4325. doi: 10.1128/JCM.01331-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Xie J, Patel M, Bakhtyar A, Ehrlich GD, Ahmed A, Earl J, Marrs CF, Clemans D, Murphy TF, Gilsdorf JR. Nontypeable Haemophilus influenzae genetic islands associated with chronic pulmonary infection. PLoS One. 2012;7:e44730. doi: 10.1371/journal.pone.0044730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69(Suppl 8):108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.