

Abstract
Aneurysms-osteoarthritis syndrome (AOS) is a recently delineated autosomal dominant disorder characterized by aneurysms, dissections, and tortuosity throughout the arterial tree in association with early onset osteoarthritis, mild craniofacial features, and skeletal and cutaneous anomalies. Previous studies have demonstrated that mutations in SMAD3, a key regulator of TGF-β signal transduction, contribute to AOS. Here, we investigated a family of three generations affected by AOS. A novel SMAD3 mutation, c.266G>A (p.C89Y), was identified and cosegregated with the affected individuals in this family. Our finding expands the mutation spectrum of SMAD3 gene and further strengthens the connection between the presence of aneurysms-osteoarthritis phenotype and SMAD3 mutations, which facilitates the understanding of the genotype-phenotype correlation of AOS.
1. Introduction
Aneurysms-osteoarthritis syndrome (AOS) is an autosomal dominant disorder caused by mutation in the SMAD3 gene encoding protein that is essential for TGF-β signal transduction [1, 2]. It is characterized by the presence of aneurysms, dissections, and tortuosity throughout the arterial tree in association with mild craniofacial, skeletal and cutaneous anomalies, and early onset osteoarthritis. AOS is a recently recognized disease and genotype-phenotype correlation remains yet to be determined. Patients may present with a wide spectrum of phenotypes, ranging from very mild (isolated bifid uvula) to severe (multiple aneurysms and dissections) [3]. Despite different genes involved, clinical features of AOS significantly overlap with other multisystem disorders such as Marfan syndrome (MFS), Loeys-Dietz syndrome (LDS), or vascular Ehlers-Danlos syndrome (vEDS) [2]. However, patients with AOS often suffer from early onset osteoarthritis, a distinguished manifestation that may direct molecular analysis [2, 4]. Moreover, SMAD3 mutation carriers are more susceptible to early sudden death due to aortic dissection and/or rupture, which often occurs in a mildly dilated aorta [5].
Located on chromosome 15q22, SMAD3 spans nine exons and encodes a 425 amino acid protein comprised of two major domains, the MH1 (MAD homology 1) domain and the MH2 (MAD homology 2) domain. To date, more than 20 pathogenic SMAD3 sequence variants have been identified, including missense, nonsense, frameshift, and splice-site mutations [1–4, 6–12]. Most disease-causing mutations are clustered within exon 6 in the MH2 domain. Haploinsufficiency of SMAD3 is thought to play a critical role in the development of AOS [2]. However, augmented TGF-β signaling is observed in certain mutation carriers, which may be attributed to compensatory increase caused by functional haploinsufficiency of SMAD3 [2].
So far, AOS mutations have been only reported in Caucasian families. In the present study, we described the clinical findings from the study of a small Chinese family affected with AOS; the novel SMAD3 p.Cys89Tyr mutation was identified in the affected individuals.
2. Patients and Methods
2.1. Patients
The pedigree of a three-generation Chinese family with AOS is shown in Figure 1(a). The family exhibited an autosomal dominant inheritance pattern. Radiographic examination of the artery tree and joints was performed in family members. The study was approved by the local ethics committee. Informed written consent was obtained from all participants.
Figure 1.

Pedigree and radiologic findings. (a) Pedigree of a Chinese family with aneurysms-osteoarthritis syndrome (AOS). Round symbols indicate female; square symbols, male; fully filled symbols, AOS; unfilled symbols, unaffected; diagonal lines, deceased; arrow, proband. (b) Computed tomography angiography (CTA) of II-2 demonstrated a bilateral common iliac artery aneurysm and abdominal aorta tortuosity. (c) CTA of II-2 revealed mural thrombosis in the right common iliac artery aneurysm. (d) Magnetic resonance image (MRI) of II-2 showed marked degenerative changes of lumbar spine and narrowing of the spinal cord.
2.2. Mutation Detection
Genomic DNA was isolated from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen) using standard protocols. All nine coding exons and exon-intron boundaries of SMAD3 were amplified by polymerase chain reaction (PCR) with primers designed by the Primer3 program (Table 1). PCR products were purified and sequenced on the ABI PRISM 3730 automated sequencer (Applied Biosystems) using the BigDye terminator cycle sequencing method. Sites of variation were identified by comparison to the SMAD3 gene GenBank reference sequence (NM_005902.3).
Table 1.
Primers for PCR.
| Exon | Forward (5′-3′) | Reverse (5′-3′) | Size |
|---|---|---|---|
| 1a | AGAGTTGAGGCGAAGTTTGG | TGAGTTTCTTGACCAGGCTCTT | 252 |
| 1b | GAGGAGAAATGGTGCGAGAAG | GATCTTTGCAAATCAGAGATGGTT | 334 |
| 2 | AAATGAGGGGAGAGAGAGCTT | ACCAACACAGGAGGTAGAACTG | 488 |
| 3 | ATCGACACTGAGCCACCTCT | AGTGTTGCTATTTCCGCTTCC | 462 |
| 4 | TGGTGTGCATGTGTGATGTC | ATCGCGGTTGCTCTACAAAT | 299 |
| 5 | CAGGGTTTTCTTTCTGCTGTG | GTTCTCAAGTTTCCCCATTCC | 352 |
| 6 | ACACCCAATGACCCAGTAGC | GAATGGAGCCACCCCATA | 269 |
| 7 | GCCATTGTGTGTGAGCAAAG | TGAGTGAGCAGAAAAGGTGAGA | 292 |
| 8 | CCAGGACTTGCTTTATCCAG | TCTTTGGTCTTTCTGCTCTTG | 350 |
| 9 | TGTCACCAAAGCAGAAAAAGC | CAATGGGTTGAGTAGAGTTCCA | 302 |
2.3. Bioinformatic Analysis
The multiple SMAD3 protein sequences across species were aligned using the program ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/) [13]. The PolyPhen2 (Polymorphism Phenotyping, http://genetics.bwh.harvard.edu/pph2/) [14], SIFT (Sorting Intolerant From Tolerant, http://sift.jcvi.org/www/SIFT_enst_submit.html) [15], and MutationTaster (http://www.mutationtaster.org/) [16] programs were utilized to predict the effects of sequence variants on the function of the protein.
3. Results
The proband (II-1) is a 52-year-old male who was admitted to our department complaining of abdominal pulsatile mass. Computed tomography angiography (CTA) scan revealed a bilateral common iliac artery aneurysm and abdominal aorta tortuosity (Figure 1(b)). The right common iliac artery aneurysm was estimated to be 5.0 cm in diameter accompanied by mural thrombosis (Figure 1(c)). He also reported a ten-year history of back pain. He was subsequently referred to magnetic resonance image (MRI) evaluation, which showed marked degenerative changes of lumbar spine and narrowing of the spinal cord (Figure 1(d)). He underwent an endovascular repair of the aneurysm and the procedure was uneventful. Family history was positive for sudden death in the proband's father (I-1) at the age of 48, for which the underlying cause remained unknown. Subsequent screening of his children revealed prominent dilation of the ascending aorta in the 22-year-old son (III-1). Neither of his mother (I-2) and daughter (III-2) presented vascular anomalies at the time of evaluation.
SMAD3 screening identified a heterozygous substitution of guanine to adenine at nucleotide 266 in the coding sequence of exon 2 (c.266G>A), in individuals II-1 and III-1 (Figure 2(a)). This variant resulted in a transformation of Cysteine into Tyrosine at amino acid position 89 (p.Cys89Tyr). It was not present in the unaffected individuals of the pedigree or in the 100 controls. Furthermore, this variant was not annotated in major databases, such as the Exome Sequencing Project, 1000 Genome, and dbSNP139. The altered amino acid is highly conserved across species (Figure 2(b)). Three programs for analyzing protein functions, PolyPhen2, SIFT, and MutationTaster, predicted that the p.C89Y variants are possibly damaging and disease causing, respectively. All three different algorithm based bioinformatics programs yield a consistent result of detrimental effect of the variant, suggesting that the site (C89) plays pivotal roles in the function of SMAD3.
Figure 2.
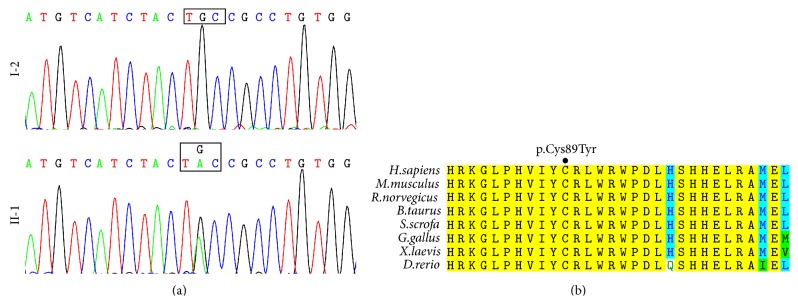
Mutation and bioinformatics analysis. (a) Sequencing results of the SMAD3 mutation. Sequence chromatogram indicates a G to A transition of nucleotide 266, resulting in a transformation of Cysteine into Tyrosine at amino acid position 89. (b) Sequence alignment of SMAD3 protein shows highly conserved amino acid Cysteine across species.
4. Discussion
To the best of our knowledge, this is the first report of a Chinese family with AOS caused by a novel SMAD3 mutation. AOS is a recently delineated autosomal dominant disorder characterized by aneurysms, dissections, and tortuosity throughout the arterial tree in association with early onset osteoarthritis, mild craniofacial features, and skeletal and cutaneous anomalies [2]. SMAD3 is the causative gene of AOS and may account for up to 2% of familial and nonfamilial thoracic aortic aneurysms and dissections (TAAD) [1]. Several lines of evidence support the notion that this variant is responsible for clinical phenotype of affected individuals in this family: (1) the sequence variant cosegregates with disease phenotype in the affected family members; (2) it is not reported in publicly available variation databases accessed in April 2015 and it is absent in ethnic-matched 200 control chromosomes; (3) the missense variant affects evolutionary highly conserved amino acid within the MH1 domain of SMAD3; (4) three computer programs which predict the possible impact of amino acid substitutions on the structure and function of human proteins (PolyPhen2, SIFT, and MutationTaster) indicate that the missense variant is pathogenic.
So far, 28 distinct exon mutations have been identified in the SMAD3 gene, including the mutation identified in this study (Table 2). The mutations spread over the entire gene with marked increased rate in exon 6. The majority of them (19 of 29) are located in the MH2 domain, which are supposed to disrupt oligomerization of SMAD3 with SMAD4- and SMAD-dependent transcriptional activation. SMAD3 mutations lead to truncated protein or substitution of highly conserved amino acids, which are predicted in silico to have a deleterious effect. Besides exon mutations, splice-site mutations and large deletion of SMAD3 gene have also been reported in AOS patients [11, 17]. With deeper understanding of the syndrome, novel mutations of SMAD3 gene will be uncovered in near future. However, how to accurately distinguish deleterious variants from neutral ones is an issue that needs to be addressed. Although functional validation is the gold standard to confirm pathogenicity, the procedure is time consuming and laborious. Different types of computational algorithms have been developed to predict the effect of amino acid substitution [18]. Three basic prediction tools utilized in this study produced consistent and reliable results, suggesting that they are useful in identifying disease associated mutations. More advanced approaches such as molecular dynamics simulation have emerged as powerful methods with high accuracy and have already shown promising results in mutation analysis of cancer, neurodegenerative disorder, cardiomyopathy disease, and so on [19–21]. Further investigation of identified SMAD3 mutations by these advanced tools is warranted to shed light on their pathogenic mechanisms.
Table 2.
Exon mutations identified in the SMAD3 gene.
| ID | Mutation | Exon | Predicted protein change | Protein domain | Reference |
|---|---|---|---|---|---|
| 1 | c.3G>A | 1 | p.Met1Ile | MH1 | [10] |
| 2 | c.266G>A | 2 | p.Cys89Tyr | MH1 | This study |
| 3 | c.313delG | 2 | p.Ala105Profs∗11 | MH1 | [3] |
| 4 | c.335C>T | 2 | p.Ala112Val | MH1 | [1] |
| 5 | c.401_405dup | 3 | p.Pro136Phefs∗52 | MH1 | [9] |
| 6 | c.539_540insC | 4 | p.Pro180Thrfs∗7 | Linker | [3] |
| 7 | c.546delT | 4 | p.Gly183Alafs∗3 | Linker | [11] |
| 8 | c.584_585insTC | 4 | p.Gln195Hisfs∗3 | Linker | [11] |
| 9 | c.652delA | 5 | p.Asn218Thrfs∗23 | Linker | [1] |
| 10 | c.668delC | 6 | p.Pro223Glnfs∗18 | Linker | [4] |
| 11 | c.715G>A | 6 | p.Glu239Lys | MH2 | [1, 8, 11] |
| 12 | c.733G>A | 6 | p.Gly245Arg | MH2 | [4] |
| 13 | c.741_742delAT | 6 | p.Thr247Profs∗61 | MH2 | [2] |
| 14 | c.742T>C | 6 | p.Phe248Leu | MH2 | [4] |
| 15 | c.782C>T | 6 | p.Thr261Ile | MH2 | [2] |
| 16 | c.788C>T | 6 | p.Pro263Leu | MH2 | [3] |
| 17 | c.836G>A | 6 | p.Arg279Lys | MH2 | [1] |
| 18 | c.859C>T | 6 | p.Arg287Trp | MH2 | [2, 11] |
| 19 | c.860G>A | 6 | p.Arg287Gln | MH2 | [4] |
| 20 | c.862_871 + 1dup | 6 | p.Arg292Aspfs∗53 | MH2 | [4] |
| 21 | c.887T>C | 7 | p.Leu296Pro | MH2 | [11] |
| 22 | c.1045G>C | 8 | p.Ala349Pro | MH2 | [3] |
| 23 | c.1080dupT | 8 | p.Glu361∗ | MH2 | [3] |
| 24 | c.1102C>T | 8 | p.Arg368∗ | MH2 | [4] |
| 25 | c.1170_1179del | 9 | p.Ser391Alafs∗7 | MH2 | [12] |
| 26 | c.1179_1180dupC | 9 | p.Cys394Leufs∗4 | MH2 | [4] |
| 27 | c.1208C>T | 9 | p.Pro403Leu | MH2 | [6] |
| 28 | c.1259G>A | 9 | p.Arg420His | MH2 | [7] |
| 29 | c.1267A>G | 9 | p.Ser423Gly | MH2 | [4] |
The SMAD3 reference sequence used was NM_005902.3, in which the A of the ATG translation initiation codon was nucleotide 1.
It is noteworthy that significant overlap of clinical features is observed between AOS and other aorta aneurysm syndromes, especially LDS (caused by mutations in TGFBR1 or TGFBR2) [5]. Similar to AOS, LDS is typically characterized by the triad of hypertelorism, cleft palate or bifid uvula, widespread arterial aneurysms, and tortuosity [22]. It is therefore challenging for clinicians to conduct specific gene screening according to aforementioned phenotype. In this study, we also performed mutation analysis of TGFBR1 and TGRBR2 in the proband. No pathologic mutation was identified in both genes. van de Laar et al. found the invariable presence of osteoarthritis at a young age in all patients with SMAD3 mutations. Indeed, symptomatic osteoarthritis was the presenting complaint for them to seek medical advice [2]. In contrast, Regalado et al. reported a marked decreased incidence of osteoarthritis in AOS individuals. The diagnosis of osteoarthritis is based on interviews and medical records rather than systematic radiological evaluation, which is assumed to be responsible for the underestimate of incidence [1]. Aubart et al. reported skeleton involvement in 100% of SMAD3 mutation carriers on X-ray or CT-scan study and recommended aorta screening in patients suffering from atypical osteoarthritis [4]. Recently, a patient with the SMAD3 mutation was demonstrated to have multiple aneurysms and rheumatoid arthritis [9]. In this study, the presence of both aneurysms and osteoarthritis prompted us to perform SMAD3 screening in this family and find a causative mutation. Our results further strengthened the connection between this special phenotype and SMAD3 mutation.
Given that mutation analysis of SMAD3 was negative for the proband's mother, the variant, p.Cys89Tyr, was likely to be inherited from his father. His father was reported to be in good health until he suffered sudden death at the age of 48. The underlying cause could not be determined since postmortem examination was not performed. We highly speculated that his father carried this mutation and died of aneurysm rupture or dissection due to SMAD3 mutation. Due to rapid aneurysmal growth and occurrence of dissections in only mildly dilated arteries, AOS is an aggressive disease with substantial mortality [5]. The estimated median survival in AOS is shorter than that in treated patients with MFS [23]. The proband developed aneurysm in the common iliac artery, which is a predilection site for aneurysm formation in AOS patients [24]. Given its aggressive behavior of AOS, early diagnosis and treatment are necessary. Endovascular repair seemed to be the first choice of treatment since it reduced the risk of major complication [24, 25]. Consistent with the observation, we performed stents implantation and achieved good results regarding the proband's treatment.
In conclusion, we identified a novel SMAD3 sequence variant, segregating with the AOS phenotype in a Chinese family. This finding expands the mutation spectrum of SMAD3 gene and highlights the importance of screening patients with aneurysms as well as early onset osteoarthritis for SMAD3 mutation, which will facilitate identification of at-risk family members and early intervention.
Acknowledgments
The authors greatly thank the family who participated in this study. This work was supported by Nanjing Municipal Science and Technology Commission (BL2012035), Medical Science and Technology Development Foundation, Nanjing Department of Health (YKK13085), and Natural Science Foundation of Jiangsu Province, China (BK20140103).
Conflict of Interests
The authors of this paper declare that they have no conflict of interests.
References
- 1.Regalado E. S., Guo D.-C., Villamizar C., et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circulation Research. 2011;109(6):680–686. doi: 10.1161/circresaha.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van de Laar I. M. B. H., Oldenburg R. A., Pals G., et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nature Genetics. 2011;43(2):121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 3.van de Laar I. M. B. H., van der Linde D., Oei E. H. G., et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. Journal of Medical Genetics. 2012;49(1):47–57. doi: 10.1136/jmedgenet-2011-100382. [DOI] [PubMed] [Google Scholar]
- 4.Aubart M., Gobert D., Aubart-Cohen F., et al. Early-onset osteoarthritis, Charcot-Marie-Tooth like neuropathy, autoimmune features, multiple arterial aneurysms and dissections: an unrecognized and life threatening condition. PLoS ONE. 2014;9(5) doi: 10.1371/journal.pone.0096387.e96387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Linde D., van de Laar I. M. B. H., Bertoli-Avella A. M., et al. Aggressive cardiovascular phenotype of aneurysms-osteoarthritis syndrome caused by pathogenic SMAD3 variants. Journal of the American College of Cardiology. 2012;60(5):397–403. doi: 10.1016/j.jacc.2011.12.052. [DOI] [PubMed] [Google Scholar]
- 6.Martens T., Van Herzeele I., De Ryck F., et al. Multiple aneurysms in a patient with aneurysms-osteoarthritis syndrome. The Annals of Thoracic Surgery. 2013;95(1):332–335. doi: 10.1016/j.athoracsur.2012.05.085. [DOI] [PubMed] [Google Scholar]
- 7.Proost D., Vandeweyer G., Meester J. A., et al. Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Human Mutation. 2015 doi: 10.1002/humu.22802. [DOI] [PubMed] [Google Scholar]
- 8.Panesi P., Foffa I., Sabina S., Ait Ali L., Andreassi M. G. Novel TGFBR2 and known missense SMAD3 mutations: two Case reports of thoracic aortic aneurysms. The Annals of Thoracic Surgery. 2015;99(1):303–305. doi: 10.1016/j.athoracsur.2014.02.068. [DOI] [PubMed] [Google Scholar]
- 9.Berthet E., Hanna N., Giraud C., et al A case of rheumatoid arthritis associated with SMAD3 gene mutation: a new clinical entity? The Journal of Rheumatology. 2015;42(3):p. 556. doi: 10.3899/jrheum.140645. [DOI] [PubMed] [Google Scholar]
- 10.Fitzgerald K. K., Bhat A. M., Conard K., Hyland J., Pizarro C. Novel SMAD3 mutation in a patient with hypoplastic left heart syndrome with significant aortic aneurysm. Case Reports in Genetics. 2014;2014:4. doi: 10.1155/2014/591516.591516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campens L., Callewaert B., Mosquera L. M., et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities inverted question mark results of comprehensive testing in a cohort of 264 patients. Orphanet Journal of Rare Diseases. 2015;10(1):p. 9. doi: 10.1186/s13023-014-0221-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wischmeijer A., Van Laer L., Tortora G., et al. Thoracic aortic aneurysm in infancy in aneurysms-osteoarthritis syndrome due to a novel SMAD3 mutation: further delineation of the phenotype. American Journal of Medical Genetics, Part A. 2013;161(5):1028–1035. doi: 10.1002/ajmg.a.35852. [DOI] [PubMed] [Google Scholar]
- 13.Larkin M. A., Blackshields G., Brown N. P., et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 14.Adzhubei I. A., Schmidt S., Peshkin L., et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng P. C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Research. 2001;11(5):863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz J. M., Cooper D. N., Schuelke M., Seelow D. Mutationtaster2: mutation prediction for the deep-sequencing age. Nature Methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 17.Hilhorst-Hofstee Y., Scholte A. J. H. A., Rijlaarsdam M. E. B., et al. An unanticipated copy number variant of chromosome 15 disrupting SMAD3 reveals a three-generation family at serious risk for aortic dissection. Clinical Genetics. 2013;83(4):337–344. doi: 10.1111/j.1399-0004.2012.01931.x. [DOI] [PubMed] [Google Scholar]
- 18.Kumar A., Rajendran V., Sethumadhavan R., Shukla P., Tiwari S., Purohit R. Computational SNP analysis: current approaches and future prospects. Cell Biochemistry and Biophysics. 2014;68(2):233–239. doi: 10.1007/s12013-013-9705-6. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A., Rajendran V., Sethumadhavan R., Purohit R. Roadmap to determine the point mutations involved in cardiomyopathy disorder: a Bayesian approach. Gene. 2013;519(1):34–40. doi: 10.1016/j.gene.2013.01.056. [DOI] [PubMed] [Google Scholar]
- 20.Kumar A., Purohit R. Use of long term molecular dynamics simulation in predicting cancer associated SNPs. PLoS Computational Biology. 2014;10(4) doi: 10.1371/journal.pcbi.1003318.e1003318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamaraj B., Rajendran V., Sethumadhavan R., Kumar C. V., Purohit R. Mutational analysis of FUS gene and its structural and functional role in amyotrophic lateral sclerosis 6. Journal of Biomolecular Structure and Dynamics. 2015;33(4):834–844. doi: 10.1080/07391102.2014.915762. [DOI] [PubMed] [Google Scholar]
- 22.Loeys B. L., Schwarze U., Holm T., et al. Aneurysm syndromes caused by mutations in the TGF-β receptor. The New England Journal of Medicine. 2006;355(8):788–798. doi: 10.1056/nejmoa055695. [DOI] [PubMed] [Google Scholar]
- 23.Pyeritz R. E. Marfan syndrome: 30 years of research equals 30 years of additional life expectancy. Heart. 2009;95(3):173–175. doi: 10.1136/hrt.2008.160515. [DOI] [PubMed] [Google Scholar]
- 24.van der Linde D., Verhagen H. J. M., Moelker A., et al. Aneurysm-osteoarthritis syndrome with visceral and iliac artery aneurysms. Journal of Vascular Surgery. 2013;57(1):96–102. doi: 10.1016/j.jvs.2012.06.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burke C., Shalhub S., Starnes B. W. Endovascular repair of an internal mammary artery aneurysm in a patient with SMAD-3 mutation. Journal of Vascular Surgery. 2014 doi: 10.1016/j.jvs.2014.01.049. [DOI] [PubMed] [Google Scholar]