

Abstract
Epidermolysis bullosa acquisita is a rare, acquired, autoimmune subepidermal blistering disease of the skin, characterised by blisters and erosions, especially in trauma-prone sites and extensor skin surface, scarring with formation of milia, skin fragility and nail dystrophy. Epidermolysis bullosa acquisita is extremely rare in childhood and it has been reported to be frequently associated with Crohn's disease. Furthermore, autoantibodies against type VII collagen have been found in a large number of patients with Crohn's disease without epidermolysis bullosa acquisita. We report a case of a 17-year-old boy affected by Crohn's disease who presented with milia on infiltrated erythematous plaques over the back of the hands. The diagnosis of epidermolysis bullosa acquisita was confirmed by histopathology, direct and indirect immunofluorescence analysis and ELISA.
Background
Crohn's disease may be associated with a variety of skin diseases, which have been classified according to their pathogenesis into the following categories: (1) specific cutaneous manifestation or granulomatous cutaneous lesions with similar histological features of inflammatory bowel disease (IBD); (2) reactive cutaneous manifestation of IBD related to immunological mechanisms triggered by common antigens shared by gut bacteria and skin; (3) secondary skin manifestations due to complications or adverse effects of IBD treatments; and (4) dermatoses associated with IBD.1 2 Cutaneous or metastatic Crohn's disease is very rare and is characterised by the presence of granulomatous skin lesions with histopathological features similar to those of intestinal lesions.3 Reactive cutaneous manifestations related to immunological mechanisms include erythema nodosum, pyoderma gangrenosum, Sweet's syndrome, pyodermatitis vegetans and leucocytoclastic vasculitis.1 Skin lesions secondary to malabsorption induced by IBD are rare and include acrodermatitis enteropathica, pellagra and scurvy.4 Many dermatoses have been described to be associated with IBD, notably psoriasis, urticaria, lichen planus, amyloidosis, vitiligo and epidermolysis bullosa acquisita (EBA).2 3
EBA is a rare, acquired, autoimmune subepidermal blistering disease of the skin, characterised by blisters and erosions, especially in trauma-prone sites and extensor skin surfaces, scarring with formation of milia, skin fragility and nail dystrophy.5 EBA patients have tissue-bound and circulating IgG antibodies directed against type VII collagen, a major component of anchoring fibrils, responsible for holding together the dermal and epidermal compartments.6
Case presentation
We report the case of a 17-year-old Italian boy with asymptomatic lesions on the back of his hands. The patient reported that he had noted the presence of the skin manifestation 1 year before (figure 1A, B). His family history was negative for other dermatological diseases, whereas his medical history was remarkable for Crohn's disease with involvement of the distal ileum, the entire colon and rectum, with anal fistulas, treated with azathioprine, infliximab and mesalazine for more than 2 years. Moreover, the patient had a history of trauma due to a motorcycle accident that occurred 1 year prior. The physical examination showed multiple whitish-yellow papules, 1–3 mm in diameter, on well defined, oval erythematous oedematous plaques over the back of the hands. This clinical picture was consistent with formation of milia on atrophic scarring areas. The cutaneous lesions were asymptomatic, but interfered with the patient's social life. No topical or systemic medications had been used. On the second visit made after 2 months, we observed erosions in the same area as the previous lesions (figure 2).
Figure 1.
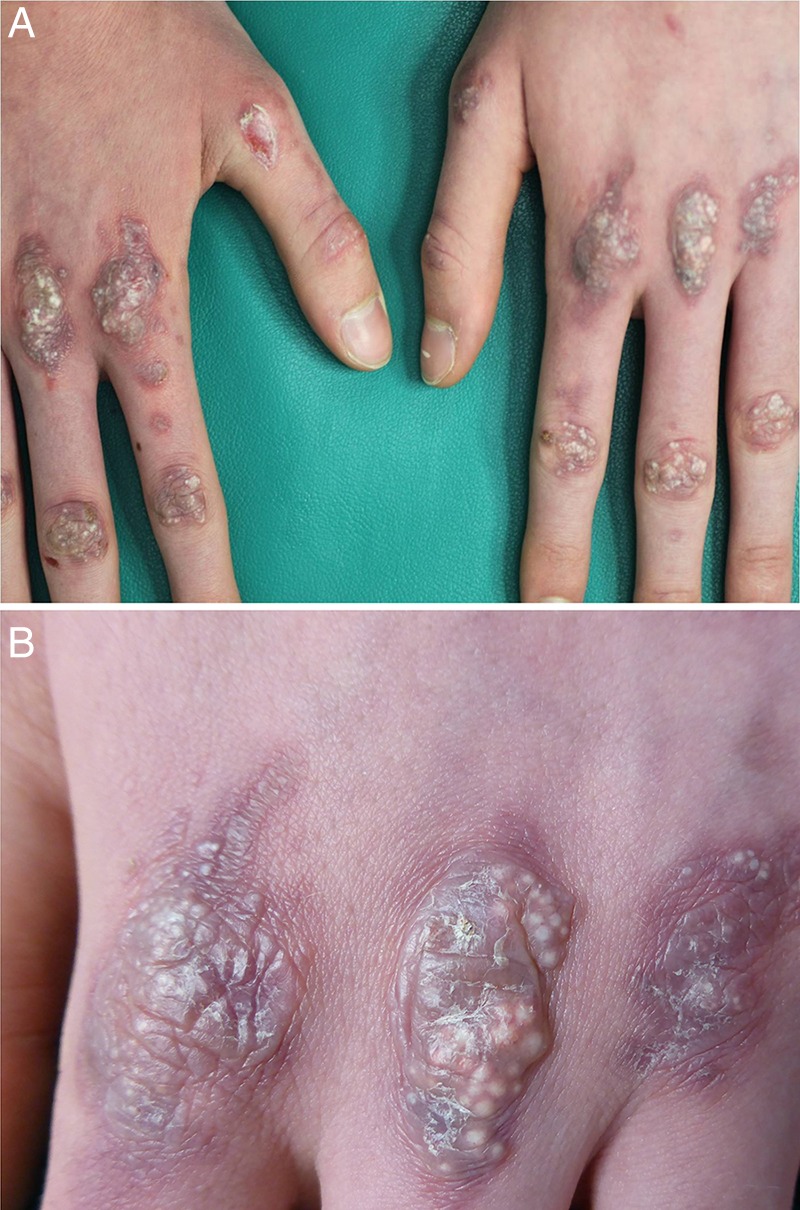
(A) Asymptomatic scarring areas on the back of the hands of a 17-year-boy appeared approximately 1 year before and (B) formation of milia on infiltrated erythematous plaques on the back of the metacarpal–phalangeal joints.
Figure 2.
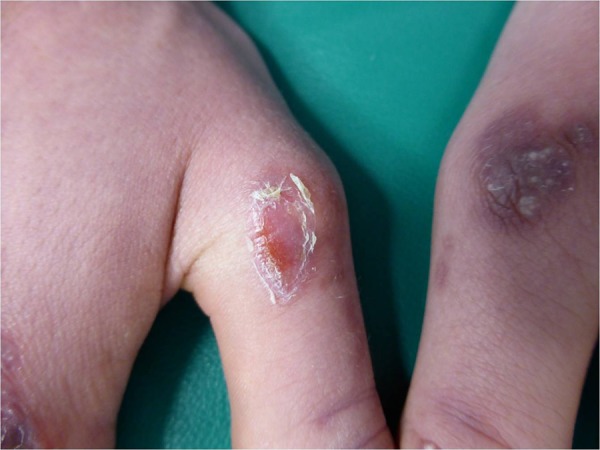
Erosion in the same areas as scars and milia.
Investigations
Complete blood cell count, erythrocyte sedimentation rate, C reactive protein level, creatine kinase level, antinuclear antibodies, antiextractable nuclear antigens antibodies, urocoproporphyrins and liver and kidney function tests were normal except for a mild anaemia (13.3 mg\dL). Nikolsky’s sign was negative. Examination of a skin biopsy specimen revealed epidermal cysts filled with keratin and subepidermal blisters with eosinophil infiltration in the superficial layer of dermis and fibrinoid necrosis of collagen. Direct immunofluorescence of perilesional skin showed a linear depositions of IgG and C3 at the dermoepidermal junction. By indirect immunofluorescence microscopy on salt split-skin, circulating autoantibodies binding on the dermal side were detected. IgG autoantibodies (40.6 U/mL) reacted with a recombinant form of collagen type VII by enzyme-linked immunoassay. On the basis of clinical, histopathological and immunological findings, the diagnosis of EBA was established.
Differential diagnosis
On the basis of the clinical appearance, we considered the following diagnosis:
Foreign body granulomas
Cutaneous calcinosis
Porphyria cutanea tarda
Gouty tophi
Cutaneous sarcoidosis
Gottron papules
Deep mycosis.
Treatment
Dapsone 50 mg daily was started. Furthermore, we suggested dermabrasion of milia and psychological support.
Outcome and follow-up
Unfortunately, after a few months of treatment, the patient stopped medical treatment, because, in his opinion, it was ineffective; he did not return to our Clinic.
Discussion
EBA is a rare autoimmune bullous disease characterised by the presence of autoantibodies directed against the non-collagenous 1 domain of the type VII collagen.5 A number of published reports suggest that EBA may be associated with various systemic diseases, notably IBD, systemic lupus erythematosus, amyloidosis, thyroiditis, multiple endocrinopathy syndrome, rheumatoid arthritis, pulmonary fibrosis, chronic lymphocytic leukaemia, thymoma, diabetes, multiple myeloma and other diseases in which an autoimmune pathogenesis has been implicated.7 EBA is very rare, with an estimated incidence of 0.25 new cases/million inhabitants/year,8 and usually affects elderly people.9 Only a few cases of childhood onset of EBA have been reported.9 10 Clinical features of childhood EBA differ somewhat from those of adult EBA.10 Children over the age of 5 are more likely to present with the classic type of EBA, whereas younger patients more frequently present with the inflammatory type.10 An association between EBA and IBD has been well documented, particularly for Crohn's disease, usually with gastrointestinal symptoms preceding the onset of blistering. In this regard, approximately 30% of patients with EBA have Crohn's disease.11 Ulcerative colitis is also associated with EBA, but with a lower frequency than Crohn's disease.12 In the majority of cases, the onset of gastrointestinal symptoms precedes EBA.12 Although these findings suggest that chronic intestinal inflammation in IBD predisposes for autoimmunity against type VII collagen, the pathogenetic mechanisms are still unclear.13 Probably exposure of type VII collagen to inflammation in the bowel creates antibodies that can recognise this antigen in its different sites. Alternatively, type VII collagen expressed in the colonic mucosa may be altered by the chronic inflammation, revealing cryptic epitopes and/or generating neoepitopes. A further possibility is that the immune-response directed against pathogens or commensal intestinal flora may be responsible for the initial activation of T and B cells, which are cross-reactive with type VII collagen epitopes.12
Furthermore, autoantibodies against type VII collagen have been found in up to 68% of patients with Crohn's disease.12 Interestingly, the isotypes of the IgG autoantibodies against type VII collagen show different distribution patterns in EBA and IBD. In EBA, there are IgG1 and IG4 subclasses, whereas in IBD, IgG3 autoantibodies are generally observed.12 14 This finding suggests that progression towards skin blistering diseases is probably associated with generation of IgG1 and IgG4 autoantibodies against type VII collagen.14 Diagnostic criteria for EBA consist of typical clinical presentation (skin fragility, trauma-induced blisters, scarring, milia and nail dystrophy), typical histopathological findings (subepidermal bullae with inflammatory infiltrate), demonstration of IgG and C3 deposits at the dermal–epidermal junction by direct immunofluorescence microscopy, IgG linear binding at the dermal side of salt-split skin by indirect immunofluorescence microscopy and detection of circulating autoantibodies against type VII collagen by ELISA.15 In our case, the patient initially presented with multiple scarring areas without any history of blisters or erosions. The classical cutaneous lesions of EBA are tense blisters and erosions over trauma-prone surfaces such as the backs of the hands, knuckles, elbows, knees, sacral areas and toes, which heal with scarring. The differential diagnosis of EBA includes porphyria cutanea tarda and pseudoporphyria cutanea tarda, which can be ruled out because our patient did not show other features of these conditions. Although bullous lesions were initially absent, pearl-like milia cysts formation within the scarring areas suggested the diagnosis of EBA. Milia are a hallmark of EBA, which should direct dermatologists towards the diagnosis of this pathological skin condition.
In our case, histopathology showed a clear separation between the epidermis and dermis, an epithelioid granuloma consistent with milia and an inflammatory infiltrate. Direct immunofluorescence demonstrated an IgG linear deposit along the dermal–epidermal junction. Finally, ELISA revealed the presence of circulating autoantibodies directly against type VII collagen, confirming the diagnosis of EBA.
Although there are no guidelines for the treatment of EBA, conventional therapy includes high potency topical steroids, oral prednisone and dapsone.16 Colchicine has also been used as an alternative therapeutic approach for EBA as single or combined therapy with corticosteroids, but it may cause significant gastrointestinal adverse effects including diarrhoea, nausea and vomiting.17
We decided to treat our patient with dapsone because of his young age and to avoid the adverse effects of colchicine, in particular, diarrhoea, in a patient with an altered bowel habit because of Crohn's disease. The use of dapsone in EBA is controversial, although there are several case reports suggesting the use of dapsone in EBA.18 The exact action mechanism of dapsone in autoimmune bullous diseases is not fully understood; it is plausible that its action is related to inhibition of neutrophils’ adherence to autoantibodies as well as interleukin 8 release.19 20
To the best of our knowledge, this is one of the few cases of EBA in a young boy affected by Crohn's disease. It confirms that EBA is frequently associated with IBD and should always be considered in the differential diagnosis of skin manifestations associated with Crohn's disease.
Learning points.
Crohn's disease may be associated with a large number of cutaneous manifestations.
The very rare cases of epidermolysis bullosa acquisita (EBA) are frequently associated with Crohn's disease.
EBA may, rarely, affect young people, and its clinical presentation differs from that of adults.
The gastrointestinal symptoms of Crohn's disease are usually observed before the onset of EBA.
EBA should be considered as a possible complication of Crohn's disease.
Footnotes
Contributors: IR, AF and IZ were responsible for conception and design, acquisition of data or analysis and interpretation of data, and drafting the article or revising it critically for important intellectual content. MA was involved in drafting the article or revising it critically for important intellectual content and final approval of the version published.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Huang BL, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front Physiol 2012;3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine JS, Burakoff R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol 2011;7:235–41. [PMC free article] [PubMed] [Google Scholar]
- 3.Georgiou G, Pasmatzi E, Monastirli A et al. Cutaneous manifestations of inflammatory bowel disease. Hosp Chron 2006;1:158–68. [Google Scholar]
- 4.Trost LB, McDonnell JK. Important cutaneous manifestations of inflammatory bowel disease. Postgrad Med J 2005;81:580–5. 10.1136/pgmj.2004.031633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woodley DT, Chang C, Saadat P et al. Evidence that anti-type VII collagen antibodies are pathogenetic and responsible for the clinical, histological, and immunological features of epidermolysis bullosa acquisita. J Invest Dermatol 2005;124:958–64. 10.1111/j.0022-202X.2005.23702.x [DOI] [PubMed] [Google Scholar]
- 6.Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol 2012;30:60–9. 10.1016/j.clindermatol.2011.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roenigk HH, Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita: report of three cases and review of all published cases. Arch Dermatol 1971;103:1–10. 10.1001/archderm.1971.04000130003001 [DOI] [PubMed] [Google Scholar]
- 8.Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol 2002;147:476–80. 10.1046/j.1365-2133.2002.04919.x [DOI] [PubMed] [Google Scholar]
- 9.Callot-Mellot C, Bodemer C, Caux F et al. Epidermolysis bullosa acquisita in childhood. Arch Dermatol 1997;133:1122–6. 10.1001/archderm.1997.03890450070008 [DOI] [PubMed] [Google Scholar]
- 10.Yang B, Wang C, Wang N. Childhood epidermolysis bullosa acquisita: report of a Chinese case. Pediatr Dermatol 2012;29:614–17. 10.1111/j.1525-1470.2011.01509.x [DOI] [PubMed] [Google Scholar]
- 11.Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol 2013;38:225–30. 10.1111/ced.12114 [DOI] [PubMed] [Google Scholar]
- 12.Chen M, O'Toole EA, Sanghavi J et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Chron's disease have autoantibodies to type VII collagen. J Invest Dermatol 2002;118:1059–64. 10.1046/j.1523-1747.2002.01772.x [DOI] [PubMed] [Google Scholar]
- 13.Hundorfean G, Neurath MF, Sitaru C. Autoimmunity against type VII collagen in inflammatory bowel disease. J Cell Mol Med 2009;14:2393–403. 10.1111/j.1582-4934.2009.00959.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mihai S, Chiriac MT, Herrero-Gonzalez JE et al. IgG4 autoantibodies induce dermal-epidermal separation. J Cell Mol Med 2007;11:1117–28. 10.1111/j.1582-4934.2007.00081.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otten JV, Hashimoto T, Hertl M et al. Molecular diagnosis in autoimmune skin blistering conditions. Curr Mol Med 2014;14:69–95. 10.2174/15665240113136660079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirtschig G, Murrel D, Wojnarowska F et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev 2003;(1):CD004056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JH, Kim SC. Epidermolysis bullosa acquisita. J Eur Acad Dermatol Venereol 2013;27:1204–13. 10.1111/jdv.12096 [DOI] [PubMed] [Google Scholar]
- 18.Arpey CJ, Elewski BE, Moritz DK et al. Childhood epidermolysis bullosa acquisita: report of three cases and review of literature. J Am Acad Dermatol 1991;24:706–14. 10.1016/0190-9622(91)70107-D [DOI] [PubMed] [Google Scholar]
- 19.Thuong Nguyen V, Kadunce DP, Hendrix JD et al. Inhibition of neutrophil adherence to antibody by dapsone: a possible therapeutic mechanism of dapsone in the treatment of IgA dermatosis. J Invest Dermatol 1993;100:349–55. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt E, Reimer S, Kruse N et al. The IL-8 release from cultured human keratinocytes, mediated byantibodies to bullous pemphigoid autoantigen 180, is inhibited by dapsone. Clin Exp Immunol 2001;124:157–62. 10.1046/j.1365-2249.2001.01503.x [DOI] [PMC free article] [PubMed] [Google Scholar]