

Abstract
Beneficial effects of intracerebral transplantation of mesenchymal stromal cells (MSC) and their derivatives are believed to be mediated mostly by factors produced by engrafted cells. However, the mesenchymal cell engraftment rate is low, and the majority of grafted cells disappear within a short post-transplantation period. Here, we hypothesize that dying transplanted cells can affect surrounding tissues by releasing their active intracellular components. To elucidate the type, amounts, and potency of these putative intracellular factors, freeze/thaw extracts of MSC or their derivatives were tested in enzyme-linked immunosorbent assays and bioassays. We found that fibroblast growth factor (FGF)2 and FGF1, but not vascular endothelial growth factor and monocyte chemoattractant protein 1 levels were high in extracts despite being low in conditioned media. Extracts induced concentration-dependent proliferation of rat cortical neural progenitor cells and human umbilical vein endothelial cells; these proliferative responses were specifically blocked by FGF2-neutralizing antibody. In the neuropoiesis assay with rat cortical cells, both MSC extracts and killed cells induced expression of nestin, but not astrocyte differentiation. However, suspensions of killed cells strongly potentiated the astrogenic effects of live MSC. In transplantation-relevant MSC injury models (peripheral blood cell-mediated cytotoxicity and high cell density plating), MSC death coincided with the release of intracellular FGF2. The data showed that MSC contain a major depot of active FGF2 that is released upon cell injury and is capable of acutely stimulating neuropoiesis and angiogenesis. We therefore propose that both dying and surviving grafted MSC contribute to tissue regeneration.
Introduction
Transplantations of mesenchymal stromal cells (MSC) and their derivatives are being proposed as a treatment for various degenerative disorders of central nervous system (CNS). The therapeutic effects of MSC transplantation into the CNS are thought to be mostly due to the secretion of soluble factors, which provide tissue protective, regenerative, and immunomodulating stimuli [1–3] from living donor cells. One of paradoxes of such an explanation is that the engraftment rates of MSC in the CNS are low [4,5]; however, therapeutic benefits have been observed to continue long after the grafted cells can no longer be detected. A variety of conflicting data have accumulated to explain the poor engraftment of transplanted MSC. While some reports implicate triggering of an innate and subsequent adaptive immune response to explain graft loss, others find similar rates of graft cell loss irrespective of human leucocyte antigen matching status [6,7]. Other studies have found that allogeneic MSC do not elicit a significant immune response (reviewed in [8]). It has also been reported that intracellularly labeled MSCs, either live or dead, transplanted into the adult brain, can transfer labels to the surrounding and distant recipient's cells, and the labels become incorporated into these cells [9,10]. This suggests that intracellular contents of the graft can be “recycled” by the surrounding tissue. How this affects the brain microenvironment in particular, and the therapeutic outcome in general, is unclear.
Fibroblast growth factor (FGF)2 is a major growth factor for stem cells, one of the most potent inducers of angiogenesis, an essential wound healing mediator, and a major player in the development and regeneration of the nervous system (reviewed in [11]). Five FGF2 isoforms are translated from a unique FGF2 mRNA by alternative translation initiation: an 18 kDa low molecular weight (LMW) isoform and high molecular weight (HMW) isoforms comprising molecular weights of 22, 22.5, 24, and 34 kDa. LMW FGF2 is mostly cytoplasmic and is secreted, while the HMW isoforms are predominantly nuclear, however, either form can be found in the nucleus, cytoplasm, or extracellular matrix (ECM) under certain conditions. All isoforms lack a signal peptide to direct secretion through the endoplasmic reticulum-golgi pathway. Early studies demonstrated that mechanically wounded monolayers of endothelial cells release high levels of FGF2 [12,13]. Based on these studies and the lack of signal peptide for secretion, cell death, or even sub-lethal injury has been described as a major mechanism for FGF2 release [14]. Accordingly, FGF2 was nominated as a “wound hormone for rapidly initiating the cell growth required for routine maintenance of tissue integrity and/or repair after injury” [15].
While many reports document the expression of FGF2 mRNA by MSC and demonstrate the presence of intracellular protein [11,12,16], very few reports provide measurements of FGF2 secretion because the concentration of secreted FGF2 is very low [17,18]. Perhaps for this reason, FGF2 has not been considered to be a primary candidate mediating the regenerative effects of implanted MSC on surrounding neural tissue.
SB623, an MSC derivative, is currently being tested in a Phase 1/2a clinical trial for safety and efficacy in chronic stroke. These cells are derived from human bone marrow MSC using transient transfection with a vector encoding the human Notch1 intracellular domain followed by G418 selection and subsequent expansion. This manufacturing process produces a cell population that demonstrates superior angiogenic and neuropoietic properties in vitro as compared to the parental MSC [19–21]. The neuropoietic effects of SB623 have been attributed to the increased expression, and correspondingly, increased secretion, of FGF1, FGF2, and bone morphogenetic proteins (BMPs) [19,22].
Herein, we hypothesize that cell injury and the subsequent release of intracellular content could be of particular relevance to the intracerebral implantation of MSC; and to explore this hypothesis, we test the type, amounts, and potency of these putative intracellular factors. Quantity of several growth factors released from either MSCs or SB623 by secretion versus mechanical cell injury was measured and compared. We demonstrated that contents released by mechanical cell injury were highly active in stimulating the proliferation of both NPC and endothelial cells. Furthermore, the mitogenic activity of these intracellular contents was shown to be due to release of intracellular FGF2. We modeled alternative, non-mechanical cell injury that could lead to MSC death after intracerebral implantation and demonstrated that substantial amounts of FGF2 are also released in these models. Finally, we showed that cocultures of mechanically injured and live mesenchymal cells work synergistically to change the differentiation of neural progenitors.
Materials and Methods
MSC and SB623 cell preparation
MSC and SB623 cell preparation and their characterization were described previously [21]. MSC and SB623 from seven donors were used in this study.
Preparation of extracts and conditioned media
MSC, SB623, or other cell lines were used for cell extract and conditioned media (CM) preparation. The other cell lines were as follows: human foreskin fibroblasts (HFF), purchased from ATCC and Cellular Engineering Technologies (CET) Inc. human embryonic cell lines ReNcell and ENStemA from EMD Millipore, and human umbilical vein endothelial cells (HUVEC) from Lonza and Life Technologies. Cryopreserved cell aliquots were thawed, washed, resuspended in basal medium for embryonic neuronal cells (NeuroBasal, NB; Life Technologies), and washed twice. These cells were used for both preparation of extracts and CM.
For preparation of extracts designated as E0, 2×106 cells were frozen in 4 mL of NB at −80°C for 1–2 h, and then thawed, resuspended in a total of 10 mL NB medium, and the suspension was cleared by centrifugation at 3,000 rpm for 15 min. The supernatants were aliquoted and stored at −80°C. For preparation of CM, 2×106 cells were plated into a T75 flask, in α-minimum essential medium (Mediatech, Inc.) supplemented with 10% fetal bovine serum (FBS; HyClone) and penicillin/streptomycin (Life Technologies) and cultured overnight. Next day, the medium was changed to NB for 1 h, then discarded and replaced with fresh 10 mL of NB for 24 h. The CM was removed, centrifuged at 3,000 rpm for 15 min, aliquoted, and stored at −80°C. The flask with the cell layer was frozen and thawed, cell remnants extracted with 10 mL NB, centrifuged, and the supernatants were aliquoted and stored for CM normalization (designated as extract E1). In preliminary experiments, it was determined that no cells survived the freeze/thaw procedure. Note that unless indicated otherwise, E0s and CMs were produced using the same proportion: 1 million cells/5 mL NB.
Rat embryonic cortical cell proliferation assay
Ninety six-well plates (Corning, Inc.) were coated with Ornithine/Fibronectin (Orn/Fn; both from Sigma Aldrich) as described in [23]. Rat embryonic E18 cortex pairs were purchased from BrainBits; and the neural cells isolated as described in Aizman et al. [23]. Assay medium consisted of NB supplemented with B27 and 0.5 mM l-alanyl-l-glutamine (GlutaMAX) (NB/B27/GLX; all from Invitrogen). Neural cells were plated at 6.7×103 cells/well and various concentrations (0%–75% range) of E0 or CM were added to triplicate wells. In some experiments, neutralizing anti-FGF2 antibody clone bFM1 (Millipore) and control Mouse IgG1 (R&D Systems) were added at 2 μg/mL. Wells containing medium, but no cells, were used as blank. Neural cells were cultured for 5 days. To quantify proliferation, 5-bromo-2-deoxyuridine (BrdU) labeling was carried out for 2 h and the plates processed using cell proliferation enzyme-linked immunosorbent assay (ELISA), BrdU (Colorimetric) from Roche Diagnostics GmbH according to the manufacturer's instructions. Standards were made by serial dilutions of the anti-BRDU reagent starting from 1:1,000. The highest standard value was arbitrarily set as 100 and results of colorimetric analyses were expressed in these units. Color development was quantified using SpectraMax Plus plate reader (Molecular Devices).
HUVEC proliferation assay
HUVEC were cultured in endothelial growth medium, EGM™, supplemented with Bovine Brain Extract (both from Lonza) and 2% FBS for 2–4 passages, aliquoted, and stored cryopreserved. For the assay, 96-well plates were coated with 40 μg/mL of Rat tail Collagen I from Life Technologies for 2 h, then aspirated, dried, and washed or stored at−20°C until use. The assay medium was Medium 199 (Life Technologies) supplemented with 0.5% FBS. HUVEC, either freshly thawed or after overnight culturing, were plated at 2.5×103/well in the presence of various dilutions of extracts and CM using NB alone as a negative control, in triplicates. In some experiments, bFM1 and the control mouse IgG1 were used (at 2 μg/mL), as well as recombinant humanVEGF165 (rVEGF; R&D Systems) and FGF2 (rFGF2; Peprotech). After 3 days of culturing, cells were labeled with BRDU for 2 h; and BRDU incorporation was quantified as described above.
ECM-based quantitative neuropoiesis assay
A neuropoiesis assay was done as described in Aizman et al. [19], using CellBIND Surface 96-well plates (Corning, Inc.) coated with MSC-derived ECM as a substrate for cell growth. Rat nestin and glial fibrillary acidic protein (GFAP), and human glyceraldehyde 3 phosphate dehydrogenase (GAP) expression were quantified using quantitative reverse transcriptase-polymerase chain reaction with TaqMan assays from Life Technologies [23].
Enzyme-linked immunosorbent assays
The following Quantikine immunoassays were purchased from R&D Systems: basic FGF, high sensitivity basic FGF, vascular endothelial growth factor (VEGF), and acidic FGF. The MCP-1 ELISA kit was purchased from Boster Biological Technology. ELISAs were performed according to manufacturers' instructions, except that in FGF2 ELISAs the samples were incubated overnight. (This gave similar results to the 2 h-incubation recommended by the manufacturer—data not shown). Optimal dilutions for FGF2 detection as determined in preliminary experiments were 1/10 for E0 and 1/2 or no dilution for CM.
Lactate dehydrogenase activity assay for cell number control
Lactate dehydrogenase (LDH) activity as a surrogate for cell number was detected in cell extracts at 1:2 and 1:4 dilutions using an LDH Cytotoxicity Detection Kit (Clontech Laboratories) and averaged. Bovine LDH (Sigma Aldrich) was used to make standards on each plate.
Cytotoxicity assays
Peripheral blood mononuclear cells (PBMC) were obtained from buffy coat preparations of whole blood, which were purchased from the Stanford Blood center. After purification using Ficoll-Paque Plus (GE Healthcare) according to the manufacturer's instruction, the lymphocyte/monocyte/platelet fraction was collected and washed using slow speed centrifugation (600 rpm for 20 min) to remove the majority of platelets. PBMC were cultured for either 1 or 7 days with or without interleukin (IL)2 (10 ng/mL). PBMC served as effectors in the cytotoxicity assays, where they were cocultured with mesenchymal cells at either 10- or 30-fold excess for 5 or 18 h. PBMC alone and MSC or SB623 alone, and the medium only served for background measurements. MSC or SB623 alone were also plated for determination of total LDH activity. All conditions were repeated five times. After culturing, the plate was centrifuged at 1,000 rpm for 5 min and 25 μL/well of supernatant were removed in three out of five replicas for measurement of LDH activity using LDH Cytotoxicity Detection Kit. The cytotoxicity was assessed as a specific release of LDH activity in effector-target cocultures and expressed as percentage of total LDH activity released by target cells lysed in 1% Triton according to the formula provided in the manufacturer's protocol. The remaining duplicates were used in the FGF2 Quantikine assay and the percent specific release of FGF2 was calculated the same way as percent specific release of LDH described above.
Model of high cell density/hypoxia/insufficient nutrients
To model the intracerebral microenvironment post-implantation, MSC or SB623 cells were plated in round-bottom wells of 96-well plate in NB/B27/GLX at 0.35×106/350 μL/well and tightly sealed with PCR tape, which prevents gas exchange. The medium became rapidly acidic indicating a hypoxic environment. The vast majority of cells remained non-adherent. The contents of the wells were harvested at various time points and centrifuged. Supernatants were collected (300 μL/well, designated here as CM), pellets were harvested into 300 μL of NB (for the release of intracellular content); and all samples were frozen. After all time points were collected, all samples were then thawed and cleared at 200 g for 10 min in an Eppendorf centrifuge. LDH activity and FGF2 concentrations were determined in the CMs and extracts. Note that in this instance, CM and extracts were generated using 1 million cells/1 mL NB proportion.
Immunofluorescence
Glass coverslips (Fisher Scientific) were coated with Orn/Fn as described above. Rat cortical cells were plated onto the coverslips at 2.5×104 cells/well in 24-well plates, in NB/B27/GLX. SB623-derived extract (20%) was added or not. Cells were grown for 7 days, and then labeled with 10 μM of BRDU (Sigma-Aldrich) for 4 h. Cultures were then fixed with 2% paraformaldehyde for 20 min, permeabilized with 0.5% TritonX100 and treated with DNase (MP Biomedicals) in the buffer containing 150 mM NaCl and 4.2 mM MgCl2 for 1 h at 37°C. The cultures were then post-fixed with cold methanol, blocked, and incubated with anti-BRDU monoclonal antibody (BD Pharmingen), and with either goat antibody to rat nestin (R&D Systems) or to doublecortin (Dcx; Santa Cruz Technologies) overnight at 4°C. Then, cultures were washed and incubated with Cy3-conjucated AffiPure donkey anti-mouse IgG and DyLight 488-conjugated AffiPure donkey anti-goat F(ab′)2 fragments (both from Jackson Immunoresearch), washed and mounted with ProLong Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (Life Technologies). Fluorescent microscopy was carried using Eclipse50i (Nikon) and a Nikon DXM1200C digital camera.
Statistical analysis
Error bars indicate standard deviations. Two sample equal variance unpaired t-test was used to calculate P-values. The difference between two values was considered statistically significant when P<0.05. No multiple comparison adjustments were made.
Results
Release of FGF2 upon mechanical cell injury versus secretion
The release of FGF2 from SB623 and their parental MSC by either secretion or mechanical cell rupture was measured. Figure 1 represents averaged results from cell lots obtained from seven donors. Cryopreserved MSC and SB623 were thawed, washed, and subjected to one freeze/thaw cycle. The released cell contents were extracted (E0 extracts); and the amount of FGF2 was quantified (Fig. 1A). On average, 3.9 and 7.2 ng of FGF2 were released from 106 MSC and SB623, respectively. One freeze/thaw cycle was sufficient to kill all the cells (as tested with Trypan blue and cell plating), while each additional freeze/thaw cycle decreased FGF2 concentration by about 20% (not shown). CM conditioned by the same number of either MSC or SB623 cells contained only∼0.02 ng of FGF2 (Fig. 1B). To control for potential differences in cell metabolic activity, LDH was measured in E0 and also in cell extracts obtained after the production of CM by plated MSC and SB623 (E1) (Fig. 1C). LDH activity differed between E0 and E1 (0.3 vs. 0.13 U/106 cells, respectively), but not between MSC and SB623, indicating that metabolic activity dropped in starving cells compared with cells ruptured after cryopreservation, but was similar between MSC and SB623. The comparison between FGF2 contents of CM and E0 indicated that FGF2 was predominantly intracellular, while very little was secreted. This difference appeared to be true for FGF1, but not for VEGF and MCP1 (Table 1).
FIG. 1.

Fibroblast growth factor (FGF)2 content of mesenchymal stromal cells (MSC) and SB623 cell extracts and conditioned media (CM). Total amounts of FGF2 in extracts (A) or CM (B) obtained from 1 million cells were calculated based on FGF2-enzyme-linked immunosorbent assay (ELISA) (A) or high sensitivity (HS)-FGF2-ELISA (B). (A) *P<0.05; No statistically significant difference in (B). (C) Lactate dehydrogenase (LDH) activity released from 1 million cells into extracts prepared from cryopreserved cells (E0) or from cells that were cultured to produce CM (E1). No statistically significant difference between MSC and SB623. Bars represent the average across seven cell lots. Error bars represent standard deviation.
Table 1.
Detection of Various Mesenchymal Stromal Cells Factors in Conditioned Media and Freeze/Thaw Extracts (E0)
| CM | E0 | Difference (E0/CM) | |||
|---|---|---|---|---|---|
| Average (ng/106 cells) | CV (%) | Average (ng/106 cells) | CV (%) | Fold | |
| FGF2 | 0.017 | 94 | 3.9 | 64 | 230 |
| FGF1 | LLD | — | 1.1 | 49 | — |
| VEGF | 0.4 | 121 | 0.002 | 23 | 0.005 |
| MCP1 | 0.003 | 85 | 0.001 | 89 | 0.3 |
Amounts of factors produced by 1 million MSC. Average measurements of seven cell lots are shown for FGF2 and of two cell lots are shown for other factors.
CM, conditioned media; FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor; LLD, lower limit of detection; CV, coefficient of variation; MSC, mesenchymal stromal cells.
FGF2 contents were also tested in other mesenchymal (HFF and HUVEC) and non-mesenchymal (neural stem cell line, ENStem-A) human cells. As shown in Table 2, HFF released more FGF2, while HUVECs and human neural stem cell lines released less FGF2, than MSC.
Table 2.
Fibroblast Growth Factor 2 Content in Extracts from Different Cell Types
| Average (ng/106 cells) | Standard deviation | n lots | |
|---|---|---|---|
| Human NPC lines | 0.5 | 0.08 | 2 (ReNcell, ENStem) |
| HUVEC | 0.7 | 0.04 | 2 |
| HFF | 8.9 | 1.1 | 2 |
| MSC | 3.9 | 2.5 | 7 |
| SB623 | 7.2 | 2.7 | 7 |
NPC, neural progenitor cells; HUVEC, human umbilical vein endothelial cells; HFF, human foreskin fibroblasts.
Cell extracts promote neural cell proliferation
Rat embryonic cortical cell populations contain a large proportion of NPC that proliferate in response to FGF2. Therefore, we stimulated rat cortical cells with dilutions of MSC-derived E0 and CM varying from 0% to 75% and conducted proliferation assays using BRDU incorporation (Fig. 2A). The proliferation was increased by E0, but not by CM in a concentration-dependent manner. The proliferative response to E0 was diminished in the presence of the neutralizing anti-FGF2 (bFM1) mAb, while the control antibody had no effect (Fig. 2B).
FIG. 2.

Proliferative response of cortical Nestin-positive cells to extracts and the role of FGF2. (A) Proliferation in response to various concentrations of extract (E0) and CM from MSC. (B) Effect of FGF2 neutralization on cell proliferation stimulated by SB623-derived E0 at either 15% or 5% (equivalent to 0.2 and 0.07 ng/mL FGF2, respectively). *P<0.05; **P<0.01. Neutralizing anti-FGF2 (clone bFM1) or isotype control (mouse IgG1) were added at 2 μg/mL. (C, D) Immunostaining of rat cortical cells stimulated or not with 20% SB623-derived extract. After 7 days, cells cultures were labeled with 5-bromo-2-deoxyuridine (BRDU) and stained for either BRDU and Doublecortin (Dcx) (C) or for BRDU and Nestin (Nes) (D). We concluded that cells predominantly labeled with BRDU and increased in numbers in extract-treated cultures were nestin-positive cells.
Besides NPC, the embryonic rat cortical cell population contains immature neurons. To identify, which subpopulation proliferated in response to extracts, the cells were cultured with or without SB623 extract for 5 days and then labeled with BRDU. The subsequent immunostaining (Fig. 2C, D) revealed that in treated cultures, there was no increase in DCX+- or BRDU+/DCX+ cells; in contrast, there was a drastic increase in NPC numbers (Nestin+ cells) and practically all of them were also BRDU+. We concluded that after 5-day culturing, NPC were the main subpopulation proliferating in response to the extracts.
Extracts promote HUVEC proliferation
FGF2 is a very potent angiogenic factor. Therefore, extracts were tested for their ability to induce proliferation of HUVEC. On day 2, MSC-E0 strongly induced proliferation of HUVEC, while MSC-CM and NB medium had no effect (Fig. 3A). In a separate experiment, HUVEC were incubated with rVEGF, rFGF2, or SB623-E0 (15%) with or without FGF2-neutralizing and control antibodies (Fig. 3B). The response to E0 was inhibited by neutralizing bFM1, but not by control mouse IgG1, indicating that HUVEC proliferation in this assay was driven by FGF2. Notably, the activity of both native and recombinant FGF2 was similar in this assay; indeed, when the background was subtracted, the response induced by 15% E0 (which corresponded to the final FGF2 concentration of 0.2 ng/mL in this E0 preparation) was approximately four times less than the response induced by 1 ng/mL rFGF2.
FIG. 3.
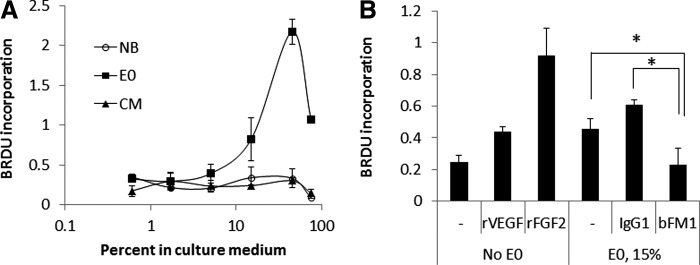
Proliferative response of human umbilical vein endothelial cells (HUVEC) to extracts and the role of FGF2. (A) HUVEC proliferation in response to various dilutions of either MSC extract (E0), CM, or neurobasal medium after 2 day-culturing. (B) Proliferative responses of HUVEC to 15% SB623-extract (E0) (final FGF2 concentration 0.2 ng/mL), rVEGF (10 ng/mL), and rFGF2 (1 ng/mL), and to FGF2-neutralization in E0. bFM1 and IgG1 are anti-FGF2 neutralizing antibody and mouse isotype control antibody, respectively, both at 2 μg/mL. *P<0.05. No statistically significant difference between E0 alone and E0+IgG1.
Neuropoiesis assay: comparison to activity of extracts and dead and live cells
The neuropoiesis assay [23] quantifies growth and differentiation of rat embryonic cortical cells promoted by small numbers of human MSC in direct cocultures. This sensitive assay was used to (i) assess the activity of extracts and (ii) compare the activity of extracts to the activity of live and dead cells. For these experiments, a working suspension of either MSC or SB623 in NB was divided into three aliquots. One aliquot was left intact; cells were alive (denoted “A”). Two aliquots were frozen and then thawed, which made them suspensions of dead cells (denoted “D”). One of these two aliquots was then cleared by centrifugation, becoming the extract (denoted “E”). All three aliquots were plated at identical dilutions, which corresponded to 500, 250, and 125 of live MSC or SB623 cells/well. Cortical cells (5,000 cells/well) were added to all wells. After culturing, expression of rat nestin, rat GFAP, and human GAP were determined (Fig. 4A–C, correspondingly). It should be noted that extracts in this experiment had 40 times lower strength than our standard extract preparations: indeed, an extract from 500 cells in 100 μL culture medium corresponded to 2.5% dilution of E0 prepared as described in the section “Materials and Methods” (106 cells/5 mL medium).
FIG. 4.

Neuropoietic activity of extracts in comparison to the activity of dead and alive cells. Neuropoiesis assay was performed with rat cortical cells. Rat cells were plated into cell-derived extracellular matrix-coated wells and incubated with SB623-derived samples. The samples were produced as following: the SB623 cell suspension was aliquoted; one aliquot was used as live cells (A, alive), another was used after freeze/thaw (D, dead), and another was cleared after freeze/thaw by centrifugation (E, extract). Numbers of human cells or their equivalent per well are indicated on X-axis. After culturing, relative expression of genes was accessed using quantitative reverse transcriptase-polymerase chain reaction: rat Nestin (A), rat glial fibrillary acidic protein (GFAP) (B), and human glyceraldehyde 3 phosphate dehydrogenase (GAP) (C) expression levels were analyzed from the same wells. In another experiment (D, E), cell suspensions of live and dead cells were mixed in indicated proportions and their effect on rat GFAP expression was compared to effects of live or dead cells alone. *P<0.005. (E) Human GAP expression was analyzed in same wells as in (D) to confirm relative numbers of live human cells.
As expected, Nestin expression was induced by extracts; however, the response was substantially lower than that elicited by the corresponding live cell suspension (Fig. 4A). The suspension of dead cells induced slightly stronger Nestin response than did the suspension of live cells. In contrast, GFAP was induced by live cells, but not by extracts, and only slightly by dead cells (Fig. 4B). The absence of human GAP expression in cultures with dead cells confirmed that there were no surviving human cells (Fig. 4C). These differences in Nestin expression can be explained by our prior observation that MSC coculture triggers incremental Nestin expression by supporting the proliferation of Nes+GFAP+ precursors [23]. While extracts are an abundant source of easily extractable cytosolic FGF2, dead cell preparations contain also cell remnants (nuclei, for example), which may further release FGF2 and provide additional stimulation of cell proliferation and Nestin expression. The extract-mediated induction of Nestin expression observed in Fig. 4A likely reflects an increase in numbers of Nes+GFAP− NPC (presumably stimulated by high levels of FGF2 in the extract); while, in contrast to cultures with live cells, differentiation and proliferation of Nes+GFAP+ precursors did not take place.
Since it is known that shortly after intracerebral implantation the majority of grafted cells die, we tested an “implantation-relevant combination”: dead (D) and live cell (A) samples were mixed, 3:1 (D/A), and this mixed sample was used in comparison with D and A samples (same total cell numbers). As expected, Nestin expression was induced similarly by all three samples (data not shown). The GFAP induction driven by D/A sample, however, showed a synergy between corresponding A and D samples (Fig. 4D). This was not caused by excessive presence of live human cells in mixed D/A preparation as demonstrated by similar human GAP levels (Fig. 4E). This synergy was observed with both MSC- or SB623-derived samples. Thus, live MSC or SB623, their extracts, or cell suspensions of cells killed by a single freeze/thaw cycle could all promote neural precursor growth; while robust astrogenesis required the presence of live MSC or SB623.
FGF2 is released in cytotoxic injury mediated by PBMC
The data above suggested that MSC or SB623 contain large amounts of intracellular FGF2, and that this FGF2 is biologically active when released by a membrane rupture due to a freeze/thaw procedure. However, it was unclear whether mesenchymal cells can release substantial amounts of intracellular FGF2 due to other types of cell injuries that may be incurred naturally following intracerebral implantation.
When cells are injected into the brain, damage to small brain vessels may occur. This disruption of the vasculature may result in exposure of the implanted cells to local PBMC and subsequent cytotoxic effects. Preliminary experiments were conducted to establish in vitro conditions, in which PBMC cytotoxicity toward MSC could be observed. Although there was high degree of variability in the cytotoxic effect of PBMC with different MSC or SB623 lots, we observed that in all assays, cytotoxicity was stronger at 18 h than at 5 h post exposure and also greater when PBMCs were pre-cultured for 7 days, as opposed to 1 day. The presence of IL2 in PBMC cultures did not affect their cytotoxicity. Therefore, PBMC-mediated cell lysis and target cell release of FGF2 were assessed in 18 h-cocultures of PBMCs (pre-cultured for 7 days without IL2) and target cells (either MSC or SB623). An example of these data is shown in Fig. 5A. The specific cell lysis was directly dependent on PBMC:target cell ratios and varied from 30% to 90% at 30:1 PBMC/target cell ratio for different donors of MSC, SB623, and PBMC. Specific FGF2 release was expressed the same way as was the specific cell lysis. The percentage of specific FGF2 release was lower than that of LDH by about 2–2.5 times, but nevertheless correlated well with the specific cell lysis. Although in this experiment specific lysis in MSC-PBMC cocultures was higher than that in SB623-PBMC cocultures, more FGF2 was released in latter case, due to higher intracellular levels in SB623 (Fig. 5B). This together with higher spontaneous release of FGF2 from SB623 cells in the absence of PBMC lead to approximately equal specific FGF2 release for MSC- and SB623-PBMC cocultures. The result showed that substantial amounts of FGF2 can be released by both MSC and SB623 as a result of the cytotoxic effects of PBMC.
FIG. 5.
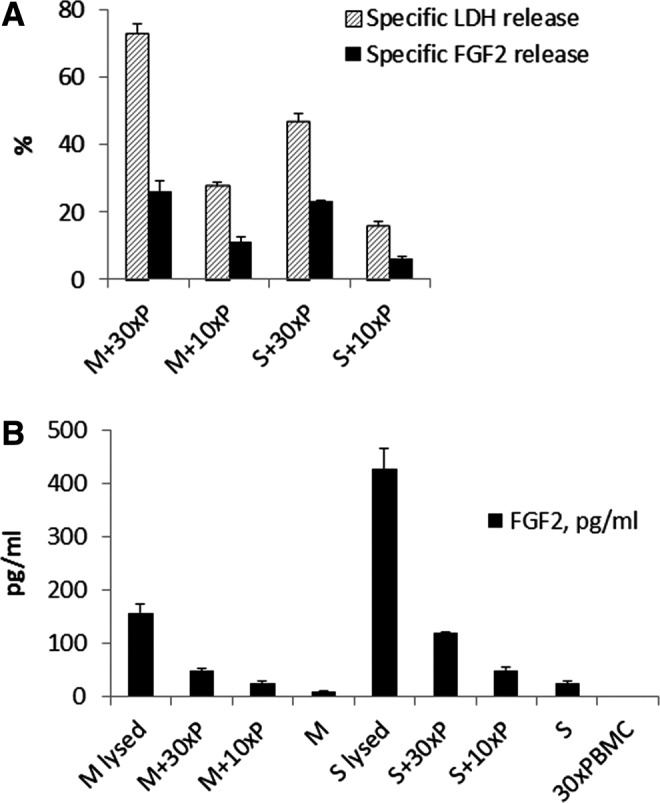
Peripheral blood mononuclear cells (PBMC)-mediated cytotoxic injury of MSC and SB623, and FGF2 release in cocultures. (A) MSC (M) or SB623 (S) were cocultured with 10- or 30-fold excess of PBMC (10×P or 30×P, correspondingly). Release of LDH and FGF2 into the culture medium by these cocultures or by separately plated mesenchymal cells or PBMC was measured. Calculations of specific LDH release (ie, specific cell lysis) and specific FGF2 release were done the same way. (B) FGF2 concentrations measured for calculating specific FGF2 release (in A) are shown.
FGF2 is released in high cell density/hypoxia/insufficient nutrients cultures
When MSC or their derivatives are transplanted into a peri-infarct zone, they are deposited at high density into a hostile environment with limited diffusion of oxygen and nutrients. To model this environment in vitro, MSCs or SB623 cells were plated at very high density in round-bottom wells of 96-well plates in NB/B27/GLX and incubated sealed to prevent gas-exchange. The media became rapidly acidic indicating a hypoxic environment. Cells tended to adhere homotypically and very few cells were able to attach to the bottom of the well. Of note, even after 5 days of culturing in this harsh environment, these cultures still contained a few living cells that were able to attach, grow, and proliferate, if replated under normal growth conditions (data not shown). The contents of the wells were harvested at several time points, centrifuged to separate CM from cells and debris, and the pellets were freeze/thawed to release the intracellular contents from surviving cells. Measurement of FGF2 and LDH in medium (“CM”) and after mechanical damage (“intracellular”) showed that while intracellular FGF2 content quickly dropped within 20 h of initial plating, levels of released FGF2 (in CM) were quite steady between 4 h and 2 days; with substantially higher FGF2 levels released from SB623, than from MSC (around 2 vs. 0.5 ng/1 million cells, respectively) (Fig. 6). A decrease in the level of FGF2 in the CM levels was detected on day 5. The LDH pattern was different to that of FGF2: intracellular LDH activity remained high longer than did FGF2. This result indicated that dying cells release high levels of intracellular FGF2 when placed in harsh culture conditions, while surviving cells reduce their intracellular FGF2 levels. It appeared that FGF2 released into the media remained stable for some time despite these conditions. We also noticed that SB623 typically survived better than MSCs in these harsh conditions as judged by higher levels of intracellular LDH at later time points.
FIG. 6.
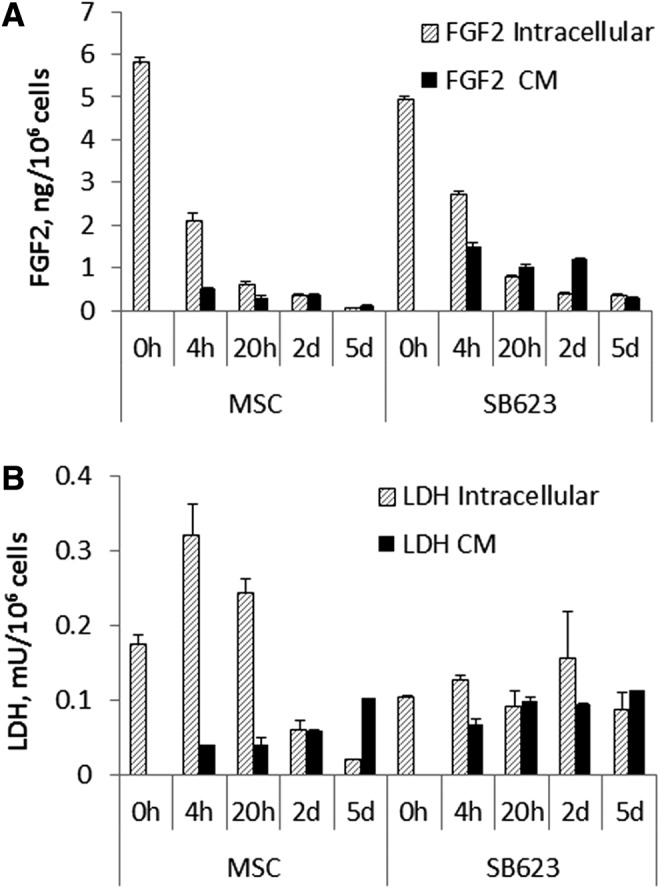
FGF2 release by MSC and SB623 in high cell density/hypoxia/insufficient nutrients cultures. MSC or SB623 were plated in round 96-well plates at 1×106 cells/mL, 350 μL/well, and tightly closed to prevent gas exchange. At the indicated time points, the well contents were collected. FGF2 (A) and LDH activity (B) were measured in both culture medium (CM) and cells (intracellular) after releasing intracellular contents by freeze/thaw treatment. The results are expressed per million cells.
Discussion
Our in vitro data provide support for the notion that damage and death of MSC during or soon after intracerebral implantation can provide a major regenerative stimulus to endogenous cells within the surrounding brain tissue. This appears to be largely due to the release of FGF2-rich intracellular contents from the donor cells. When ruptured, both MSCs and their derivative, SB623, release intracellular FGF2 at levels that were hundreds of times higher than levels released into CM by secretion (Fig. 1A vs. B). FGF2 levels released by mechanical injury from MSC and SB623 were much higher than those released from several other cell types, including endothelial cells and embryonic stem cell derivatives.
Importantly, MSC extracts were active in terms of their effect on relevant types of endogenous CNS cells. Specifically, the extracts induced concentration-dependent proliferation of both neural progenitors and endothelial cells. Both neuropoiesis and angiogenesis are essential for regeneration of ischemic brain tissue (reviewed in [24]). The activity of extracts was remarkable: extracts diluted to 5%–15% or less could induce significant cell proliferation in our assays. Since we were extracting 1 million damaged cells in 5 mL of medium, this suggests that the intracellular content of 1 million MSCs could be still mitogenic even when diluted in volumes as large as 100 mL. Conversely, damage to even a low percent of implanted cells could produce a strong mitogenic signal for endogenous cells. FGF2-neutralizing antibody prevented the proliferation of both neural progenitors and endothelial cells stimulated by MSC extracts (Figs. 2 and 3) suggesting that these effects are largely mediated by FGF2. An early study on the distribution of 1 ng of 125I-FGF2 injected into the rat striatum highlights the fate of the exogenous FGF2 in the brain [25]. The radioactive FGF2 was found to diffuse through the injection site, quickly accumulating in the ependymal cell layer of ipsilateral and even contralateral ventricles, along adjacent fiber tracks, in the corpus callosum, and bound to the basement membrane of the vasculature. Remarkably, the radioactive label remained stable and disappeared slowly during a 7-day observation period. Together with our data these observations may imply that FGF2 from damaged MSC transplants could diffuse long distances in a short time, where it can induce proliferation of local or distant neural precursor cells and stimulate angiogenesis.
The proliferation of endothelial cells is an important part of angiogenesis (reviewed in [26]). We found that FGF2 appeared to be more potent than VEGF (a hallmark angiogenic factor secreted by MSC [27,28]) in inducing HUVEC proliferation, either in recombinant (Fig. 3B) or native forms (as a component of the cell extract). This was evidenced by the observation that CM containing around 80 pg/mL of VEGF (Table 1) did not induce HUVEC proliferation, while an extract diluted to∼70 pg/mL FGF2 did (Fig. 3A).
Thus, we have demonstrated that a lethal mechanical cell injury such as freeze/thawing, releases highly active FGF2 from MSC and SB623. In vivo, some MSCs would certainly be injured during implantation. Extrapolating from data in Fig. 2A, injury to only 1% of the grafted MSCs would result in the immediate release of about 0.2–0.4 ng FGF2 (assuming 5 million cells/graft).
FGF2 release as a result of various, lethal and non-lethal, cell membrane injuries has been demonstrated in various cell types of predominantly mesenchymal origin. Beside classical works that have demonstrated that endothelial cells release FGF2 upon mechanical injuries [12,13,15], endothelial cells have been shown to release FGF2 upon irradiation [29], osmotic shock [30], and treatment with complement proteins [31]. Vascular smooth muscle cells have been shown to release FGF2 upon strong mechanical strain [32] or nitric oxide [33]; skeletal myofibers released it upon puncture [34], and cardiomyocytes upon beating [35]. In all of these studies the released FGF2 was implicated in autocrine growth stimulation. The use of MSC and their derivatives in various implantation procedures brings them into direct contact with other tissues. This has two important implications: (i) the surrounding microenvironment can be a source of nonmechanical cell injury and (ii) this cell injury can release FGF2 that has a potent paracrine effect on the endogenous tissues.
We attempted to model the conditions of the microenvironment into which MSCs and SB623 are implanted to observe whether biologically relevant amounts of FGF2 would be released. Damage of cerebral microvessels during implantation can expose the grafted cells to PBMC-mediated cytotoxicity [36,37]. Therefore, cytotoxicity detected in PBMC-MSC cocultures may serve as a model of implantation-associated MSC damage. We found that 20%–70% of MSCs or SB623 may be killed as a result of PBMC-mediated cytotoxicity and thus a proportional percentage of intracellular FGF2 could be released this way (Fig. 5). When we compared the percentage of total FGF2 released to the percentage of cell death (as evidenced by released LDH activity), the former was lower. This disparity could be explained by the possibility of higher FGF2 protein turnover than LDH and/or due to the fact that percentages were calculated based on two different parameters: protein content (FGF2) and enzymatic activity (LDH).
Another possible mechanism of cell damage following intracerebral implantation could be related to local conditions of high cell density, hypoxia, and insufficient nutrients diffusion to cells in the injection bolus. This is a likely scenario given the fact that highly metabolically active cells are implanted into the peri-infact area; a region of the brain, which is characterized by chronic hypoperfusion [38,39]. Our in vitro model of these conditions (Fig. 6) demonstrated that: (i) FGF2 was released into the medium at high levels; (ii) despite massive cell death in these conditions, a number of mesenchymal cells were able to survive preserving some FGF2 intracellularly until day 5; (iii) despite these harsh culture conditions, massive cell death, and presumably associated proteolysis, FGF2 could still be detected in the CM at high levels past day 2.
In summary, these data suggest that when MSCs die soon after implantation, they release high levels of FGF2. The importance of FGF2 for treating cerebral ischemia has been demonstrated in multiple animal studies. Cells genetically modified to enhance FGF2 secretion were shown to be more efficient than nonmodified controls in improving neurological outcome after middle cerebral artery occlusion [40,41] and recombinant FGF2 or FGF2-encoding vector delivered by various routes improved neurological outcome [42–44]. However, in a clinical trial for acute stroke, recombinant FGF2 (Trafermin) failed to show statistically significant improvement [45], which underlines the importance of delivery route, therapeutic window, and the context, in which factor is presented.
In addition to the importance of route and timing of administration it appears that cell dose may also be critical for therapeutic efficacy. In a recent meta-analysis of preclinical studies of MSC for ischemic stroke Vu et al. showed that behavioral effect sizes for MSC administration are consistently very high and that there is an inverse correlation between behavioral effect size and MSC dose [46]. At least in some instances this could be explained by very high amounts of FGF2 released from dying MSC and saturating response levels at higher doses. Conversely, lower doses may provide both sufficient FGF2 to trigger the regeneration response and better cell survival, which could augment the astrogenic response seen with combinations of dead and alive cells in our experiments.
Various studies have demonstrated clinically relevant biological activity of cell-free preparations of MSCs. Some of these studies have shown positive effects of MSC lysates or extracts for liver and kidney failure and wound healing [47–49] and apoptotic MSC for hypertrophic scare prevention [50], however, none of these studies measured levels of FGF2 in these preparations. Although we observed profound stimulating effects of cell-free extracts, we also noticed that a small numbers of surviving MSC may work in concert or even synergize with the dying majority of implanted cells by providing differentiating stimuli to the progeny of the proliferating neural precursors. In particular, we demonstrated a synergistic effect on astrocytic differentiation of a subpopulation of live cells within a population of dead cells (Fig. 4D). This synergy could be explained by a delayed stimulation that live cells could provide, presumably mediated by BMPs [19,23], to precursors generated in response to the intracellular FGF2 content from freeze/thawed (dead) MSC.
A better understanding of how implanted MSC mediate their regenerative effects on endogenous nervous tissue can help optimize the parameters for successful clinical outcomes. Here, we provide in vitro data supporting the notion that one of major stimuli for neuroregeneration could be produced by damaged transplanted cells due to the release of FGF2-rich intracellular content into the surrounding neural tissue. In vivo testing should establish if this cell injury-induced release of FGF2 is indeed an important mechanism by which MSC exert their neuroregenerative effects, in addition to the secretion of soluble and insoluble factors by living cells.
Acknowledgments
The authors are grateful to Dr. George Martin for critical reading of the article and valuable suggestions, Dr. Ernest Yankee for his intellectual contribution and support, and Rouzbeh Shooshtarian for providing MSC and SB623 for this study.
Author Disclosure Statement
All authors are employees of SanBio.
References
- 1.Caplan AI. and Dennis JE. (2006). Mesenchymal stem cells as trophic mediators. J Cell Biochem 98:1076–1084 [DOI] [PubMed] [Google Scholar]
- 2.Joyce N, Annett G, Wirthlin L, Olson S, Bauer G. and Nolta JA. (2010). Mesenchymal stem cells for the treatment of neurodegenerative disease. Regen Med 5:933–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Venkat P, Zacharek A. and Chopp M. (2014). Neurorestorative therapy for stroke. Front Hum Neurosci 8:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bang OY, Lee JS. and Lee PH. (2005). Autologous mesenchymal stem cell transplantation in stroke patients. Ann Neurol 57:874–882 [DOI] [PubMed] [Google Scholar]
- 5.Isakova IA, Baker K, Dufour J, Gaupp D. and Phinney DG. (2006). Preclinical evaluation of adult stem cell engraftment and toxicity in the CNS of rhesus macaques. Mol Ther 3:1173–1184 [DOI] [PubMed] [Google Scholar]
- 6.Westrich J, Yaeger P, He C, Stewart J, Chen R, Seleznik G, Larson S, Wentworth B, O'Callaghan M, et al. , (2010). Factors affecting residence time of mesenchymal stromal cells (MSC) injected into the myocardium. Cell Transplant 19:937–948 [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Tredget EE, Liu C. and Wu Y. (2009). Analysis of allogenicity of mesenchymal stem cells in engraftment and wound healing in mice. PLoS One 4(9):e7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffin MD, Ryan AE, Alagesan S, Lohan P, Treacy O. and Ritter T. (2013). Anti-donor immune responses elicited by allogeneic mesenchymal stem cells: what have we learned so far?. Immunol Cell Biol 91:40–51 [DOI] [PubMed] [Google Scholar]
- 9.Coyne TM, Marcus AJ, Woodbury D. and Black IB. (2006). Marrow stromal cells transplanted to the adult brain are rejected by an inflammatory response and transfer donor labels to host neurons and glia. Stem Cells 24:2483–2492 [DOI] [PubMed] [Google Scholar]
- 10.Burns TC, Ortiz-González XR, Gutiérrez-Pérez M, Keene CD, Sharda R, Demorest ZL, Jiang Y, Nelson-Holte M, Soriano M, et al. , (2006). Thymidine analogs are transferred from prelabeled donor to host cells in the central nervous system after transplantation: a word of caution. Stem Cells 24:1121–1127 [DOI] [PubMed] [Google Scholar]
- 11.Yu PJ, Ferrari G, Galloway AC, Mignatti P. and Pintucci G. (2007). Basic fibroblast growth factor (FGF-2): the high molecular weight forms come of age. J Cell Biochem 100:1100–1108 [DOI] [PubMed] [Google Scholar]
- 12.Gajdusek CM. and Carbon S. (1989). Injury-induced release of basic fibroblast growth factor from bovine aortic endothelium. J Cell Physiol 139:570–579 [DOI] [PubMed] [Google Scholar]
- 13.McNeil PL, Muthukrishnan L, Warder E. and D'Amore PA. (1989). Growth factors are released by mechanically wounded endothelial cells. J Cell Biol 109:811–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Amore PA. (1990). Modes of FGF release in vivo and in vitro. Cancer Metastasis Rev 9:227–238 [DOI] [PubMed] [Google Scholar]
- 15.Muthukrishnan L, Warder E. and McNeil PL. (1991). Basic fibroblast growth factor is efficiently released from a cytolsolic storage site through plasma membrane disruptions of endothelial cells. J Cell Physiol 148:1–16 [DOI] [PubMed] [Google Scholar]
- 16.Brunner G, Gabrilove J, Rifkin DB. and Wilson EL. (1991). Phospholipase C release of basic fibroblast growth factor from human bone marrow cultures as a biologically active complex with a phosphatidylinositol-anchored heparan sulfate proteoglycan. J Cell Biol 114:1275–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunner G, Nguyen H, Gabrilove J, Rifkin DB. and Wilson EL. (1993). Basic fibroblast growth factor expression in human bone marrow and peripheral blood cells. Blood 81:631–638 [PubMed] [Google Scholar]
- 18.Benavente CA, Sierralta WD, Conget PA. and Minguell JJ. (2003). Subcellular distribution and mitogenic effect of basic fibroblast growth factor in mesenchymal uncommitted stem cells. Growth Factors 21:87–94 [DOI] [PubMed] [Google Scholar]
- 19.Aizman I, Tirumalashetty BJ, McGrogan M. and Case CC. (2014). Comparison of the neuropoietic activity of gene-modified versus parental mesenchymal stromal cells and the identification of soluble and extracellular matrix-related neuropoietic mediators. Stem Cell Res Ther 5:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dao M, Tate CC, McGrogan M. and Case CC. (2013). Comparing the angiogenic potency of naïve marrow stromal cells and Notch-transfected marrow stromal cells. J Transl Med 11:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aizman I, Tate CC, McGrogan M. and Case CC. (2009). Extracellular matrix produced by bone marrow stromal cells and by their derivative, SB623 cells, supports neural cell growth. J Neurosci Res 87:3198–3206 [DOI] [PubMed] [Google Scholar]
- 22.Tate CC, Fonck C, McGrogan M. and Case CC. (2010). Human mesenchymal stromal cells and their derivative, SB623 cells, rescue neural cells via trophic support following in vitro ischemia. Cell Transplant 19:973–984 [DOI] [PubMed] [Google Scholar]
- 23.Aizman I, McGrogan M. and Case CC. (2013). Quantitative microplate assay for studying mesenchymal stromal cell-induced neuropoiesis. Stem Cells Transl Med 2:223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiong Y, Mahmood A. and Chopp M. (2010) Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs 11:298–308 [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez AM, Carman LS, Ong M, Ray J, Gage FH, Shults CW. and Baird A. (1994). Storage, metabolism, and processing of 125I-fibroblast growth factor-2 after intracerebral injection. Brain Res 665:285–292 [DOI] [PubMed] [Google Scholar]
- 26.Liekens S, Clercq ED. and Neyts J. (2001). Angiogenesis: regulators and clinical applications. Biochem Pharmacol 61:253–270 [DOI] [PubMed] [Google Scholar]
- 27.Liew A. and O'Brien T. (2012). Therapeutic potential for mesenchymal stem cell transplantation in critical limb ischemia. Stem Cell Res Ther 3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madrigal M, Rao KS. and Riordan NH. (2014). A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J Transl Med 12:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Witte L, Fuks Z, Haimovitz-Friedman A, Vlodavsky I, Goodman DS. and Eldor A. (1989). Effects of irradiation on the release of growth factors from cultured bovine, porcine, and human endothelial cells. Cancer Res 49:5066–5072 [PubMed] [Google Scholar]
- 30.Hartnett ME, Garcia CM. and D'Amore PA. (1999). Release of bFGF, an endothelial cell survival factor, by osmotic shock. Invest Ophthalmol Vis Sci 40:2945–2951 [PubMed] [Google Scholar]
- 31.Benzaquen LR, Nicholson-Weller A. and Halperin JA. (1994). Terminal complement proteins C5b-9 release basic fibroblast growth factor and platelet-derived growth factor from endothelial cells. J Exp Med 179:985–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng GC, Briggs WH, Gerson DS, Libby P, Grodzinsky AJ, Gray ML. and Lee RT. (1997). Mechanical strain tightly controls fibroblast growth factor 2 release from cultured human vascular smooth muscle cells. Circ Res 80:28–36 [DOI] [PubMed] [Google Scholar]
- 33.Fukuo K, Inoue T, Morimoto S, Nakahashi T, Yasuda O, Kitano S, Sasada R. and Ogihara T. (1995). Nitric oxide mediates cytotoxicity and basic fibroblast growth factor release in cultured vascular smooth muscle cells: a possible mechanism of neovascularization in atherosclerotic plaques. J Clin Invest 95:669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaye D, Pimental D, Prasad S, Mäki T, Berger HJ, McNeil PL, Smith TW. and Kelly RA. (1996). Role of transiently altered sarcolemmal membrane permeability and basic fibroblast growth factor release in the hypertrophic response of adult rat ventricular myocytes to increased mechanical activity in vitro. J Clin Invest 97:281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke MSF, Caldwell RW, Chiao H, Miyake K. and McNeil PL. (1995). Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ Res 76:927–934 [DOI] [PubMed] [Google Scholar]
- 36.Spaggiari GM, Capobianco A, Becchetti S, Mingari MC. and Moretta L. (2006). Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood 107:1484–1490 [DOI] [PubMed] [Google Scholar]
- 37.Roemeling-van Rhijn M, Reinders ME, Franquesa M, Engela AU, Korevaar SS, Roelofs H, Genever PG, Ijzermans JN, Betjes MG, et al. , (2013). Human allogeneic bone marrow and adipose tissue derived mesenchymal stromal cells induce CD8+cytotoxic T cell reactivity. J Stem Cell Res Ther 3(Suppl 6):004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brumm KP, Perthen JE, Liu TT, Haist F, Ayalon L. and Love T. (2010). An arterial spin labeling investigation of cerebral blood flow deficits in chronic stroke survivors. Neuroimage 51:995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richardson JD, Baker JM, Morgan PS, Rorden C, Bonilha L. and Fridriksson J. (2011). Cerebral perfusion in chronic stroke: implications for lesion-symptom mapping and functional MRI. Behav Neurol 24:117–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ikeda N, Nonoguchi N, Zhao MZ, Watanabe T, Kajimoto Y, Furutama D, Kimura F, Dezawa M, Coffin RS, et al. , (2005). Bone marrow stromal cells that enhanced fibroblast growth factor-2 secretion by herpes simplex virus vector improve neurological outcome after transient focal cerebral ischemia in rats. Stroke 36:2725–2730 [DOI] [PubMed] [Google Scholar]
- 41.Fujiwara K, Date I, Shingo T, Yoshida H, Kobayashi K, Takeuchi A, Yano A, Tamiya T. and Ohmoto T. (2003). Reduction of infarct volume and apoptosis by grafting of encapsulated basic fibroblast growth factor-secreting cells in a model of middle cerebral artery occlusion in rats. J Neurosurg 99:1053–1062 [DOI] [PubMed] [Google Scholar]
- 42.Watanabe T, Okuda Y, Nonoguchi N, Zhao MZ, Kajimoto Y, Furutama D, Yukawa H, Shibata MA, Otsuki Y, Kuroiwa T. and Miyatake S. (2004). Postischemic intraventricular administration of FGF-2 expressing adenoviral vectors improves neurologic outcome and reduces infarct volume after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab 24:1205–1213 [DOI] [PubMed] [Google Scholar]
- 43.Wang ZL, Cheng SM, Ma MM, Ma YP, Yang JP, Xu GL. and Liu XF. (2008). Intranasally delivered bFGF enhances neurogenesis in adult rats following cerebral ischemia. Neurosci Lett 446:30–35 [DOI] [PubMed] [Google Scholar]
- 44.Li Q. and Stephenson D. (2002). Postischemic administration of basic fibroblast growth factor improves sensorimotor function and reduces infarct size following permanent focal cerebral ischemia in the rat. Exp Neurol 177:531–537 [DOI] [PubMed] [Google Scholar]
- 45.Bogousslavsky J, Victor SJ, Salinas EO, Pallay A, Donnan GA, Fieschi C, Kaste M, Orgogozo JM, Chamorro A. and Desmet A. (2002). European-Australian Fiblast (Trafermin) in Acute Stroke Group: fiblast (trafermin) in acute stroke: results of the European-Australian phase II/III safety and efficacy trial. Cerebrovasc Dis 14:239–251 [DOI] [PubMed] [Google Scholar]
- 46.Vu Q, Xie K, Eckert M, Zhao W. and Cramer SC. (2014). Meta-analysis of preclinical studies of mesenchymal stromal cells for ischemic stroke. Neurology 82:1277–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mishra PJ, Mishra PJ. and Banerjee D. (2012). Cell-free derivatives from mesenchymal stem cells are effective in wound therapy. World J Stem Cells 4:35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parekkadan B, van Poll D, Suganuma K, Carter EA, Berthiaume F, Tilles AW. and Yarmush ML. (2012). Mesenchymal stem cell-derived molecules reverse fulminant hepatic failure. PLoS One 2:e941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiao J, Milwid JM, Yarmush ML. and Parekkadan B. (2011). A mesenchymal stem cell potency assay. Methods Mol Biol 677:221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu S, Jiang L, Li H, Shi H, Luo H, Zhang Y, Yu C. and Jin Y. (2014). Mesenchymal stem cells prevent hypertrophic scar formation via inflammatory regulation when undergoing apoptosis. J Invest Dermatol 134:2648–2657 [DOI] [PubMed] [Google Scholar]