

Abstract
Human pluripotent stem cells (hPSCs) are powerful tools for regenerative therapy and studying human developmental biology, attributing to their ability to differentiate into many functional cell types in the body. The main challenge in realizing hPSC potential is to guide their differentiation in a well-controlled manner. One way to control the cell differentiation process is to recapitulate during in vitro culture the key events in embryogenesis to obtain the three developmental germ layers from which all cell types arise. To achieve this goal, many techniques have been tested to obtain a cellular cluster, an embryoid body (EB), from both mouse and hPSCs. Generation of EBs that are homogeneous in size and shape would allow directed hPSC differentiation into desired cell types in a more synchronous manner and define the roles of cell–cell interaction and spatial organization in lineage specification in a setting similar to in vivo embryonic development. However, previous success in uniform EB formation from mouse PSCs cannot be extrapolated to hPSCs possibly due to the destabilization of adherens junctions on cell surfaces during the dissociation into single cells, making hPSCs extremely vulnerable to cell death. Recently, new advances have emerged to form uniform human embryoid bodies (hEBs) from dissociated single cells of hPSCs. In this review, the existing methods for hEB production from hPSCs and the results on the downstream differentiation of the hEBs are described with emphases on the efficiency, homogeneity, scalability, and reproducibility of the hEB formation process and the yield in terminal differentiation. New trends in hEB production and directed differentiation are discussed.
Introduction
Human pluripotent stem cells (hPSCs) such as embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs) that are able to differentiate into cell types of all three somatic germ layers represent a powerful cell source for regenerative therapy and studying human developmental biology. Beyond the capability for self-renewal and multilineage differentiation, more recent generation of hiPSCs from patient cells through reprogramming has mitigated the concerns such as ethical rejections and requirement for immunosuppression therapy that surround the uses of hESC derivatives. Clinical and biopharmaceutical translation of hPSCs are highly contingent upon the ability to produce cells of desired phenotypes in high purity and large quantity [1]. To date, the utility of these cells has not been carried out to its full potential due to the lack of standardized protocols to direct their lineage-specific differentiation. Although a growing body of protocols exists, describing directed differentiation of hPSCs into specific lineages, significant barriers, including the variance between starting populations, scalability, reproducibility, and culture definition (eg, substrate, media, feeders, and eventual cell lineage of interest), have impeded the industrial and clinical translation of current differentiation protocols.
In vitro differentiation of hPSCs often requires the formation of embryoid bodies (EBs), which represents the onset of directed differentiation of hPSCs toward specific lineages [2–5]. EBs are three-dimensional (3D) hPSC aggregates that can differentiate into cells of all three germ layers (endoderm, ectoderm, and mesoderm) [3]. Many events in the in vitro lineage-specific differentiation process within the EBs recapitulate those seen in vivo in the developing embryo [6], which justifies the uses of EBs as a model system to simulate the in vivo differentiation of hPSCs under in vitro culture conditions, and mechanistically examine hPSC differentiation programs/lineage commitment during embryogenesis as an alternative to the whole embryo approach [7]. In addition, in vitro formed EBs have opened access to early precursor cell populations that are not accessible in vivo [8]. EBs have been shown to effectively initiate lineage-specific differentiation of hPSCs toward many lineages, such as cardiac [9], neural [10,11], hematopoietic [12], and pancreatic β cells [13]. Although EB permits the generation of cells arising from all three primary germ layers, the differentiation outcomes are highly dependent upon the endogenous parameters of EBs, including the media composition [14], the cell numbers, the size, and the morphology of EBs [9,15]. For example, EB viability and the yield in terminal differentiation vary in a size-dependent manner [16]. While too small EBs did not survive well during the differentiation procedures, too large EBs underwent core necrosis [16]. A wide distribution in the EB size introduces a source of variability in their downstream differentiation [17], which depends on the immediate microenvironment perceived by individual cells in the EBs, that is, the position of cells relative to others in the EBs. This effect is more pronounced when EBs exceed a certain size range: cells at the peripheral of the differentiating EBs tend to differentiate into the primitive endoderm, while the cells at the center of the EBs tend to give rise to primitive ectoderm cells [18]. When cultured in chondrogenic medium, small EBs exhibited higher propensity toward chondrogenesis, yet medium and large EBs shifted their potential toward hematopoietic and endothelial differentiation [19–21]. EBs of different morphology (eg, cystic-, bright cavity-, and dark cavity-type EBs) also exhibited different differentiation propensities [15]. Cystic EBs primarily comprised the endoderm lineage population, while both types of cavity EBs consisted of cells from all three germ layers [15]. The fate specifications of the cells in the differentiating EBs may be fine-tuned through cell–cell interactions within the aggregates [22]. There may exist an ideal size range for the best viability and differentiation of EBs toward desired lineages.
To harness the full potential of human embryoid bodies (hEBs) as a powerful platform to direct hPSC differentiation into desired cell types in large quantities and define the roles of cell–cell interaction and spatial organization in lineage specification in a setting similar to in vivo embryonic development, a highly efficient scalable system capable of reproducibly generating synchronous hEBs that are homogeneous in size and morphology is the key. In this review, we will go over the existing methods for hEB production from hPSCs and the results on the downstream differentiation. The major techniques to induce hEB formation are outlined in Fig. 1. The two conventional methods, that is, suspension culture (Fig. 1a, b) and hanging drop (HD) (Fig. 1c), were covered in the review by Kurosawa in 2007 [23], who also described advances on developing bioreactors for EB production (Fig. 1d, e) up to then. To build upon Kurosawa's review, which focused more on EB formation from mouse ESCs due to the limited knowledge of hESCs, we have, in the current review, highlighted the newly emerged technologies such as low-adherence microwells that may allow quick cell aggregation for uniform EB formation from dissociated hPSCs (Fig. 1f). For the techniques with which success has not been demonstrated for homogeneous EB formation from dissociated human PSCs, results from mouse PSCs are discussed.
FIG. 1.

The major techniques for the formation of embryoid bodies (EBs).
Methods for EB Formation
Conventional methods
The key to healthy hPSC cultures is to maintain cell pluripotency by preventing spontaneous differentiation. The presence of antidifferentiation factors, such as basic fibroblast growth factor (bFGF), leukemia inhibitory factor, or those released from feeder cells, are crucial for maintaining hPSC pluripotency. EB represents the onset of differentiation; therefore, antidifferentiation factors need to be withdrawn before the induction of EB formation. Since hPSCs are anchorage-dependent cells, the principle involved in EB production deals with deprivation of cell attachment to the culture surfaces and promoting cell aggregation while remaining in suspension. Two conventional methods are used: suspension culture and HD. EBs are generally formed in suspension culture in nonadherent cell culture dishes that allow spontaneous aggregation in solution. The HD method involves dispensing equal numbers of cells in gravity-induced, physically separated cell aggregates that are suspended from the lid of a Petri dish.
Suspension culture
Suspension culture with or without a feeder layer was first tested in as early as 1985 by Doetschman et al. for spontaneous EB formation from mouse ESCs [24]. Suspension culture was first tested on EB formation from hESCs in 2000 by Itskovitz-Eldor et al. [13]. For hPSCs, both feeder-based and feeder-free/floating culture systems are used. In feeder-based culture, healthy undifferentiated hESC colonies that were cultured on feeder cell layers (eg, mouse embryo fibroblasts) were enzymatically or mechanically dissociated into medium-sized aggregates, followed by scraping of adherent hESC colonies to suspend cell clumps before transferring the clumps into suspension culture in low-adherence vessels [13]. Overnight culture results in EB formation in suspension, characterized by colonies of a round shape and with clear borders. Following the initial cellular aggregation of hESCs, the hEBs became cavitated over time and eventually turned into cystic hEBs with central cavities [13]. The enzymatic or mechanical dissociation method is apparently associated with problems of a wide range of size variation, irregular shapes, and the tendency of EB agglomeration to form larger colonies. The method is not reproducible, producing EBs of heterogeneous sizes and morphologies that are asynchronous in their downstream differentiation, which may compromise the utility of the ESC-derived cells in many applications [13,15].
Instead of using confluent hPSC colonies as the input cell population, dissociated single-cell suspension of hPSCs represents a better starting population for EB formation, which allows strict control over EB sizes by seeding defined numbers of cells for the formation of discrete EBs. Singularized hPSCs also eliminate any pre-existing organizations that may be present in the colonies, which may bias the downstream differentiation [25]. One challenge when using dissociated single-cell suspension of hPSCs is to maintain cell viability. hPSCs have exhibited extremely low viability when they are present in the form of dissociated single-cell suspension [13,26]. Treatment with the p160 Rho-associated coiled-coil kinase (ROCK) inhibitor (ROCKi, Y-27632) has been widely used to promote survival of dissociated hESCs after passages [27] and assist EB formation from dissociated single-cell suspension of hESCs [27–29]. Nonetheless, ROCKi is a xeno-factor and has been shown to bias cell fate toward residual pluripotency and inhibit differentiation in neural differentiation studies, reducing the utility of the derived cells for potential clinical applications [30]. To facilitate cell aggregation in suspension for EB formation, nonadhesive culture surfaces were developed [12,31], and soluble factors that promote cell–cell interactions were administered in the culture media [32]. Recently, a serum-free, animal product-free defined medium, mTeSR™1, has been developed to support the feeder-independent culture and EB formation from dissociated hESCs [33,34], making it possible to derive clinically relevant human cell lineages from hPSCs in a reproducible manner. In feeder-free suspension culture in nonadherent dishes (eg, extremely hydrophobic or extremely hydrophilic surfaces) (Fig. 2) in the absence of antidifferentiation factors such as bFGF, hESCs formed 3D multicellular aggregated EBs [15]. At 10 days in suspension culture, the EBs became morphologically classified into three types: cystic-, bright cavity-, and dark cavity-type EBs [15]. The organization of the EBs ranged from cystic structures filled with fluid (cystic EBs) to spheroids with loosely aggregated cores (bright cavity EBs) and to spheroids with dense cores like solid balls (dark cavity EBs). The morphologically classified EBs possess different differentiation capacities and are associated with different differentiation stages, for example, cystic EBs exhibited greater propensity toward endoderm lineages, and both types of the cavity EBs displayed specification potential for lineages associated with all three germ layers. Among the three EB types analyzed, bright cavity EBs were the most pluripotent with equal potential for all three germ layers, while cystic EBs experienced the fastest induction of differentiation that was enriched for endoderm lineages. In addition to the morphology, EB sizes critically determine lineage specifications. These early studies suggest that directing the EB formation process by governing the microenvironment factors may produce subsets of EBs that are enriched for specific lineage differentiation. However, the suspension culture technique does not allow control over the distributions of EB classifications and therefore the differentiation efficacy for target lineage production. In addition, microdissection methods were applied to isolate regions of EBs formed in suspension culture for enrichment toward specific lineages [35,36]. The efficiency of these methods to enrich EBs for target lineage specifications is only moderate, and the involvement of labor-intensive procedures prevents scale-up.
FIG. 2.
Suspension culture in bacterial-grade nonadherent dishes. Human embryonic stem cell suspension in a nonadherent dish made of highly hydrophobic or highly hydrophilic materials. To survive, the anchorage-dependent cells spontaneously aggregate, giving rise to three-dimensional (3D) spheroids.
Evolving of culture vessels, for example, round-bottomed, low-adherence 96-well plates, and low-adherence conical tubes have facilitated cell aggregation during EB formation from defined numbers of dissociated hPSCs (Fig. 3). EBs produced through forced aggregation of known numbers of dissociated hESCs in round-bottomed, low-adherence 96-well plates by centrifugation underwent reproducible hematopoietic differentiation into blood cells in an input cell number-dependent manner at 8–12 days after EB formation when transferred to tissue culture-treated plates [12]. In contrast, dissociated hESCs failed to aggregate to form stable EBs in flat-bottomed wells even after centrifugation [12]. Burridge et al. tested the formation of EBs from several lines of hESCs using a high-throughput, forced aggregation system that was based upon the centrifugation of known numbers of dissociated hESCs in V-bottomed 96-well plates (ie, V-96FA) and evaluated the downstream cardiomyogenic differentiation [37]. Their results were consistent with those from the Ng group on the ability of the V-96FA system to promote reproducible and homogeneous EB formation and synchronous directed differentiation. The data also highlighted interline variability among hESC lines in terms of the cardiomyogenic potential as well as the amenability to growth factor induction of cardiomyocyte differentiation. Evseenko et al. examined the roles of extracellular matrix (ECM) components in cell aggregation in round-bottomed, low-adherence 96-well plates to form EBs from single-cell suspension of hESCs [32]. In the presence of a purified protein complex comprising human laminin-511 and nidogen-1, but without the feeder layer or exogenous chemicals, hESCs in single-cell suspension consistently assembled into uniform EBs that were later induced to differentiate into endodermal, ectodermal, and mesodermal derivatives [32]. Their findings suggest ECM protein-based adhesion mechanisms in regulating cell aggregation during EB formation from single hESCs. Low-adherence, polypropylene conical tubes (Fig. 4) were also used to promote cell sedimentation and aggregation during EB formation, but had only been tested on mouse ESCs [38,39]. Round- and V-bottomed 96-well plates and conical tube cultures allow media change without disturbance on the cells for prolonged culture and differentiation within the same vessels. Different from the 96-well plates, there is a lack of control over the EB sizes in the conical tube cultures. Scale-up EB production is difficult with all these culture vessels.
FIG. 3.

For this type of suspension culture, round-bottom, low-adherence 96-well plates are used. From these plates, it is possible to obtain EBs from a known number of cells: The round bottom promotes cell–cell contact and subsequent aggregation. In this way, it is possible to investigate the number of cells necessary to make a good quality EB in both size and morphology.
FIG. 4.

A polypropylene conical tube enables use of a large amount of cells. In 1 mL media, 2×104 cells can be placed. They promote sediment on the bottom of the tube. To cultivate the cells, the conical tube was put into an incubator with the cap loosely closed to supply oxygen. A 5-day culture was required to obtain good EBs for further differentiation.
EBs formed in suspension cultures have generally demonstrated a high degree of heterogeneity in their downstream differentiation patterns [18], reducing the overall efficiency for directed differentiation into a specific lineage in high fidelity. Optimization of the differentiation protocols of EBs toward specific lineages requires the incorporation of the appropriate microengineering technology to obtain a monodisperse synchronous EB population that has a differentiation status enriched for desired lineages.
Enhancement of static suspension culture for EB homogeneity
Although suspension-based EB derivation is simple, fast, labor efficient, scalable, and cell friendly, it offers little control over EB size and homogeneity. Upon formation, the EBs may agglomerate to form bigger clumps, resulting in a wide size distribution [40]. Current attempts to enhance homogeneity in EB size and shape are based upon increased stirrer speeds [40] or applying rotation to the suspension culture [41]. EBs that were formed in these hydrodynamically enhanced suspension cultures from hPSCs were further differentiated into a variety of cell lineages, including neural cells [13], cardiomyocytes [13,42], hepatic and pancreatic cells [41,43], vascular cells [44], chondrocytes [19–21], germ cells [45], hematopoietic cells [13], and endothelial cells [44,46]. Nonetheless, concerns were raised surrounding the shear stresses and hydrodynamic forces that were associated with the motions, which may alter cell viability, aggregation, proliferation, and differentiation.
EBs in suspension tend to agglomerate, dissociate, as well as adhere to the culture dish, which contribute to low EB formation efficiency and poor viability [47]. To eliminate these problems, hESC single-cell suspension was encapsulated/cultured in a 3D semisolid matrix, such as a hydrogel, to physically isolate individual cells as well as provide a niche-like milieu for EB formation from individual hESCs [48]. When compared with the regular suspension culture in Petri dishes, EB formation in the agarose-based 3D environment demonstrated higher efficiency with enhanced stability of individual EBs and the lack of EB agglomeration into large clusters or fragmentation into small unproliferative clusters or single cells during prolonged in vitro culture. Similarly, the addition of 1% methylcellulose to the culture media of hESC suspension creates semisolid media, which is the key to isolate the cells and develop single-cell-cloned EBs in suspension (Fig. 5). Common problems of the semisolid culture system for EB formation include low EB yield due to the intrinsic instability of single hiPSCs during prolonged culture, the hindrance of mass transport necessary for effective soluble factor treatments, as well as the complicated process for differentiated cell retrieval [48]. It remains interesting to see whether the 3D semisolid culture would improve the yield in lineage-specific terminal differentiation of the hEBs.
FIG. 5.
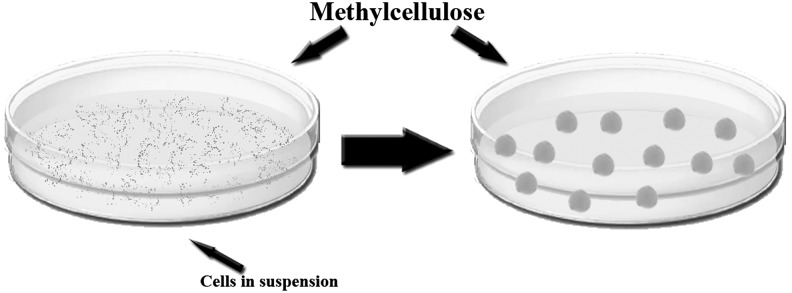
Single-cell suspension culture in 1% semisolid methylcellulose media. The semisolid media keeps the cells isolated and allows single-cell-cloned EB formation. When the EBs are transferred to standard media, they fuse into a larger structure in a reproducible manner.
Hanging drops
HDs were first developed for EB formation from mouse ESCs in 2002 by Dang et al. [49]. In the HD method, droplets of cell suspension are placed on an invertible substrate, for example, the internal face of the lid of a Petri dish. When the lid is full of drops, it is flipped over onto the bottom of the same Petri dish filled with phosphate-buffered saline to avert drying (Fig. 1c). The resultant Petri dish is put in an incubator for 2 days for EB formation in the drops. The EBs will then be recovered from the drops and put into a nonadherent plate for suspension culture (Fig. 6), which compacts the EBs. The HD method allows easy control of the EB sizes. It is noteworthy that there are numerous technical difficulties with the HD method, such as the maintenance of trace amount of culture media for each droplet without disturbing the EBs, loss of EBs when picking up using the pipette, controlling the shape after harvesting the EBs, attachment of premature EBs onto Petri dishes, and the upper size limit of the EBs due to the volume limitation of the droplet allowing fluid tension to adhere the droplet to the lid [23]. In particular, the number of EBs formed per dish (about 100 per 10-cm plate) is limited by surface area, which hinders scale-up production [50]. Although the HD method allows the generation of uniform EBs from murine PSCs for small-scale applications, it is difficult to be used for automation due to the cumbersome and time-consuming process. Furthermore, the reproducibility may be an issue due to the pipetting-dependent delay between first and last HD preparations [51]. The formed EBs are not spherical due to the cell spreading and the satellite aggregation of PSCs. In the conventional HD culture, it is impossible to change media during the culture if growth of the EB is needed. However, by direct observation of the drops using a stereomicroscope, it is possible to estimate the growth state of the EBs. If necessary, growth factors may be incorporated into the media used to make the drops to improve EB formation or to initiate the differentiation process directly from the drops, driving the cell fate from the first moments of their differentiation.
FIG. 6.
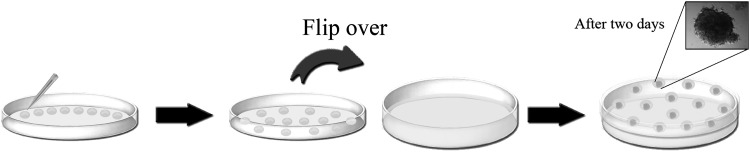
In hanging drop culture, droplets of cell suspension are seeded onto the internal surface of the lid of a Petri dish, followed by the inversion of the lid onto the bottom of the same dish filled with phosphate-buffered saline to avoid drying. After 2 days, it is possible to obtain EB formation on the lid.
To date, due to its laborious nature, which is further confounded by the two-step process (ie, EB formation on the lid, and then transfer to suspension culture for EB maturation), the HD method has been largely abandoned for hPSCs. Success of the HD method for uniform EB formation from singularized hPSCs has not been reported. The experience of our group with HD culture of dissociated hESCs in the presence of ROCKi failed to produce cell aggregates (unpublished observation). With hESCs, it is possible to use the HD technique to produce EBs with large clumps derived from colonies. In such cases, an entire colony will be scraped to use as a clump for the HD culture. However, these clumps of hESCs should be uniform in size to ensure consistency in further differentiation. Following the HD culture, hEBs may undergo suspension culture for further compaction before being used in a differentiation protocol. A few studies have shown differentiation of HD-produced human EBs to several cell types, including hematopoietic cells [52], cardiomyocytes [53], and hepatocytes [54]. These investigations indicate propensities of the HD-produced EBs to progress toward cell types of all three germ layers.
New methods
The common problems with conventional methods for EB formation such as static suspension culture or HD are the lack of control over the homogeneity of the environmental factors that individual cells are exposed to and are not amenable for scalable production. Recently, new techniques have emerged to enhance the production of uniform and synchronous EBs on a large scale in a reproducible manner. These techniques include bioreactor cultures, hydrogel embedding systems, and the microwells.
Bioreactor cultures and the rotary cell culture system
Bioreactors of various designs have been developed since early 2000s to induce EB formation and differentiation in a well-defined scalable manner [55,56]. Bioreactors offer the advantages of easy scale-up EB production, controllable culture parameters, and labor-efficient processing. The scaling-up is highly dependent upon the design of the bioreactors [57]. A variety of scalable dynamic culture systems, including spinner flasks, stirred bioreactors, and rotary cultures, have been tested in EB formation from hESCs [6,58–60]. To control cell aggregation and therefore EB sizes and the mass transport in the culture, stirring/agitation is oftentimes applied. For example, direct seeding of hESC suspension into a spinner flask equipped with an impeller would enable formation of EBs (Fig. 7) that may be subsequently differentiated into cardiomyocytes [61] or endothelial cells for vasculogenesis and angiogenesis [60]. Through mixing in 3D systems, these dynamic cultures have enhanced the physiochemical uniformity of the culture condition (eg, oxygenation, pH, and the mass transport), reduced EB agglomeration [52], improved cell viability, and facilitated circulation of exogenous factors in the culture, however, at the expense of introducing an additional variable of shear stress on the cells. Low-speed stirring results in extensive EB agglomeration that hinders the mass transport, while high-speed stirring is damaging to the cells [60]. A stirrer speed of 100 rpm has been shown to result in smaller and more uniform EB formation than a speed of 60 rpm [60]. In addition to the stirrer speed, EB formation also depends on the impeller type. The bulb-type impeller resulted in more homogeneous and reproducible EBs when compared with the paddle-type impeller [60]. To date, EBs with uniform size distribution have not been achieved with stirring bioreactors. Large clumps form when the cells are directly inoculated into the bioreactors. To address this problem, either preformed EBs have been added to the stirred suspension culture [59] or the cells have been enclosed within agarose hydrogel spheres of controlled size [52]. Systems that combine different types of bioreactors [60], encapsulation with bioreactors [62], or static suspension culture followed by bioreactors [60] have been attempted to increase the EB formation and directed terminal differentiation without compromising the self-renewal properties of ES cells. It is conceivable though that the hydrodynamic conditions present in the bioreactors may alter cell pluripotency and differentiation, and medium supplementation alone is not sufficient to efficiently drive the lineage-specific differentiation of EBs in suspension bioreactors.
FIG. 7.

Through a direct injection of stem cells into a spinner flask with a paddle impeller, it is possible to obtain EB formation.
Another class of bioreactors is the rotary cell culture system (RCCS) [52,63]. The RCCS is a horizontally rotated, bubble-free, disposable culture vessel with diffusion gas exchange (Fig. 8). The system provides a reproducible, complex, 3D, in vitro culture system with large cell masses. During cell growth, the rotation speed can be adjusted to compensate for increased sedimentation rates. The RCCS and its unique environment of low shear forces, high mass transfer, and microgravity leads to EB formation close to a stationary point. The RCCS may provide hydrodynamic culture conditions for many cell types, including PSCs. In the RCCS, hESC suspension is subjected to constant circular motion, which enhances cellular incorporation during EB formation, and produces more uniform EBs with higher efficiency of lineage-specific directed differentiation when compared with the EBs generated in static suspension cultures or under HD conditions [6,64,65]. The RCCS also allows scalable production of EBs and EB-derived cells.
FIG. 8.

The rotary cell culture system is a horizontally rotated, bubble-free, disposable culture vessel with diffusion gas exchange. The system provides a reproducible, complex, 3D in vitro culture system with large cell masses. During cell growth, the rotation speed can be adjusted to compensate for increased sedimentation rates.
A simpler version of the rotary suspension culture involves nontissue, culture-treated, polystyrene (ie, bacteriological grade) Petri dishes placed on an orbital rotary shaker. The resulting EBs were maintained for up to 7 days in suspension [64]. Depending upon the rotation mode, there are two types of RCCSs, that is, slow-turning lateral vessel (STLV) and high-aspect rotating vessel (HARV). The STLV appeared to be more efficient in producing EBs of a defined size range than the HARV, which tends to generate very large clusters [52]. When using an STLV to produce EBs from hESCs, Come et al. observed formation of more uniform hEBs in both shape and size compared with the conventional static culture. Concurrently, the yield of neuroinductive differentiation of the formed hEBs was substantially higher [66]. Although bioreactor cultures may provide a more uniform environment to sustain EB formation and increase yield in directed differentiation, the large volume of bioreactors (eg, minimally 100 mL) makes the bioreactor-based process laborious and expensive. For example, it is difficult to use bioreactors to screen medium compositions for directed differentiation of EBs and examine multiple samples in parallel. This has posed a compelling need for a miniaturized culture system that allows quick EB formation while mimicking the central attributes of a large volume of bioreactors. The microwell technology to be described next may provide a practical solution.
Microwell technology
Microwell-based technologies have been previously used to create patterned substrates, on which cell spheroids/clusters with controlled size, shape, and uniformity may form cocultures with supporting cells to study heterotypic cell–cell interactions [63] or interactions between hESCs and supporting murine embryonic fibroblast feeder cells [67]. These studies have demonstrated the utility of microwells in improving the homogeneity of culture conditions, specifically in templating cell spheroids/clusters with enhanced control over sizes and shapes. The microwell approach for EB formation was first introduced by Ungrin et al. in 2008 [29] and has ever since been extensively adopted for homogeneous EB formation from dissociated hPSCs. EBs formed using the microwell approach are oftentimes referred to as spin EBs since in addition to heavy dependence of hEB formation on the presence of ROCKi, most protocols have applied centrifugation as a means to force cell aggregation [12,37]. Although centrifugation may avoid exposure of hPSCs to the ROCKi xeno-factor, it is not conducive to high-throughput automated production of hEBs and may alter the stem characteristics of stem cells and their differentiation potential [68].
In the microwell approach, microwells/microcavities of a volume in the range of several microliters were created on nonadherent, micropatterned polydimethylsiloxane (PDMS) surfaces to allow controlled cell aggregation and formation of one discrete EB per microwell [69] (Fig. 9). By controlling the input cell numbers in each microwell, EB size can be well controlled. In addition, this approach allows controlled delivery of soluble and bound cues to the cells, recovery of the EBs for maturation and further experiments, and reproducible high-throughput large-scale production of uniform-sized EBs. The obstacle for scale-up, however, resides in the cost of the single-use microwell-based consumables. Alternatively, low-cost, time-efficient, in-house mass production methods of microwell plates using off-the-shelf materials are desirable. Attempts have been directed toward creating nonadhesive agarose/Dulbecco's modified Eagle's medium (DMEM) microwells using soft lithography strategy from different templates [69]. Agarose is desirable for microwell fabrication, attributing to its moldability, transparency, and noncell-adhesive properties. Silicone masters with defined patterned surfaces were created using the soft lithography technique. A second soft lithography step allowed solidification of the agarose-DMEM hydrogel, and through replica molding, about 800 corner cube apertures representing mirror-inverted replicas of the silicone master topography were simultaneously fabricated on the agarose surfaces to host EB formation. hESCs with controlled inoculation densities were seeded into each agarose microwell in the presence of ROCKi and allowed to aggregate. Although cell aggregation started within 10 h, it required about 20 h to complete. The resultant EBs were uniform in size, which were correlated with the initial seeding densities, and were able to retain pluripotency after the short-term aggregation. However, this approach did require centrifugation and individual manipulation of the formed EBs manually and an additional plating step for further culture and maturation. The presence of the ROCKi was necessary for cell viability and aggregation for EB formation. Another important factor of cell aggregation is the geometry of the microwells. It was found that an angled geometry without horizontal interspaces was preferable since cells settling on horizontal interspaces may not contribute to aggregation and therefore die from anoikis [69]. Using the same technique, EB formation from murine PSCs was much easier when compared with that of hPSCs [69].
FIG. 9.

Micromold technology allows for creation of a mold in various materials to form EBs with uniform shape and size. The embryonic stem cells are digested with an enzymatic procedure and the resultant single-cell suspension is plated into the micromold made with 50 μL of low-melting-point agarose at various concentrations, depending on the sizes of the embryonic bodies to be produced. After 10 min, the cells fall into the microwells, and 950 g is applied for 5 min to force cell aggregation. After 2 days of incubation at 37°C, the cells form EBs. The obtained human embryoid bodies (hEBs) are transferred into a multiwell dish for subsequent differentiation or they may be shaken in an incubator at 37°C for 2 days to promote compaction. The mold is constructed based on the type of EB sought.
To date, one commercial product for uniform and synchronous EB formation from dissociated single-cell suspension of hPSCs is based upon the Aggrewell™ plates [28], which are similar to a standard 24-well dish, except that an array of microwells of inverse pyramidal configuration are textured onto the center eight wells of the dish. By seeding well-dispersed hPSC single-cell suspension at known cell density to the plate, followed by centrifugation, one discrete EB may form per microwell. The formed EBs are then transferred after 24–48 h for further culture and maturation in low-adherence plates. Using the Aggrewell plates, at very high cell seeding densities, ROCKi is not required for cell survival. The thus produced EBs were spherical, uniform in size, and remained structurally intact during transfer. EB size could be easily controlled by adjusting the density of the input cell suspension. Centrifugation is not required if ROCKi is used [29]. This technique allows ultrahigh-throughput production of size-specific aggregates starting from an hPSC single-cell suspension. However, it is time-consuming and experiences limitations in scalability. Once EBs are pooled and transferred to low-adherence plates, they tend to agglomerate to form conjoint EBs that are large and may cause center necrosis due to restricted access to oxygen and nutrients. This may also introduce additional variation in the EB size and differentiation patterns.
Parallel to the development of the Aggrewell plates, low-cost 3D culture plates and Petri dishes based on replica molding for uniform EB formation are available commercially under the trademark CellSphere™ from Biomaterials USA LLC. These presterilized, 3D hydrogel-supporting products allow for uniform hEB formation without the need for centrifugation. In addition, there are other versions of cell-repellent microwells/micromolds for EB formation. These microwells were made of different cell-repellent polymers, such as poly(ethylene glycol) (PEG), agar, collagen/matrigel, and agarose (Fig. 1f). Using the PEG microwells as templates to initiate EB formation, given precise control of the input cell numbers, and the size and shape of the microwells, homogenous spherical mouse EBs may be produced in large quantities [70]. Existing reports on the size effect of microwell-produced EBs on their directed differentiation have solely been based on mouse embryoid bodies (mEBs) [71], but not hEBs. Uniform mEBs of different sizes were formed in nonadhesive, PEG hydrogel microwell arrays of various diameters. Upon inductive differentiation, smaller mEBs exhibited greater propensity toward endothelial lineages, while cardiac differentiation was dominant in large mEBs. In line with these findings, Choi et al. documented the utility of PDMS-based concave microwells in the production of uniform mEBs that displayed defined fate specifications in a size-mediated manner [72]. Both cardiac and neuronal differentiations were enhanced in mEBs formed in large concave microwells when compared with smaller microwells. Since dissociated hPSCs survive poorly, it appears that the key to high cell survival and successful hEB formation from singularized hPSCs is quick cell aggregation and cell–cell contact during EB formation. In addition to the low adherence of the dishes or microwells, the shape of the microwells plays a role in determining cell aggregation and the overall EB formation efficiency. The round- and V-bottomed microwells promoted cell aggregation relative to flat-bottomed wells. In particular, V-bottomed wells seemed to better allow quick cell aggregation when compared with the round-bottomed ones [37]. Production of uniform-sized hEBs with controllable lineage specifications has not been achieved using the microwell technology.
To enable scalable, high-throughput, reproducible automated production of hEBs for mass generation of hPSC-derived cells, our laboratory has recently developed a simple technique based upon nonadhesive, round-bottomed, agarose microwell arrays for hEB formation from dissociated single hPSCs [3,73]. We have demonstrated robust production of homogeneous and synchronous hEBs from singularized hPSCs while eliminating both ROCKi xeno-factor and centrifugation for a total of three hPSC lines [ie, hiPSC line derived from foreskin fibroblasts (WiCell Research Institute—WB0002) and BG01V/hOG and H9 hESC lines] [3,73]. Our microwell system has allowed quick and controlled aggregation of singularized hPSCs to minimize the residential time of the cells in the form of dissociated single cells and promoted cell–cell interactions and therefore enhanced overall cell viability without the need for ROCKi and/or the rate-limiting centrifugation step during EB formation. Uniform-sized nearly spherical hEBs were formed under the no ROCKi and no centrifugation conditions and were extracted intact from the microwells for subsequent culture/differentiation. The hEBs exhibited organized internal tissue-level structures and expressed proteins distinctive for all three embryonic germ layers. When subjected to lineage-specific directed differentiation protocols, the hEBs formed in our system from all of the three tested cell lines differentiated into tissue lineages specific to each germ layer [eg, neural lineage (ectoderm specific), cardiac lineage (mesoderm specific), and pancreatic lineage (endoderm specific)], and the results were highly consistent. Our agarose microwell system offers a new avenue in automated, low-cost large-scale production of hEBs and hPSC-derived cells.
Complementary techniques to enhance EB formation and differentiation
To minimize the variations in the microenvironment within hEBs, which may lead to spontaneous and heterogeneous differentiation, Ferreira et al. incorporated biodegradable particles into the hEB structures as a means to homogeneously deliver growth factors to the cells within hEBs [74,75]. Growth factor-releasing biodegradable poly(lactide-co-glycolide) nano- and microparticles carrying regulatory factors that are known to contribute to early vascular development, including vascular endothelial growth factor (VEGF), bFGF, and placenta growth factor (PlGF), were mixed with hESCs during EB formation in round-bottomed, low-adherence 96-well plates through forced aggregation by centrifugation. It was shown that particles of a preferential size range (0.24–5 μm in diameter) were most actively uptaken by the hESCs without interference on cell viability, proliferation, EB formation, and cell organization within the EBs. The incorporated nanoparticles distributed uniformly within the EBs. Sustained release of VEGF, bFGF, and PlGF from the incorporated nanoparticles within the hEBs enhanced vascular differentiation, suggesting the utility of biodegradable particles in uniform growth factor delivery throughout EBs for homogeneous directed differentiation toward specific lineages. For the strategies to control ESC differentiation through engineering the EB microenvironment, please refer to the review by Bratt-Leal et al. [7].
Some studies examined the effect of key small biomolecules on EB formation and differentiation. According to Khoo et al. [14], a physiological glucose concentration (5.5 mM) was sufficient to sustain hESC culture as well as hEB formation, growth, and further differentiation into germ layer-specific lineages for all three germ layers. hEBs cultured at the physiological glucose concentration exhibited a similar gene expression pattern to those cultured at a high-glucose concentration, suggesting that high-glucose concentrations currently used in the hESC differentiation procedures were not necessary to support hESC culture, nor differentiation even in the long term (eg, up to Day 104) [14]. Instead of high-glucose concentrations, the use of normal physiological glucose concentration on hESC culture and differentiation also reduced cell impairment due to the exposure to high-glucose levels [14].
Mohr et al. applied a cell- and protein-repellent self-assembled monolayer (SAM) in the microwells to constrain colony growth, therefore promoting cell aggregation, cell–cell interactions, and colony characteristics for better hESC survival and proliferation [9]. hEBs formed from the hESCs in these SAM-coated microwells demonstrated relatively uniform sizes and shapes in a microwell size-dependent manner when compared with standard unstrained cultures. Subsequent cardiac differentiation of the hEBs indicated a correlation between the yield in directed cardiogenesis and the size of microwells used to form hEBs with enriched cardiomyocytes derived from hEBs formed in smaller microwells. However, since the hEB formation started with enzymatically digested hESC colonies from the microwells rather than dissociated single hESCs, precise control of the hEB size based upon the inputting cell numbers would not be possible. After all, this study provided an alternative means by using a cell- and protein-repellent SAM coating to regulate the hESC colony size for the formation of uniform-sized hEBs and demonstrated the benefit of uniform-sized hEBs of a different size range on directed downstream differentiation of hESCs.
Recently, microfluidic devices have been developed and tested using mEBs to control the downstream differentiation. In the study of Fung et al. [76], a Y-channel microfluidic device that generates parallel laminar flows of two different culture media without major intermixing has been used to subdifferentiate a single EB into more than one lineage at the same time. By placing EBs across both streams of two separate culture media resulting from laminar coflow in a microchannel, a single EB may be simultaneously induced into different lineages, or control differentiated in some areas, while remaining in undifferentiated stages in the rest of the areas. Although similar work has not been performed on hEBs, microfluidic technology may hold promise in administering differentiation conditions to EBs in a culture that leads to better controlled fate specifications of hPSCs.
In alignment with this study, Kang et al. developed a multilayer, microfluidic array platform consisting of PDMS concave microwells of a bottom layer and flat cell culture chambers of a top layer for mouse embryonic stem cells (mESC) culture and mEB formation, but generated somewhat disappointing results [77]. Upon mEB formation in the microwells of the bottom layer, the microfluidic device was inverted to allow spontaneous plating of the mEBs into the flat cell culture chambers, therefore avoiding manual mEB retrieval and replating. Cell docking in the concave microwells varied as a function of flow rates and microwell depths. At 3 days in culture in the flat cell culture chamber, structural fragmentation of some mEBs was observed. At 8 days upon neuronal induction, expression of neuronal markers by the cells in each flat chamber appeared to be quite heterogeneous, suggesting the intrinsic limitations of microfluidic array-based platforms in controlling EB formation, stability, and directed differentiation.
In Table 1, we have compared all the techniques for hEB formation from dissociated hPSCs with respect to the efficiency, homogeneity, scalability, and reproducibility of the hEB formation process and the yield in terminal differentiation. An important criterion to choose a particular technique for homogeneous hEB formation is better definition of culture components that are xeno-free (ie, free of animal-derived components). Involvement of undefined xeno-components in the protocol may introduce batch-to-batch variability that compromises the quality control during scale-up production, along with increased possibility of the presence of immunogenic materials in the differentiated cell populations and transmission of animal-origin diseases. In this regard, nonadhesive microwell-based technologies, which may allow quick aggregation of dissociated single hPSCs in the absence of ROCKi (a xeno-factor), may offer better scalability and reproducibility relative to the conventional methods that are limited in scalability and compatibility with process control strategies during mass production.
Table 1.
Techniques for Embryoid Body Formation from Human Pluripotent Stem Cells
| Technique | Description | Principle of EB size control | Downstream differentiation | Pros | Cons | References |
|---|---|---|---|---|---|---|
| Suspension culture | In low-adherence vessels, withdrawal of antidifferentiation factors such as bFGF and LIF promotes aggregation of hPSCs for EB formation in feeder-free cultures. In feeder-based cultures, additional enzymatic or mechanical dissociation is needed to release hPSC colonies to suspension for EB formation. |
Little control over EB size and homogeneity. | A high degree of heterogeneity in their downstream differentiation patterns. | Easy processing Compatible with both feeder-based and feeder-free cultures |
Low efficiency Low homogeneity Low scalability Low reproducibility due to poor control of EB size and morphology |
[13,15,35,36] |
| Hanging drops | Dispensing equal numbers of cells in gravity-induced, physically separated cell aggregates that are suspended from the lid of a Petri dish. | HD culture of dissociated hPSCs for EB formation not reported. HD culture of hESC lumps for EB formation depends on the uniformity of hESC lumps. |
EBs formed from hESC lumps in HD culture gave rise to a few lineages. | Easy control of sizes if starting from dissociated single cells. | Low efficiency Laborious Low scalability Low reproducibility due to the cumbersome and time-consuming process |
[52–54] |
| Bioreactor culture | Scalable dynamic culture systems that apply hydrodynamic conditions to control cell aggregation for EB formation. | Stirring/agitation to enhance the physiochemical uniformity of the culture condition and reduce EB agglomeration. | Bioreactor-produced EBs have been directed differentiated into a variety of lineages. | Easy scale-up EB production Controllable culture parameters Labor-efficient processing |
May introduce additional variable of shear stress on the cells Low homogeneity due to the difficulty to control EB sizes Low efficiency: laborious and expensive process due to the large volume of bioreactors |
[6,52,58–60,63–66] |
| Microwell technology | Microwells/microcavities of a volume in the range of several microliters were created on nonadherent micropatterned PDMS surfaces to allow controlled cell aggregation and formation of one discrete EB per microwell. | By controlling the input cell numbers in each microwell, EB size can be well controlled. | Production of uniform-sized hEBs with controllable lineage specifications has not been achieved using the microwell technology. | High homogeneity High reproducibility, attributing to easy control of EB sizes and morphology |
Time-consuming Limited scalability due to the cost of the single-use microwell-based consumables and the general requirement for centrifugation Limited clinical applicability of the EB-derived cells due to the potential presence of the ROCKi xeno-factor for cell viability during EB formation |
[3,12,28,29,37,69,70,73] |
EB, embryoid body; hPSC, human pluripotent stem cell; bFGF, basic fibroblast growth factor; LIF, leukemia inhibitory factor; HD, hanging drop; hESC, human embryonic stem cell; ROCKi, Rho-associated coiled-coil kinase inhibitor; hEBs, human embryoid bodies; PDMS, polydimethylsiloxane.
Discussion
Recent discovery of magic small molecules in enhancing the survival of dissociated single hPSCs and hEB formation has opened up new avenues to produce synchronous hEBs for controlled differentiation. Examples of such molecules include previously described ROCKi as well as Thiazovivin (Tzv) and Tyrintegin (Ptn). Tzv and Ptn are two small compounds that have been identified by Xu and colleagues to enhance the survival of dissociated single hESCs over 30-fold, despite inhibition of cell proliferation [78]. The mechanistic investigation of hESC death following single-cell dissociation indicated the significance of E-cadherin signaling for hESC survival. E-cadherin signaling was disrupted during enzymatic dissociation, leading to perturbation of integrin signaling and ensuing hESC death. Tzv and Ptn helped restore cell-ECM adhesion-mediated integrin signaling and E-cadherin-mediated cell–cell interaction on dissociated hESCs, an effect similar to ROCKi, therefore promoting cell survival while maintaining pluripotency. Tzv also allowed formation of aggregates from single hESC suspension through stabilization of the E-cadherin protein on the plasma membrane by inhibiting protein endocytosis. Inhibition of the ROCK pathway by Tzv was another mechanism accountable for increased hESC survival. However, addition of Tzv and Ptn to the culture media of dissociated hPSCs has not been extensively exploited as a means to enhance cell survival for rapid expansion and genetic manipulation or to produce uniform hEBs from dissociated single-cell suspension.
A number of variations have emerged that should be considered in the examination of hPSC biology, hEB formation and subsequent directed differentiation, such as interline differences. For instance, the hESC lines derived between 2001 and today exhibit substantial differences in behavior. Indeed, the spatial-temporal stage, from which the cells from the inner cell mass were extracted, has resulted in a predisposition of differentiation toward a specific germ layer (ectoderm, mesoderm, or endoderm) based upon the theory of presumptive territories, also termed gastrulation [79,80]. Results from different hPSC lines should be interpreted and compared with caution to predict the appropriate cell lines to use in the generation of specific functional lineages for therapeutic development and regenerative medicine.
New Trends and Future Directions
The lack of standardized protocol for EB formation has resulted in a surge of interests in directed differentiation of hPSCs while bypassing the EB stage. Success has been achieved with directed differentiation of hESCs or hiPSCs into hepatocyte-like cells [81–84], cardiomyocytes, or chondrogenic lineage [85]. These procedures were based upon the administration of developmental signals that prime the cells to a definitive germ layer before terminal differentiation and usually employed adherent, two-dimensional culture conditions that allow scale-up. To date, varying levels of culture definition, scalability, differentiation yield, cell maturity, and function have been demonstrated using these methods. One major obstacle, however, is the lack of consistency. One effort in directly differentiating hESCs into chondrogenic lineage without prior EB formation based upon high-density micromass culture in the presence of BMP2 yielded a mixed chondrogenic population of cells residing at various stages of the chondrogenesis pathway with the subpopulation percentages evolving over time in culture [85]. The micromass culture, in which dissociated hESCs of defined volume at a density greater than confluency were spotted in each well of 24-well tissue culture dishes, essentially created an environment that promoted cell clustering and aggregation. Although EB formation was absent, the high-density micromass culture recapitulates tissue-level contexts of in vivo differentiation during embryogenesis by generating a 3D multilayered culture to foster close cell association, cellular aggregation, and condensation for differentiation in vitro, a setting that is similar to EB formation in a microwell approach, yet it offers little control over the size, the microenvironmental uniformity of the micromass (eg, the availability of exogenous biomolecules/factors in the culture media to individual cells in the micromass), as well as the spatial and temporal patterns of cell–cell interactions in the micromass [86]. It is noteworthy that the statement holding the inherent cellular heterogeneity of EBs as the major contributing factor that challenges the generation of a homogenous population of desired lineage from hPSCs is unfounded due to the limitation in the ability of the existing techniques to produce uniform and synchronous EBs.
Early success in directed differentiation of mouse ESCs to a neuronal fate in the absence of both EB formation and coculture [87] has prompted the interest in directed differentiation of hPSCs into clinically relevant populations in sufficient numbers while bypassing the EB step. These include directed differentiation of hPSCs that are cultured as monolayers on ECM proteins [78,88] or directly cultured on supportive stromal layers [89]. This area rests on the uses of hiPSCs instead of hESCs, attributing to their ability to evade the immune system and derive patient-specific hESC equivalents. Although these EB-free differentiation protocols have resulted in the differentiation of hPSCs into a broad spectrum of lineages, they are not reproducible, nor optimized to enhance the yields for the targeted lineages. In a study by Palecek and coworkers [90], genetically unmodified hPSCs were directly differentiated without EB formation into human cardiomyocytes with a high yield of over 80% using a monolayer-based (cultured on Synthemax plates) directed differentiation platform with temporal administration of two small molecules of Wnt signaling inhibitors, such as porcupine inhibitors IWP2 or IWP4, which are known to have a biphasic effect on cardiac development. Among the three hPSC lines being tested, the differentiated populations expressed proteins that are characteristic of cardiac development, formed spontaneously contractile sheets of cardiomyocytes, and exhibited a ventricular-like action potential morphology [90]. In a follow-up study by Burridge et al. [91], testing of a similar differentiation protocol was expanded to 11 hiPSC lines at passages ranging from 20 to 83. In general, cardiomyocytes were produced in greater than 85% purity and can be further enriched to over 95% with metabolic selection. Although these results are encouraging, they are highly sensitive to the timing and dose of Wnt pathway modulation, which may require individual optimization for each cell line. A temporal assay of the subtypes of cardiomyocytes derived in a chemically defined medium containing three key components (CDM3) over the number of days of differentiation revealed a temporal differentiation profile evolving from unspecified cardiomyocyte precursors to a ventricular-like phenotype, which conforms to an essential cardiac development pattern. However, at various differentiation stages, the composition of the derived population was highly heterogeneous, consisting of cells residing at various stages of the cardiomyogenic pathway, indicative of asynchronous differentiation, which compromises reproducibility of the resulting cell population. To select a desired population in the hPSC derivatives, fluorescence-activated cell sorting may be used [46,92], but with the drawback of low throughput, which requires expansion of the selected population. Alternatively, a genetic selection based upon expression of a marker driven by a lineage-restricted promoter may be more efficient and may be combined with bioreactor systems [93,94]. Nonetheless, this requires insertion of a selection cassette into the host genome, which may alter cell characteristics.
Regardless of whether the EB step is present in the differentiation protocol, one major challenge with hPSC differentiation is to increase the yield of target cells. Under current differentiation conditions, only a subset of the hPSCs specifies into the target fates. A scalable system for homogeneous and synchronous EB production from dissociated hPSCs would allow cost-effective simultaneous delivery of molecular factors that implement exogenous factor-mediated control to predifferentiate or induce the cells and therefore help to increase the differentiation yield. Another factor to consider is the form of the hPSC-derived cells. Directed differentiation of EBs would generate clusters/aggregates of derived cells in suspension that are ready for transplantation without the need for cell harvesting or recovery, whereas directed differentiation of hPSCs without EB formation is normally based on monolayers or culture substrates that require cell retrieval through enzymatic or mechanical digestion before transplantation. The digestion procedure involved in cell retrieval from monolayer or substrate-based cultures without EB formation may damage the derived cells and compromise cell viability as well as phenotype stability.
Conclusions
hPSCs are useful to create models of human diseases and generate almost all the cell types in adult humans for cell-based therapies. hEB formation is a critical phase of the differentiation process of hPSCs. The common techniques illustrated, however, do not provide a large number of hEBs suitable for clinical applications. To realize their potential for lineage-specific differentiation, formation of highly uniform and synchronous hEBs is the key. Initiation from single-cell suspension of hPSCs may be the optimum solution to produce homogeneous and synchronous hEBs. It is important that the procedures are standardized to obtain reproducible results in hEB generation. The techniques described above have shown varying degrees of success in hEB formation with the major challenge of achieving hEBs of uniform sizes and shapes. Of all the methods discussed, microwell technology is likely to evolve into the best available tool. By controlling the size, geometry, and material properties of the low-adherence microwells, the conventional centrifugation step as well as the ROCKi can be eliminated, making it possible to develop a high-throughput process for automated large-scale production of synchronous hEBs. Delivery of soluble factors may synergistically act with the microwells to fine-tune the time course of uniform hEB formation as well as the subsequent differentiation trajectory.
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology (2014CB964600, 2012CB966300), the National Science Foundation (1055922, 0748129), the National Natural Science Foundation of China (81271369), and the William H. Goodwin Endowment.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Murry CE. and Keller G. (2008). Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell 132:661–680 [DOI] [PubMed] [Google Scholar]
- 2.Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D. and Melton DA. (2014). Generation of functional human pancreatic beta cells in vitro. Cell 159:428–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pettinato G, Wen X. and Zhang N. (2014). Formation of well-defined embryoid bodies from dissociated human induced pluripotent stem cells using microfabricated cell-repellent microwell arrays. Sci Rep 4:7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramaiahgari SC, den Braver MW, Herpers B, Terpstra V, Commandeur JN, van de Water B. and Price LS. (2014). A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch Toxicol 88:1083–1095 [DOI] [PubMed] [Google Scholar]
- 5.Takayama K, Kawabata K, Nagamoto Y, Kishimoto K, Tashiro K, Sakurai F, Tachibana M, Kanda K, Hayakawa T, Furue MK. and Mizuguchi H. (2013). 3D spheroid culture of hESC/hiPSC-derived hepatocyte-like cells for drug toxicity testing. Biomaterials 34:1781–1789 [DOI] [PubMed] [Google Scholar]
- 6.Gerecht-Nir S, Cohen S. and Itskovitz-Eldor J. (2004). Bioreactor cultivation enhances the efficiency of human embryoid body (hEB) formation and differentiation. Biotechnol Bioeng 86:493–502 [DOI] [PubMed] [Google Scholar]
- 7.Bratt-Leal AM, Carpenedo RL. and McDevitt TC. (2009). Engineering the embryoid body microenvironment to direct embryonic stem cell differentiation. Biotechnol Prog 25:43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trounson A. (2006). The production and directed differentiation of human embryonic stem cells. Endocr Rev 27:208–219 [DOI] [PubMed] [Google Scholar]
- 9.Mohr JC, Zhang J, Azarin SM, Soerens AG, de Pablo JJ, Thomson JA, Lyons GE, Palecek SP. and Kamp TJ. (2010). The microwell control of embryoid body size in order to regulate cardiac differentiation of human embryonic stem cells. Biomaterials 31:1885–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar M, Bagchi B, Gupta SK, Meena AS, Gressens P. and Mani S. (2007). Neurospheres derived from human embryoid bodies treated with retinoic acid show an increase in nestin and ngn2 expression that correlates with the proportion of tyrosine hydroxylase-positive cells. Stem Cells Dev 16:667–681 [DOI] [PubMed] [Google Scholar]
- 11.Sathananthan AH. Neural stem cells in neurospheres, embryoid bodies, and central nervous system of human embryos. Microsc Microanal 17:520–527 [DOI] [PubMed] [Google Scholar]
- 12.Ng ES, Davis RP, Azzola L, Stanley EG. and Elefanty AG. (2005). Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood 106:1601–1603 [DOI] [PubMed] [Google Scholar]
- 13.Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, Soreq H. and Benvenisty N. (2000). Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med 6:88–95 [PMC free article] [PubMed] [Google Scholar]
- 14.Khoo ML, McQuade LR, Smith MS, Lees JG, Sidhu KS. and Tuch BE. (2005). Growth and differentiation of embryoid bodies derived from human embryonic stem cells: effect of glucose and basic fibroblast growth factor. Biol Reprod 73:1147–1156 [DOI] [PubMed] [Google Scholar]
- 15.Kim JM, Moon SH, Lee SG, Cho YJ, Hong KS, Lee JH, Lee HJ. and Chung HM. (2011). Assessment of differentiation aspects by the morphological classification of embryoid bodies derived from human embryonic stem cells. Stem Cells Dev 20:1925–1935 [DOI] [PubMed] [Google Scholar]
- 16.Van Winkle AP, Gates ID. and Kallos MS. (2012). Mass transfer limitations in embryoid bodies during human embryonic stem cell differentiation. Cells Tissues Organs 196:34–47 [DOI] [PubMed] [Google Scholar]
- 17.Peerani R, Rao BM, Bauwens C, Yin T, Wood GA, Nagy A, Kumacheva E. and Zandstra PW. (2007). Niche-mediated control of human embryonic stem cell self-renewal and differentiation. EMBO J 26:4744–4755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei R, Yang J, Hou W, Liu G, Gao M, Zhang L, Wang H, Mao G, Gao H, Chen G. and Hong T. (2013). Insulin-producing cells derived from human embryonic stem cells: comparison of definitive endoderm- and nestin-positive progenitor-based differentiation strategies. PLoS One 8:e72513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koay EJ, Hoben GM. and Athanasiou KA. (2007). Tissue engineering with chondrogenically differentiated human embryonic stem cells. Stem Cells 25:2183–2190 [DOI] [PubMed] [Google Scholar]
- 20.Koyama N, Miura M, Nakao K, Kondo E, Fujii T, Taura D, Kanamoto N, Sone M, Yasoda A, et al. , (2013). Human induced pluripotent stem cells differentiated into chondrogenic lineage via generation of mesenchymal progenitor cells. Stem Cells Dev 22:102–113 [DOI] [PubMed] [Google Scholar]
- 21.Toh WS, Guo XM, Choo AB, Lu K, Lee EH. and Cao T. (2009). Differentiation and enrichment of expandable chondrogenic cells from human embryonic stem cells in vitro. J Cell Mol Med 13:3570–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pompe S, Bader M. and Tannert C. (2005). Stem-cell research: the state of the art. Future regulations of embryonic-stem-cell research will be influenced more by economic interests and cultural history than by ethical concerns. EMBO Rep 6:297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurosawa H. (2007). Methods for inducing embryoid body formation: in vitro differentiation system of embryonic stem cells. J Biosci Bioeng 103:389–398 [DOI] [PubMed] [Google Scholar]
- 24.Doetschman TC, Eistetter H, Katz M, Schmidt W. and Kemler R. (1985). The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol 87:27–45 [PubMed] [Google Scholar]
- 25.Spelke DP, Ortmann D, Khademhosseini A, Ferreira L. and Karp JM. Methods for embryoid body formation: the microwell approach. Methods Mol Biol 690:151–162 [DOI] [PubMed] [Google Scholar]
- 26.Pyle AD, Lock LF. and Donovan PJ. (2006). Neurotrophins mediate human embryonic stem cell survival. Nat Biotechnol 24:344–350 [DOI] [PubMed] [Google Scholar]
- 27.Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K. and Sasai Y. (2007). A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25:681–686 [DOI] [PubMed] [Google Scholar]
- 28.Stover AE. and Schwartz PH. (2011). The generation of embryoid bodies from feeder-based or feeder-free human pluripotent stem cell cultures. Methods Mol Biol 767:391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ungrin MD, Joshi C, Nica A, Bauwens C. and Zandstra PW. (2008). Reproducible, ultra high-throughput formation of multicellular organization from single cell suspension-derived human embryonic stem cell aggregates. PLoS One 3:e1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaddah R, Arntfield M, Runciman S, Clarke L. and van der Kooy D. (2012). Clonal neural stem cells from human embryonic stem cell colonies. J Neurosci 32:7771–7781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valamehr B, Jonas SJ, Polleux J, Qiao R, Guo S, Gschweng EH, Stiles B, Kam K, Luo TJ, et al. , (2008). Hydrophobic surfaces for enhanced differentiation of embryonic stem cell-derived embryoid bodies. Proc Natl Acad Sci U S A 105:14459–14464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evseenko D, Schenke-Layland K, Dravid G, Zhu Y, Hao QL, Scholes J, Wang XC, Maclellan WR. and Crooks GM. (2009). Identification of the critical extracellular matrix proteins that promote human embryonic stem cell assembly. Stem Cells Dev 18:919–928 [DOI] [PubMed] [Google Scholar]
- 33.Ludwig TE, Bergendahl V, Levenstein ME, Yu J, Probasco MD. and Thomson JA. (2006). Feeder-independent culture of human embryonic stem cells. Nat Methods 3:637–646 [DOI] [PubMed] [Google Scholar]
- 34.Ludwig TE, Levenstein ME, Jones JM, Berggren WT, Mitchen ER, Frane JL, Crandall LJ, Daigh CA, Conard KR, et al. , (2006). Derivation of human embryonic stem cells in defined conditions. Nat Biotechnol 24:185–187 [DOI] [PubMed] [Google Scholar]
- 35.Cho SW, Moon SH, Lee SH, Kang SW, Kim J, Lim JM, Kim HS, Kim BS. and Chung HM. (2007). Improvement of postnatal neovascularization by human embryonic stem cell derived endothelial-like cell transplantation in a mouse model of hindlimb ischemia. Circulation 116:2409–2419 [DOI] [PubMed] [Google Scholar]
- 36.Kim J, Moon SH, Lee SH, Lee DR, Koh GY. and Chung HM. (2007). Effective isolation and culture of endothelial cells in embryoid body differentiated from human embryonic stem cells. Stem Cells Dev 16:269–280 [DOI] [PubMed] [Google Scholar]
- 37.Burridge PW, Anderson D, Priddle H, Barbadillo Munoz MD, Chamberlain S, Allegrucci C, Young LE. and Denning C. (2007). Improved human embryonic stem cell embryoid body homogeneity and cardiomyocyte differentiation from a novel V-96 plate aggregation system highlights interline variability. Stem Cells 25:929–938 [DOI] [PubMed] [Google Scholar]
- 38.Imamura T, Cui L, Teng R, Johkura K, Okouchi Y, Asanuma K, Ogiwara N. and Sasaki K. (2004). Embryonic stem cell-derived embryoid bodies in three-dimensional culture system form hepatocyte-like cells in vitro and in vivo. Tissue Eng 10:1716–1724 [DOI] [PubMed] [Google Scholar]
- 39.Kurosawa H, Imamura T, Koike M, Sasaki K. and Amano Y. (2003). A simple method for forming embryoid body from mouse embryonic stem cells. J Biosci Bioeng 96:409–411 [DOI] [PubMed] [Google Scholar]
- 40.Son MY, Kim HJ, Kim MJ. and Cho YS. (2011). Physical passaging of embryoid bodies generated from human pluripotent stem cells. PLoS One 6:e19134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vosough M, Omidinia E, Kadivar M, Shokrgozar MA, Pournasr B, Aghdami N. and Baharvand H. (2013). Generation of functional hepatocyte-like cells from human pluripotent stem cells in a scalable suspension culture. Stem Cells Dev 22:2693–2705 [DOI] [PubMed] [Google Scholar]
- 42.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J. and Gepstein L. (2001). Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest 108:407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Assady S, Maor G, Amit M, Itskovitz-Eldor J, Skorecki KL. and Tzukerman M. (2001). Insulin production by human embryonic stem cells. Diabetes 50:1691–1697 [DOI] [PubMed] [Google Scholar]
- 44.Wang L, Cerdan C, Menendez P. and Bhatia M. (2006). Derivation and characterization of hematopoietic cells from human embryonic stem cells. Methods Mol Biol 331:179–200 [DOI] [PubMed] [Google Scholar]
- 45.Duggal G, Heindryckx B, Warrier S, Taelman J, Van der Jeught M, Deforce D, Chuva de Sousa Lopes S. and De Sutter P. (2015). Exogenous supplementation of Activin A enhances germ cell differentiation of human embryonic stem cellsdagger. Mol Hum Reprod 21:410–423 [DOI] [PubMed] [Google Scholar]
- 46.Wang L, Li L, Shojaei F, Levac K, Cerdan C, Menendez P, Martin T, Rouleau A. and Bhatia M. (2004). Endothelial and hematopoietic cell fate of human embryonic stem cells originates from primitive endothelium with hemangioblastic properties. Immunity 21:31–41 [DOI] [PubMed] [Google Scholar]
- 47.Reubinoff BE, Pera MF, Fong CY, Trounson A. and Bongso A. (2000). Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol 18:399–404 [DOI] [PubMed] [Google Scholar]
- 48.Stenberg J, Elovsson M, Strehl R, Kilmare E, Hyllner J. and Lindahl A. (2011). Sustained embryoid body formation and culture in a non-laborious three dimensional culture system for human embryonic stem cells. Cytotechnology 63:227–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dang SM, Kyba M, Perlingeiro R, Daley GQ. and Zandstra PW. (2002). Efficiency of embryoid body formation and hematopoietic development from embryonic stem cells in different culture systems. Biotechnol Bioeng 78:442–453 [DOI] [PubMed] [Google Scholar]
- 50.Yoon BS, Yoo SJ, Lee JE, You S, Lee HT. and Yoon HS. (2006). Enhanced differentiation of human embryonic stem cells into cardiomyocytes by combining hanging drop culture and 5-azacytidine treatment. Differentiation 74:149–159 [DOI] [PubMed] [Google Scholar]
- 51.Rungarunlert S, Techakumphu M, Pirity MK. and Dinnyes A. (2009). Embryoid body formation from embryonic and induced pluripotent stem cells: benefits of bioreactors. World J Stem Cells 1:11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dang SM, Gerecht-Nir S, Chen J, Itskovitz-Eldor J. and Zandstra PW. (2004). Controlled, scalable embryonic stem cell differentiation culture. Stem Cells 22:275–282 [DOI] [PubMed] [Google Scholar]
- 53.Beauchamp P, Moritz W, Kelm JM, Ullrich ND, Agarkova I, Anson B, Suter TM. and Zuppinger C. (2015). Development and characterization of a scaffold-free 3D spheroid model of iPSC-derived human cardiomyocytes. Tissue Eng Part C Methods. [Epub ahead of print]; DOI: 10.1089/ten.tec.2014.0376 [DOI] [PubMed] [Google Scholar]
- 54.Lu H, Wang Z, Zheng Q, Li JH, Chong XQ. and Xiao SD. (2010). Efficient differentiation of newly derived human embryonic stem cells from discarded blastocysts into hepatocyte-like cells. J Dig Dis 11:376–382 [DOI] [PubMed] [Google Scholar]
- 55.Chisti Y. (2001). Hydrodynamic damage to animal cells. Crit Rev Biotechnol 21:67–110 [DOI] [PubMed] [Google Scholar]
- 56.Wartenberg M, Donmez F, Ling FC, Acker H, Hescheler J. and Sauer H. (2001). Tumor-induced angiogenesis studied in confrontation cultures of multicellular tumor spheroids and embryoid bodies grown from pluripotent embryonic stem cells. FASEB J 15:995–1005 [DOI] [PubMed] [Google Scholar]
- 57.Portner R, Nagel-Heyer S, Goepfert C, Adamietz P. and Meenen NM. (2005). Bioreactor design for tissue engineering. J Biosci Bioeng 100:235–245 [DOI] [PubMed] [Google Scholar]
- 58.Lock LT. and Tzanakakis ES. (2009). Expansion and differentiation of human embryonic stem cells to endoderm progeny in a microcarrier stirred-suspension culture. Tissue Eng Part A 15:2051–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cameron CM, Hu WS. and Kaufman DS. (2006). Improved development of human embryonic stem cell-derived embryoid bodies by stirred vessel cultivation. Biotechnol Bioeng 94:938–948 [DOI] [PubMed] [Google Scholar]
- 60.Yirme G, Amit M, Laevsky I, Osenberg S. and Itskovitz-Eldor J. (2008). Establishing a dynamic process for the formation, propagation, and differentiation of human embryoid bodies. Stem Cells Dev 17:1227–1241 [DOI] [PubMed] [Google Scholar]
- 61.Lam AT, Chen AK, Li J, Birch WR, Reuveny S. and Oh SK. (2014). Conjoint propagation and differentiation of human embryonic stem cells to cardiomyocytes in a defined microcarrier spinner culture. Stem Cell Res Ther 5:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilson JL. and McDevitt TC. (2013). Stem cell microencapsulation for phenotypic control, bioprocessing, and transplantation. Biotechnol Bioeng 110:667–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fukuda J, Khademhosseini A, Yeo Y, Yang X, Yeh J, Eng G, Blumling J, Wang CF, Kohane DS. and Langer R. (2006). Micromolding of photocrosslinkable chitosan hydrogel for spheroid microarray and co-cultures. Biomaterials 27:5259–5267 [DOI] [PubMed] [Google Scholar]
- 64.Gerlach JC, Hout M, Edsbagge J, Bjorquist P, Lubberstedt M, Miki T, Stachelscheid H, Schmelzer E, Schatten G. and Zeilinger K. (2010). Dynamic 3D culture promotes spontaneous embryonic stem cell differentiation in vitro. Tissue Eng Part C Methods 16:115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sargent CY, Berguig GY. and McDevitt TC. (2009). Cardiomyogenic differentiation of embryoid bodies is promoted by rotary orbital suspension culture. Tissue Eng Part A 15:331–342 [DOI] [PubMed] [Google Scholar]
- 66.Come J, Nissan X, Aubry L, Tournois J, Girard M, Perrier AL, Peschanski M. and Cailleret M. (2008). Improvement of culture conditions of human embryoid bodies using a controlled perfused and dialyzed bioreactor system. Tissue Eng Part C Methods 14:289–298 [DOI] [PubMed] [Google Scholar]
- 67.Khademhosseini A, Ferreira L, Blumling J, 3rd, Yeh J, Karp JM, Fukuda J. and Langer R. (2006). Co-culture of human embryonic stem cells with murine embryonic fibroblasts on microwell-patterned substrates. Biomaterials 27:5968–5977 [DOI] [PubMed] [Google Scholar]
- 68.Ferraro GA, De Francesco F, Tirino V, Cataldo C, Rossano F, Nicoletti G. and D'Andrea F. (2011). Effects of a new centrifugation method on adipose cell viability for autologous fat grafting. Aesthetic Plast Surg 35:341–348 [DOI] [PubMed] [Google Scholar]
- 69.Dahlmann J, Kensah G, Kempf H, Skvorc D, Gawol A, Elliott DA, Drager G, Zweigerdt R, Martin U. and Gruh I. (2013). The use of agarose microwells for scalable embryoid body formation and cardiac differentiation of human and murine pluripotent stem cells. Biomaterials 34:2463–2471 [DOI] [PubMed] [Google Scholar]
- 70.Karp JM, Yeh J, Eng G, Fukuda J, Blumling J, Suh KY, Cheng J, Mahdavi A, Borenstein J, Langer R. and Khademhosseini A. (2007). Controlling size, shape and homogeneity of embryoid bodies using poly(ethylene glycol) microwells. Lab Chip 7:786–794 [DOI] [PubMed] [Google Scholar]
- 71.Hwang YS, Chung BG, Ortmann D, Hattori N, Moeller HC. and Khademhosseini A. (2009). Microwell-mediated control of embryoid body size regulates embryonic stem cell fate via differential expression of WNT5a and WNT11. Proc Natl Acad Sci U S A 106:16978–16983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choi YY, Chung BG, Lee DH, Khademhosseini A, Kim JH. and Lee SH. Controlled-size embryoid body formation in concave microwell arrays. Biomaterials 31:4296–4303 [DOI] [PubMed] [Google Scholar]
- 73.Pettinato G, Vanden Berg-Foels WS, Zhang N. and Wen X. (2014). ROCK inhibitor is not required for embryoid body formation from singularized human embryonic stem cells. PLoS One 9:e100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carpenedo RL, Sargent CY. and McDevitt TC. (2007). Rotary suspension culture enhances the efficiency, yield, and homogeneity of embryoid body differentiation. Stem Cells 25:2224–2234 [DOI] [PubMed] [Google Scholar]
- 75.Ferreira L, Squier T, Park H, Choe H, Kohane DS. and Langer R. (2008). Human embryoid bodies containing nano- and microparticulate delivery vehicles. Adv Mater 20:2285–2291 [Google Scholar]
- 76.Fung WT, Beyzavi A, Abgrall P, Nguyen NT. and Li HY. (2009). Microfluidic platform for controlling the differentiation of embryoid bodies. Lab Chip 9:2591–2595 [DOI] [PubMed] [Google Scholar]
- 77.Kang E, Choi YY, Jun Y, Chung BG. and Lee SH. Development of a multi-layer microfluidic array chip to culture and replate uniform-sized embryoid bodies without manual cell retrieval. Lab Chip 10:2651–2654 [DOI] [PubMed] [Google Scholar]
- 78.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, et al. , (2007). Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol 25:1015–1024 [DOI] [PubMed] [Google Scholar]
- 79.Stern CD. (1992). Vertebrate gastrulation. Curr Opin Genet Dev 2:556–561 [DOI] [PubMed] [Google Scholar]
- 80.Allegrucci C. and Young LE. (2007). Differences between human embryonic stem cell lines. Human Reprod Update 13:103–120 [DOI] [PubMed] [Google Scholar]
- 81.Brolen G, Sivertsson L, Bjorquist P, Eriksson G, Ek M, Semb H, Johansson I, Andersson TB, Ingelman-Sundberg M. and Heins N. (2010). Hepatocyte-like cells derived from human embryonic stem cells specifically via definitive endoderm and a progenitor stage. J Biotechnol 145:284–294 [DOI] [PubMed] [Google Scholar]
- 82.Hay DC, Fletcher J, Payne C, Terrace JD, Gallagher RC, Snoeys J, Black JR, Wojtacha D, Samuel K, et al. , (2008). Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc Natl Acad Sci U S A 105:12301–12306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S. and Duncan SA. (2010). Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51:297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sullivan GJ, Hay DC, Park IH, Fletcher J, Hannoun Z, Payne CM, Dalgetty D, Black JR, Ross JA, et al. , Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology 51:329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gong G, Ferrari D, Dealy CN. and Kosher RA. (2010). Direct and progressive differentiation of human embryonic stem cells into the chondrogenic lineage. J Cell Physiol 224:664–671 [DOI] [PubMed] [Google Scholar]
- 86.Daniels K, Reiter R. and Solursh M. (1996). Micromass cultures of limb and other mesenchyme. Methods Cell Biol 51:237–247 [DOI] [PubMed] [Google Scholar]
- 87.Ying QL, Stavridis M, Griffiths D, Li M. and Smith A. (2003). Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol 21:183–186 [DOI] [PubMed] [Google Scholar]
- 88.Yao S, Chen S, Clark J, Hao E, Beattie GM, Hayek A. and Ding S. (2006). Long-term self-renewal and directed differentiation of human embryonic stem cells in chemically defined conditions. Proc Natl Acad Sci U S A 103:6907–6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Soteriou D, Iskender B, Byron A, Humphries JD, Borg-Bartolo S, Haddock MC, Baxter MA, Knight D, Humphries MJ. and Kimber SJ. Comparative proteomic analysis of supportive and unsupportive extracellular matrix substrates for human embryonic stem cell maintenance. J Biol Chem 288:18716–18731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ. and Palecek SP. (2012). Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A 109:E1848–E1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, et al. , (2014). Chemically defined generation of human cardiomyocytes. Nat Methods 11:855–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kattman SJ, Huber TL. and Keller GM. (2006). Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell 11:723–732 [DOI] [PubMed] [Google Scholar]
- 93.Klug MG, Soonpaa MH, Koh GY. and Field LJ. (1996). Genetically selected cardiomyocytes from differentiating embronic stem cells form stable intracardiac grafts. J Clin Invest 98:216–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zandstra PW, Bauwens C, Yin T, Liu Q, Schiller H, Zweigerdt R, Pasumarthi KB. and Field LJ. (2003). Scalable production of embryonic stem cell-derived cardiomyocytes. Tissue Eng 9:767–778 [DOI] [PubMed] [Google Scholar]