

Abstract
Nanocarriers delivering prodrugs are a way of improving in vivo effectiveness and efficiency. For therapeutic efficacy, the prodrug must hydrolyze to its parent drug after administration. Based on the fact that the hydrolysis is impeded by steric hindrance and improved by sufficient polarity, in this study, we proposed the PTX-S-S-VE, the conjugation of paclitaxel (PTX) to vitamin E (VE) through a disulfide bridge. This conjugate possessed the following advantages: first, it can be encapsulated in the VE/VE2-PEG2000/water nanoemulsions because of favorable hydrophobic interactions; second, the nanoemulsions had a long blood circulation time; finally, the concentrated glutathione in the tumor microenvironment could cleave the disulfide bond to weaken the steric hindrance and increase the polarity, promoting the hydrolysis to PTX and increasing the anticancer activity. It was demonstrated in vitro that the hydrolysis of PTX-S-S-VE was enhanced and the cytotoxicity was increased. In addition, PTX-S-S-VE had greater anticancer activity against the KB-3-1 cell line tumor xenograft and the tumor size was smaller after the 4th injection. The present result suggests a new way, use of reduction, to improve the in vivo anticancer activity of a prodrug for nanocarrier delivery by unshielding the ester bond and taking off the steric block.
A prodrug improves the properties of a parent drug by linking with or being modified by a functioning or non-functioning molecular group1,2. Prodrug strategy has been used to improve aqueous solubility, enhance chemical stability, promote transport across epithelial cells, reduce pre-systemic metabolism, and increase oral absorption3,4,5,6. It is administered in an inactive form, and it must convert or hydrolyze to its active form through physiological metabolism7,8. As estimated, prodrugs accounted for about 17.8% (34 of the 191) of new chemical entities approved between 2000 and 2008, and prodrug prevalence was 15.4% among the top 100 bestselling small molecular weight drugs in 20089,10.
Paclitaxel (PTX) is widely used for the treatment of non-small cell lung cancer, breast cancer, ovarian cancer and AIDS-related Kaposi’s sarcoma11. Both Taxol®12,13 and Abraxane®14,15 are commercial PTX formulations approved by the FDA. However, due to their low drug loadings and the problems associated with the burst release, experts have applied a prodrug strategy to improve the in vivo performance of PTX16,17. Some researchers have conjugated PTX with a hydrophilic polymer18,19,20,21,22,23, but the drug loading was still very low. In addition, the half-lives of the formulations are too short, leading to poor efficacy in vivo, although this strategy has prolonged the release and exhibited promising anticancer activity in vitro. Others have conjugated PTX with a hydrophobic chain24,25,26,27, in the hope that it could improve the compatibility between cargo and carrier, facilitating encapsulation in a nanocarrier. Nevertheless, the potential anticancer activity was limited due to poor hydrolysis, as evidenced by the fact that no more than 10% of PTX-linoleic acid conjugate was hydrolyzed to PTX at 48 h28.
In our group, PTX-VE (Fig. 1) has been synthesized by conjugating PTX and vitamin E (VE) together via an ester bond, and it has been encapsulated in the VE/VE2-PEG2000/water nanoemulsions (NES) because PTX-VE, VE, and VE2-PEG2000 were molecularly matched. Based on this, we have codelivered PTX and 5-fluoroucacil in a same nanoparticle by their conjugation with the VE29. What is more, using the same strategy, theranostic (PTX, and sulforhodamine B) has been also achieved30. However, PTX-VE did not release active PTX in vitro within 48 h, resulted in negotiated cytotoxicity. Thus, the aim of this paper was to find out a substitute for PTX-VE. We initially demonstrated that its hydrolysis was impeded by steric hindrance and improved by sufficient polarity, by grafting different groups onto PTX. Then, we proposed and synthesized PTX-S-S-VE, PTX conjugated with VE via a disulfide bond, a redox sensitive bridge (Fig. 2). This bridge would be broken in tumor cells because of the sufficient glutathione (GSH) concentration (2–8 mM)31, to promote the hydrolysis. In addition, the PTX-S-S-VE can also be encapsulated and assembled as PTX-S-S-VE/VE/VE2-PEG2000/water NES for administration because of favourable hydrophobic interactions between the prodrug and the nanocarrier. The tissue distribution, in vivo antitumor activity and hemolytic activity were evaluated for PTX-S-S-VE/VE/VE2-PEG2000/water NES, with PTX-VE/VE/VE2-PEG2000/water NES and Taxol® as reference formulations.
Figure 1. Chemical structures of PTX, PTX-VE, PTX-SEE, PTX-SA, and PTX-S-S-VE.
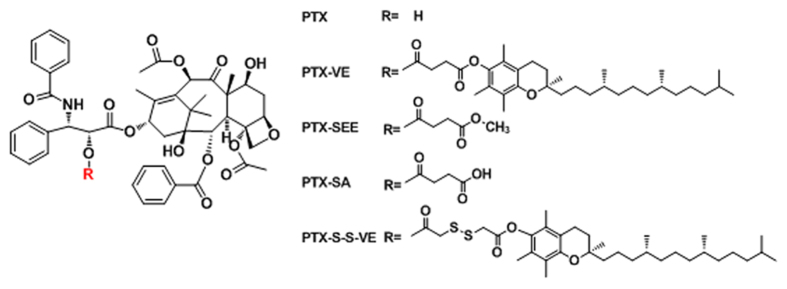
Figure 2. Schematic illustration for the enhanced hydrolysis of PTX-S-S-VE.
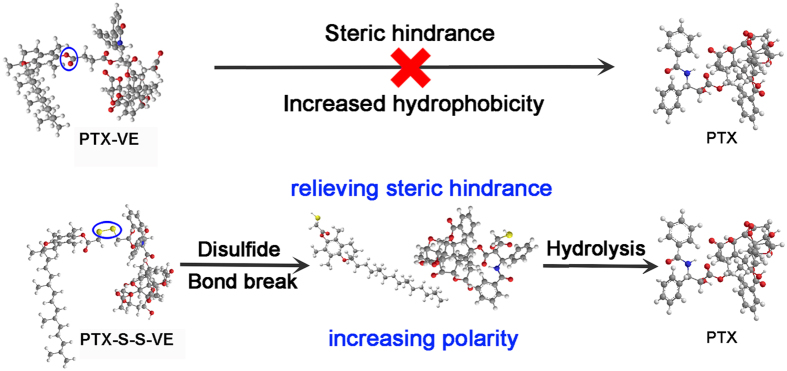
The prodrug will experience a two stepped hydrolysis to overcome the steric hindrance and polarity hurdles. The disulfide bond will be cleaved in tumor cells because the concentration of GSH in tumor cells is much higher than in blood plasma. This initial hydrolysate will then be further hydrolyzed to PTX because of the increased polarity.
Results and discussion
Proof-of-concept
In the hydrolysis study, the PTX-VE was not hydrolyzed until 48 h. The reason for the poor hydrolysis of PTX-VE might be that the hydrolytic site (C-2' hydroxyl group in PTX) is sterically shielded by the VE. To test this hypothesis, PTX-SEE (the VE group substituted by a methyl group) was synthesized. Both PTX-VE and PTX-SEE are hydrophobic. As shown in Fig. 3a, approximately 20.7% of PTX-SEE was hydrolyzed to free PTX by 48 h, a much higher percentage than for PTX-VE. This indicates that steric hindrance could be a major reason for impeded hydrolysis.
Figure 3.
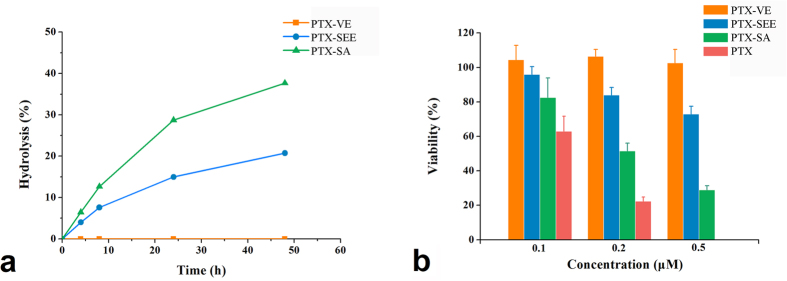
Hydrolytic release profiles in pH 7.4 PBS containing 7% DMSO and 0.1% Tween 80 (a) and in vitro cytotoxicity against KB-3-1 cell line for PTX-VE, PTX-SEE, and PTX-SA (b). These studies were carried out at 37 °C.
In addition to this, Markovic et al. established a good correlation (R = 0.9924) between solvolytic rate constants and polarity, indicating enhanced polarity could improve hydration for a prodrug32. It is reasonable to assume that the hydrolysis rate is dependent on polarity to some extent. Therefore, we conjugated PTX and succinic acid directly to produce PTX-SA. In comparison with PTX-SEE, PTX-SA had increased polarity, which was evidenced by the retention time (6.7 min for PTX-SA and 31.7 min for PTX-SEE) in a reversed phase chromatography system (Figure S6). As expected, PTX-SA had a higher hydrolytic profile than PTX-SEE, suggesting that sufficient polarity might be the other reason for hydrolysis.
Therefore, decreased steric hindrance and enhanced polarity favor increased hydrolysis.
Cytotoxicity demonstration
For PTX, the hydroxyl group at the C-2' position is important for its anticancer activity and it must therefore be exposed to the external environment to have its effect16,17. Thus, the hydrolytic rate for a PTX prodrug may therefore influence its in vitro cytotoxicity. In this research, the cytotoxicity was investigated on the KB-3-1 cell line using the MTS method. Free PTX, PTX-SA, PTX-SEE, and PTX-VE were added to the cells, respectively, with concentrations ranging from 0.1–0.5 μM. As shown in Fig. 3b, PTX-VE had no inhibitory effect when concentration was up to 0.5 μM because of the negligible hydrolysis, and its IC50 value was as high as 2.67 μM. For PTX-SEE, the inhibition effect was greater, and the viability was about 72.8% when cells treated with 0.5 μM of PTX-SEE. In the case of PTX-SA, the IC50 was 0.252 μM. In addition, free PTX had the strongest inhibition effect. The cytotoxicity did correlated well with the hydrolytic rate.
Rational design for PTX-S-S-VE
In a previous study, we have developed PTX-VE/VE/VE2-PEG2000/water NES and demonstrated sufficiently high drug loading and improved pharmacokinetics, because the PTX-VE was subjected to strong interactions with the VE and VE2-PEG2000. However, the cytotoxicity of PTX-VE was very low because of poor hydrolysis. Therefore, in this study, we would like to develop a substitute for PTX-VE, by maintaining the VE group and changing the linker, in order to improve the hydrolysis.
On the basis of the analysis above and considering the redox nature of the tumor microenvironment33, we undertook to synthesize PTX-S-S-VE (PTX conjugated with VE via a disulfide bond) because the sufficient GSH34,35 in tumor cells will cleave the bridge and promote the hydrolysis to free PTX. As shown in Fig. 4, the hydrolysis rate was accelerated with GSH concentration increased. In addition, compared with PTX-VE, the in vitro hydrolysis was improved and its anticancer activity was increased.
Figure 4. In vitro hydrolytic profiles and cytotoxicity against KB-3-1 cell line for PTX-S-S-VE (data are expressed as the mean ± SD, n = 5 wells for each formulation at each concentration).

These studies were carried out at 37 °C.
Preparation, characterization and storage stability of the NES
PTX-VE/VE/VE2-PEG2000 NES has been prepared because of favorable affinity for PTX-VE, VE, and VE2-PEG2000. In this study, PTX-S-S-VE/VE/VE2-PEG2000 NES was also prepared successfully. The particle size, zeta potential, and morphology were characterized. As shown in Fig. 5, for both NES, the particles were of similar spherical shape and size (100–150 nm). Consistent with the TEM images, the dynamic light scattering results showed that both NES were about 130 nm (Table 1, and Figure S8–9). The small PDI indicated that the particles were of a narrow size range. It was demonstrated that the PTX-S-S-VE/VE/VE2-PEG2000 NES was physically stable for at least 12 months (Table S1). Therefore, the PTX-S-S-VE/VE/VE2-PEG2000 NES was suitable for in vivo studies.
Figure 5. TEM images of PTX-VE/VE/VE2-PEG2000/water (a) and PTX-S-S-VE/VE/VE2-PEG2000/water (b) NES.
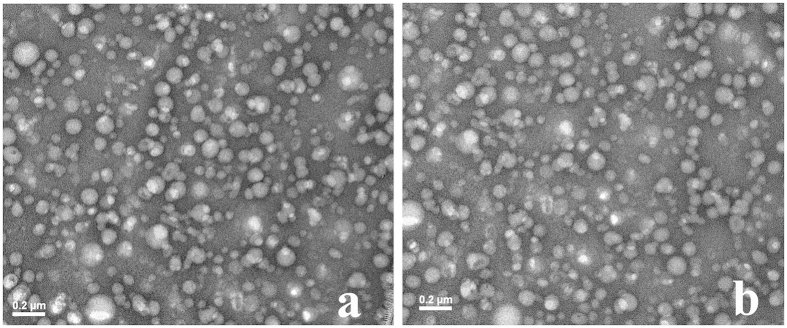
Table 1. Particle sizes and zeta-potentials of PTX-VE/VE and PTX-S-S-VE/VE NES (data are expressed as the mean ± SD, n = 3 for each measurement).
| Formulation | Z-average (nm) | PDI | Zeta-potential (mV) |
|---|---|---|---|
| PTX-VE NES | 134.6 ± 0.6 | 0.175 ± 0.012 | –4.03 ± 0.27 |
| PTX-S-S-VE NES | 128.1 ± 0.4 | 0.164 ± 0.027 | –3.99 ± 0.26 |
Tissue distribution
To evaluate the in vivo performance, the CD1 mice, in the biodistribution study, were administrated intravenously a single dose equivalent of 20 mg/kg PTX. The mice were sacrificed at 4 h and the tissues were harvested. The biodistribution of free PTX and PTX prodrugs are shown in Fig. 6a,b, respectively. For PTX-VE NES, free PTX was only found in the liver, with only a small amount being found in other tissues. This was because of the low cleavage rate of the ester bond, and indicated similar in vivo hydrolytic behavior to that seen in vitro. On the other hand, PTX-VE NES showed high prodrug accumulation, including higher than that of PTX-S-S-VE NES, with liver (3794.0 ± 1241.6 ng/g) being the major distribution tissue followed by spleen (1753.1 ± 666.7 ng/g) and then kidney (1351.4 ± 179.3 ng/g). The reason for this could be attributed to matching properties of VE oil and the VE group in PTX-VE, as well as PTX-VE being less susceptible to hydrolysis than PTX-S-S-VE. Importantly, it exhibited comparable prodrug distribution in tumors for PTX-VE NES and PTX-S-S-VE NES. In the case of PTX-S-S-VE NES, greater free PTX accumulation was seen than for Taxol® in most tissues. The distribution was statistically higher than Taxol® in plasma (p < 0.05), liver (p < 0.05), spleen (p < 0.05), kidney (p < 0.05) and tumor (p < 0.05, Student’s t-test, paired, two sided). This could be because PTX-S-S-VE and VE are molecularly matched resulting in longer circulation of the PTX-S-S-VE NES in the blood and hence longer in vivo retention time than for the Taxol®. In addition, the GSH in blood, despite being of much lower concentration than in the tumor, could start the hydrolytic program to some extent, leading to enhanced hydrolysis. The in vivo distribution results indirectly reflect the hydrolytic properties of the prodrugs.
Figure 6.
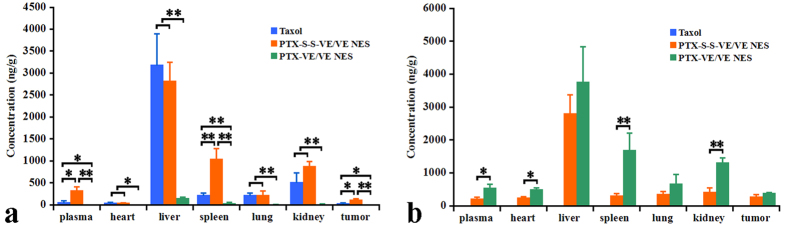
Tissue distribution of parent drug (a) and prodrugs (b) at 4 h after intravenous administration of Taxol®, PTX-VE/VE/VE2-PEG2000/water and water NES PTX-S-S-VE/VE/VE2-PEG2000/ at a PTX equivalent single dose of 20 mg/kg at 4 h (p < 0.05 [*] and p < 0.01 [**]).
Tumor growth inhibition
To evaluate the in vivo therapeutic effect of the formulations, tumor growth inhibition was studied in a KB-3-1 cells subcutaneous model in Balb/C nude mice. As shown in Fig. 7, the control group showed very rapid tumor growth. There was no significant reduction of the tumor volume in mice treated with PTX-VE/VE/VE2-PEG2000 NES (p > 0.05, Student’s t-test, paired, two sided). However, the mean tumor volume was slightly smaller than for the controls. Not surprisingly, the greatest antitumor activity was observed when the mice were treated with PTX-S-S-VE/VE/VE2-PEG2000 NES. For this formulation, the tumors were diminished before the fifth injection. Similarly, Taxol® also showed a strong tumor inhibition effect and only one mouse was found to still bear the tumor by the 15th day. In addition, no weight loss occurred in all groups (Fig. S10).
Figure 7. Inhibition of tumor growth by Taxol®, PTX-VE/VE/VE2-PEG2000/water and PTX-S-S-VE/VE/VE2-PEG2000/water NES in KB-3-1 cell subcutaneous xenografts.
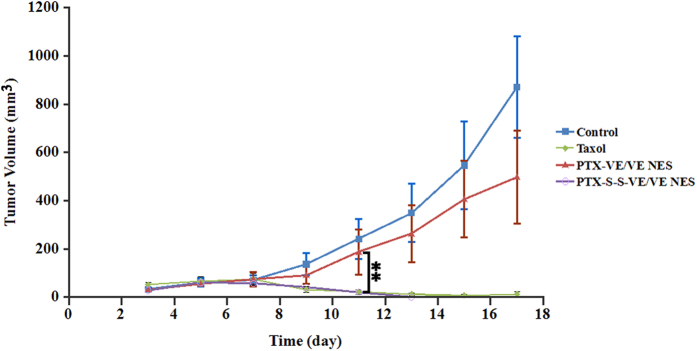
The formulations were administered intravenously at an equivalent dose of 5 mg/kg of PTX every second day for 5 injections in total (data are expressed as the mean ± SD, n = 5 mice for each formulation) (p < 0.05 [*] and p < 0.01 [**]).
In vivo toxicity
Histopathological examination
In order to determine if the accumulation of the formulations is able to damage normal tissues, the histopathological of the heart, liver, spleen, lung, and kidney for each treatment group was examined. The results were shown in Fig. S11. Lesions were not found in the PTX-VE/VE and PTX-S-S-VE/VE NES treated group. However, it is clear that the kidney, in Taxol® treated group, suffers from dilatation and congestion for renal interstitial vascular.
Measurement of serum level
AST and ALT are two tests usually used to evaluate the main liver function. In this study, the ALT and AST were significantly elevated in the group treated with Taxol® but not with the PTX-S-S-VE/VE/VE2-PEG2000/water NES (Table 2), indicating PTX-S-S-VE/VE/VE2-PEG2000 NES is much better tolerated than Taxol®.
Table 2. Kidney and liver function parameters (AST, ALT, and BUN) in the different experimental group. Values are expressed as mean ± SD.
| AST(U/L) | ALT(U/L) | BUN(mg/dL) | |
|---|---|---|---|
| Taxol® | 398.6 ± 88.7 | 140.2 ± 44.5 | 21.8 ± 2.2 |
| PTX-VE NEs | 198.0 ± 35.7* | 35.2 ± 6.3* | 24.2 ± 1.3 |
| Normal Range | 54–298 | 17–77 | 8–33 |
*p < 0.05 versus Taxol® as a control.
Hemolysis study
It is acknowledged that hemolysis is a major drawback for surfactant based formulations, such as Taxol®. In this study, TPGS and VE2-PEG, which can each also cause hemolysis, were used for stabilizing the NES. Therefore, to evaluate the safety of the two NES, we studied the hemolysis effect by incubating erythrocytes with the formulations (Taxol®, PTX-VE/VE/VE2-PEG2000 NES, PTX-S-S-VE/VE/VE2-PEG2000 NES, and Abraxane®) for 1 h at different PTX concentrations. Figure 8 shows hemolysis results for the formulations at various concentrations. At 0.0293 μM of PTX, all formulations were nonhemolytic. As the concentration was increased, hemolysis appeared. At the highest concentration tested (0.117 μM), 91.3% of the erythrocytes were hemolyzed for the Taxol® group, while only 19.5%, 15.6%, and 12.9% of erythrocytes were hemolyzed for PTX-S-S-VE/VE/VE2-PEG2000 NES, PTX-VE/VE/VE2-PEG2000 NES, and Abraxane®, respectively. Therefore, it can be suggested that both of the prodrug NES formulations are safer than Taxol® in terms of membrane toxicity.
Figure 8. Hemolytic activity of Taxol®, PTX-VE/VE/VE2-PEG2000/water NES, PTX-S-S-VE/VE/VE2-PEG2000/water NES, and Abraxane® at 37 °C for 1 h (data are expressed as the mean ± SD, n = 3 mice for each measurement) (p < 0.05 [*] and p < 0.01 [**]).

Material and Methods
Materials
PTX was purchased from Lc Laboratories (Woburn, MA). PTX injection (Taxol®) was manufactured by Ben Venue laboratories, Inc. (Bedford, OH). TPGS was purchased from Eastman Chemical Company (Anglesey, U.K.). (+)-a-Tocopherol (VE), D-a-Tocopherol succinate, N, N-dicyclohexylurea tert-butyl-2-aminoethylcarbamate, trifluoroacetic acid, tert-butyl methyl ether were purchased from Sigma-Aldrich Corporation (St. Louis, MO).
Synthesis of PTX-VE, PTX-SA, PTX-SEE, and PTX-S-S-VE
Synthesis of PTX-VE
PTX (100.0 mg, 0.117 mmol) was reacted with D-α-tocopherol succinate (62.0 mg, 0.117 mmol) in the presence of N, N’-dicyclohexylcarbodiimide (48.2 mg, 0.234 mmol) and a catalytic amount of dimethylaminopyridine (DMAP) in anhydrous dichloromethane (8 mL) at room temperature under nitrogen atmosphere for 7 h. The resulting mixture was filtered to remove N, N-dicyclohexylurea (DCU) and the filtrate was dried under vacuum. The residue was purified using silica gel column chromatography, eluting with a chloroform-methanol solution with gradually increasing methanol content. The eluting solvent was removed under vacuum to give 95.9 mg of PTX-VE conjugate with the total yield of 60%. The purity was 99.2%, analyzed by the HPLC method.
Synthesis of PTX-SA
A solution of PTX (200.0 mg, 0.234 mmol), succinic anhydride (46.9 mg, 0.468 mmol), a catalytic amount of DMAP, and DCM (5 mL) was stirred for 3 h at room temperature. The product was purified using silica gel column chromatography, eluting with a solution of chloroform and methanol (200:1, V/V) to obtain the target compound PTX-SA as white solid (145.0 mg, yield: 65.0%). The purity was 98.8%, analyzed by the HPLC method.
Synthesis of PTX-SEE
Monomethyl succinate (34.0 mg, 0.257 mmol) and dichloromethane (5 mL) were stirred in an ice water bath. DCC (53 mg, 0.257 mmol) and a catalytic amount of DMAP were added. The solution turned to suspension. After 5 min, PTX (200.0 mg, 0.234 mmol) was added. After 2 h, the suspension was filtered to remove DCU and then purified by silica gel column chromatography. The elution solution was chloroform and methanol (200:1, V/V) and the target compound was obtained as a white solid (110 mg, yield: 44.5%). The purity was 99.0%, analyzed by the HPLC method.
Synthesis of PTX-S-S-VE
A solution of dithiodiglycolic acid (2 g, 11.0 mmol) and anhydrous acetic anhydride (30 mL) was stirred at 30 °C under nitrogen atmosphere for 2 h. Acetic anhydride was removed at room temperature under high vacuum with the addition of toluene three times. The residue was added to a mixed solution of dichloromethane (20 mL), VE (1.0 g, 2.3 mmol) and a catalytic amount of DMAP, and stirred for 5 min at room temperature. The product was purified by silica gel column chromatography, eluting with a solution of hexane, ethyl acetate and acetic acid. The eluting solvent was removed under vacuum to give 0.757 g of acid VE-S-S-COOH with a yield of 55.3%. The acid (0.5 g, 0.84 mmol) and dichloromethane (10 mL) were stirred in an ice water bath. DCC (0.198 g, 1.0 mmol) and DMAP (13.8 mg, 0.01 mmol) were added. The solution changed to a suspension. After 5 min, PTX (0.72 g, 0.84 mmol) was added and the mixture was stirred for a further 2 h. After filtration to remove DCU, the solution was purified by silica gel column chromatography, eluting with a solution of chloroform and methanol (200:1) to obtain the target compound PTX-S-S-VE as white solid (0.814 g, yield: 67.7% ). The purity was 99.4%, analyzed by the HPLC method.
HPLC analysis
The HPLC system was equipped with a Waters 2487 Dual λ Absorption Detector and a Waters 600 pump (Waters Corporation, Milford, US). The separation was carried out on an ODS HYPERSIL (3 μm, 2.1 mm × 100 mm) column (Thermo Scientific, USA) at a flow rate of 0.5 mL/min. Compound concentration was determined at a ultraviolet wavelength of 228 nm and the injection volume was 20 μL. Gradient elution was applied with a mobile phase of acetonitrile (ACN) and water. The initial mobile phase composition was 40% ACN from 0–36 min, and a linear gradient was applied to reach a composition of 100% ACN after a further 1 min. This was maintained for 30 min and then returned to initial conditions within a further 1 min and equilibrated for 9 min. Total run time was 77 min.
To analyze the hydrolysis of the prodrugs (PTX-VE and PTX-S-S-VE), the initial mobile phase composition (40% ACN) was maintained from 0–12 min, then a linear gradient was applied to reach a composition of 100% ACN after a further 1 min. This was maintained for 23 min and then returned to initial conditions within a further 1 min.
Hydrolysis
Hydrolytic studies were carried out at 37 °C in pH 7.4 PBS containing 7% DMSO and 0.1% Tween 80. The prodrugs (PTX-SA, PTX-SEE, PTX-VE, PTX-Br2-C16 and PTX-S-S-VE), equivalent to 11.7 μM of PTX, were added to 100 mL of the medium and placed in a shaking table at a rotating rate of 100 rpm/min. At predetermined time intervals (4, 8, 24 and 48 h), 1 mL of the samples were drawn from the medium and filtered through acetate membranes (0.22 μm, GE Water & Process Technologies, Trevose, PA). The filtrate was mixed with an equal volume of acetonitrile and analyzed using the HPLC method described earlier. The chromatograms were presented as Fig. S7.
Cytotoxicity
MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-car-boxymethoxyphenyl)-2-(4-sulfophenyl)-2H- tetrazolium, inner salt, was used to study the cytotoxicity of the prodrugs following the manufacture’s protocol. Generally, the KB-3-1 cells were seeded in 96 wells at a density of 1 × 104 cells per well. After incubation for 24 h, the cells were treated with PTX, PTX-SA, PTX-SEE, PTX-VE, or PTX-S-S-VE at various concentrations for 48 h. Sterile water treated wells were used as controls. After the incubation, the media was replaced with a mixture of fresh cell culture medium and MTS-based CellTiter 96® AQueous One Solution Cell Proliferation Assay Reagent (Promega Corporation, Madison, WI). The plate was then incubated at 37 °C for a further 2 h. Absorbance was determined at 480 nm using a plate reader. Cell viability was calculated as ODtest/ODcontrol × 100%.
Preparation, characterization and storage stability study of the NES
PTX-VE, PTX-S-S-VE, VE, TPGS and VE2-TPGS were dissolved in chloroform. PTX-VE (0.70 μM) (or PTX-S-S-VE [0.70 μM]), VE (20 μM), TPGS (1.8 μM) and VE2-TPGS (0.34 μM) were added to an Eppendorf Safe Lock TubeTM (1.5 mL) (Next Advance Inc., Averill Park, NY) and the chloroform removed under nitrogen flow and further by vacuum pumping. Water (600 μL) was then added. The mixture was probe sonicated for about 1 min, and the initial NES were mixed with 0.5 g of beads (one third × 1.0 mm in diameter and two thirds × 0.5 mm in diameter). The NES were placed in a BBY24M Bullet Blender Storm (Next Advance Inc., Averill Park, NY) for homogenization.
The particle size and zeta potential of the NES were determined using a Malvern ZetaSizer® Nano ZS (Westborough, MA) three times. In addition, the morphology was observed with a JEOL 100CX II TEM (Tokyo, Japan).
The PTX-S-S-VE/VE/VE2-PEG2000/water NES was stored at 25 °C for 12 months. At predetermined time intervals, samples were withdrawn and evaluated, including the physical appearance, particle size, polydispersity index (PDI), and encapsulation efficiency (EE). These studies were performed in triplicate and data were expressed as mean ± SD.
Animal studies
All animal research work was approved by the University of North Carolina at Chapel Hill’s Institutional Animal Care and Use Committee and were performed in accordance with relevant guidelines and regulation.
Biodistribution
Balb/C nude mice (6–8 weeks) were used to evaluate the in vivo biodistribution of the formulations. The xenograft models were established by subcutaneous injection of KB-3-1 (5 × 106) cells into the right flanks of the mice. Fifteen mice were randomly divided into three groups (n = 5 for each group) to receive Taxol®, PTX-VE/VE/VE2-PEG2000/water or PTX-S-S-VE/VE/VE2-PEG2000/water NES, respectively. The formulations were administered intravenously via the tail vein at a single dose of 20 mg/kg PTX equivalent. After 4 h, the mice were sacrificed, and the plasma, heart, liver, spleen, lungs, kidney, and tumor were collected. The tissues were homogenized with 300 μL of HPLC initial mobile phase (ACN:water = 40:60, w/w) using the BBY24M Bullet Blender Storm. The supernatant was obtained by centrifuging the tubes. The PTX and/or prodrugs (PTX-VE and PTX-S-S-VE) were extracted using methyl tert-butyl ether, with PTX-SEE (11.7 μM) as internal standard. The organic phase was transferred to a glass tube and evaporated under nitrogen flow. The residues were redispersed using the initial mobile phase 40:60 (ACN:water, v/v). Analysis was carried out by the HPLC method described earlier.
In vivo anticancer effect
The KB-3-1 tumor bearing nude mice were randomly divided into four groups (n = 5 for each group) to intravenously receive PTX equivalent doses (5 mg/kg) of Taxol®, PTX-VE NES, PTX-S-S-VE NES or physiological saline. The treatments were started on the third day after inoculation, and tumor sizes and body weights were monitored every second day. Tumor volume was calculated as V = 0.5×(L × W2), where V represents tumor volume, L the larger perpendicular diameter and W the smaller one.
Histopathological examination
The histopathological examinations were conducted on the organs of the sacrificed mice. The tissues, including heart, liver, spleen, lung, and kidney, were collected, rinsed with normal saline, fixed in 10% formalin, and then stained with hematoxylin and eosin. Finally, the sections were observed under a microscope for histopathological evaluations.
Measurement of serum level
Ten CD1 mice were used for the analysis of serum biochemistry. The mice were randomly divided into two groups (n = 5 for each group) to receive Taxol® and PTX-S-S-VE/VE/VE2-PEG2000/water NES, respectively. The blood samples were collected by enucleating the eyeball after mice received 5 times 10 mg/kg PTX equivalent every second day, and the serum was obtained by coagulating and then centrifuging. Subsequently, the serum samples were analyzed using an AU640 blood biochemical analyzer (Olympus Co., Tokyo, Japan). The serum biochemistry analysis involved the determination of aspartate aminotransferase (AST), alanine transaminase (ALT), and blood urea nitrogen (BUN).
Hemolysis
To test the potential toxicity of the NES, hemolysis was evaluated. Blank blood was obtained by orbital collection from the CD1 mice. Erythrocytes were immediately obtained by centrifuging the whole blood and washing twice with PBS (pH 7.4) to remove plasma and serum. The isolated erythrocytes (3 × 1010 cells) were resuspended and incubated with the formulations at various concentrations at 37 °C for 1 h. The samples were then centrifuged for 10 min at 16000 g, and 100 μL supernatants were diluted with an equal volume of PBS and measured for optical density (OD) at 570 nm. A complete hemolytic sample, which was used as a control, was prepared by sonicating blood for 3 min. The percentage of hemolysis was calculated as (ODtest – ODformulation) /ODcontrol × 100%36.
Statistical analysis
All data were expressed as mean ± SD and were analyzed statistically using a one-way analysis of variance (ANOVA) or a two tailed Student’s t-test. The differences were considered statistically significant if the p value was less than 0.05 (p < 0.05).
Conclusion
In this study, by giving a comparison of hydrolytic release and cytotoxicity for the three conjugates (PTX-VE, PTX-SA, and PTX-SEE), we have found that polarity and steric hindrance are two major factors influencing anticancer activity. Thus, we have designed a programmably hydrolyzable PTX prodrug, PTX-S-S-VE, which undergoes two subsequent hydrolysis steps and releases active drug. Like the previous PTX-VE, PTX-S-S-VE can also be encapsulated in VE/VE2-PEG2000/water NES for intravenous administration. The in vivo distribution for the two NES formulations varied a lot. In nude mice, PTX-S-S-VE/VE/VE2-PEG2000/water NES had better anticancer activity against KB-3-1 cell line tumors than PTX-VE/VE/VE2-PEG2000/water NES. Both the hemolysis activity was reduced compared with Taxol®. In conclusion, the programmably hydrolyzable PTX prodrug that we have produced demonstrates a bright future for controlling drug release in the treatment of cancer.
Additional Information
How to cite this article: Fu, Q. et al. Programmed Hydrolysis in Designing Paclitaxel Prodrug for Nanocarrier Assembly. Sci. Rep. 5, 12023; doi: 10.1038/srep12023 (2015).
Supplementary Material
Acknowledgments
This work was supported by the National Cancer Institute - National Institutes of Health (5R01CA149387), by Specific Research Grant (Young Scientist) for Science Foundation of Shenyang Pharmaceutical University (QNJJ2013504), by Doctoral Research Funding of Liaoning Province (No. 20141066), by the General Project in Education Department of Liaoning Province (No. L2014379), by the Career Development Program for Young Teachers in Shenyang Pharmaceutical University, and by the National Nature Science Foundation of China (No. 81273450, No. 81373336). This paper is dedicated to the memory of Professor Feng Liu, PhD, 1955-2014 University of North Carolina at Chapel Hill.
Footnotes
Author Contributions Q.F. and Y.W. wrote the main manuscript text; Y.M. And D.Z. Did data collection; J.K.F. corrected the language; X.Y. participated in data analysis; D.L. synthesized all the chemical compounds; Z.H. and F.L. Did study conception and design. All authors reviewed the manuscript.
References
- Jana S., Mandlekar S. & Marathe P. Prodrug design to improve pharmacokinetic and drug delivery properties: challenges to the discovery scientists. Curr Med Chem 17, 3874–3908 (2010). [DOI] [PubMed] [Google Scholar]
- Majumdar S., Duvvuri S. & Mitra A. K. Membrane transporter/receptor-targeted prodrug design: strategies for human and veterinary drug development. Adv Drug Deliv Rev 56, 1437–1452 (2004). [DOI] [PubMed] [Google Scholar]
- Singh Y., Palombo M. & Sinko P. J. Recent trends in targeted anticancer prodrug and conjugate design. Curr Med Chem 15, 1802–1826 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller C. E. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem Biodivers 6, 2071–2083 (2009). [DOI] [PubMed] [Google Scholar]
- Yan Z. et al. Bifunctional peptidomimetic prodrugs of didanosine for improved intestinal permeability and enhanced acidic stability: synthesis, transepithelial transport, chemical stability and pharmacokinetics. Mol Pharm 8, 319–329 (2011). [DOI] [PubMed] [Google Scholar]
- Sun Y. et al. Synthesis, transport and pharmacokinetics of 5’-amino acid ester prodrugs of 1-beta-D-arabinofuranosylcytosine. Mol Pharm 6, 315–325 (2009). [DOI] [PubMed] [Google Scholar]
- Sabit H. et al. Cytomegalovirus protease targeted prodrug development. Mol Pharm 10, 1417–1424 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S. et al. Targeted delivery of a model immunomodulator to the lymphatic system: comparison of alkyl ester versus triglyceride mimetic lipid prodrug strategies. J Control Release 177, 1–10 (2014). [DOI] [PubMed] [Google Scholar]
- Stella V. J. Prodrugs: Some thoughts and current issues. J Pharm Sci 99, 4755–4765 (2010). [DOI] [PubMed] [Google Scholar]
- Rautio J. Prodrug design and intellectual property. In Prodrugs and targeted delivery: towards better ADME properties. (Wiley-VCH, New York, 2011). [Google Scholar]
- Fonseca C., Simoes S. & Gaspar R. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Control Release 83, 273–286 (2002). [DOI] [PubMed] [Google Scholar]
- Steed H. & Sawyer M. B. Pharmacology, pharmacokinetics and pharmacogenomics of paclitaxel. Pharmacogenomics 8, 803–815 (2007). [DOI] [PubMed] [Google Scholar]
- Straub J. A. et al. Intravenous hydrophobic drug delivery: a porous particle formulation of paclitaxel (AI-850). Pharm Res 22, 347–355 (2005). [DOI] [PubMed] [Google Scholar]
- Fu Q. et al. Nanoparticle albumin-bound (NAB) technology is a promising method for anti-cancer drug delivery. Recent Pat Anticancer Drug Discov. 4, 262–272 (2009). [DOI] [PubMed] [Google Scholar]
- Wong J. et al. Suspensions for intravenous (IV) injection: a review of development, preclinical and clinical aspects. Adv Drug Deliv Rev 60, 939–954 (2008). [DOI] [PubMed] [Google Scholar]
- Zhu Q., Guo Z., Huang N., Wang M. & Chu F. Comparative molecular field analysis of a series of paclitaxel analogues. J Med Chem 40, 4319–4328 (1997). [DOI] [PubMed] [Google Scholar]
- Sun L., Simmerling C. & Ojima I. Recent advances in the study of the bioactive conformation of taxol. ChemMedChem 4, 719–731 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald R. B., Choe Y. H., McGuire J. & Conover C. D. Effective drug delivery by PEGylated drug conjugates. Adv Drug Deliv Rev 55, 217–250 (2003). [DOI] [PubMed] [Google Scholar]
- Li C. et al. Synthesis and evaluation of water-soluble polyethylene glycol-paclitaxel conjugate as a paclitaxel prodrug. Anticancer Drugs 7, 642–648 (1996). [DOI] [PubMed] [Google Scholar]
- Skwarczynski M., Hayashi Y. & Kiso Y. Paclitaxel prodrugs: toward smarter delivery of anticancer agents. J Med Chem 49, 7253–7269 (2006). [DOI] [PubMed] [Google Scholar]
- Chen C. et al. A prodrug strategy based on chitosan for efficient intracellular anticancer drug delivery. Nanotechnology 25, 255101 (2014). [DOI] [PubMed] [Google Scholar]
- Jiang T. et al. Amphiphilic polycarbonate conjugates of doxorubicin with pH-sensitive hydrazone linker for controlled release. Colloids Surf B Biointerfaces 111, 542–548 (2013). [DOI] [PubMed] [Google Scholar]
- Jung B. et al. Tumor-binding prodrug micelles of polymer-drug conjugates for anticancer therapy in HeLa cells. Journal of Materials Chemistry 22, 9385–9394 (2012). [Google Scholar]
- Lundberg B. B., Risovic V., Ramaswamy M. & Wasan K. M. A lipophilic paclitaxel derivative incorporated in a lipid emulsion for parenteral administration. J Control Release 86, 93–100 (2003). [DOI] [PubMed] [Google Scholar]
- Yoong S. L. et al. Enhanced cytotoxicity to cancer cells by mitochondria-targeting MWCNTs containing platinum(IV) prodrug of cisplatin. Biomaterials 35, 748–759 (2014). [DOI] [PubMed] [Google Scholar]
- Yang L. et al. Synthesis, nanosizing and in vitro drug release of a novel anti-HIV polymeric prodrug: chitosan-O-isopropyl-5’-O-d4T monophosphate conjugate. Bioorg Med Chem 18, 117–123 (2010). [DOI] [PubMed] [Google Scholar]
- Nikanjam M., Gibbs A., Hunt C., Budinger T. & Forte T. Synthetic nano-LDL with paclitaxel oleate as a targeted drug delivery vehicle for glioblastoma multiforme. J Control Release 124, 163–171 (2007). [DOI] [PubMed] [Google Scholar]
- Ke X. Y. et al. The therapeutic efficacy of conjugated linoleic acid - paclitaxel on glioma in the rat. Biomaterials 31, 5855–5864 (2010). [DOI] [PubMed] [Google Scholar]
- Ma Y. et al. Combinational delivery of hydrophobic and hydrophilic anticancer drugs in single nanoemulsions to treat MDR in cancer. Mol Pharm 11, 2623–2630 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. et al. Theranostic nanoemulsions: codelivery of hydrophobic drug and hydrophilic imaging probe for cancer therapy and imaging. Nanomedicine (Lond) 9, 2773–2785 (2014). [DOI] [PubMed] [Google Scholar]
- Yuan L., Chen W., Hu J., Zhang J. Z. & Yang D. Mechanistic study of the covalent loading of paclitaxel via disulfide linkers for controlled drug release. Langmuir 29, 734–743 (2013). [DOI] [PubMed] [Google Scholar]
- Markovic B. D. et al. Investigation of solvolysis kinetics of new synthesized fluocinolone acetonide C-21 esters--an in vitro model for prodrug activation. Molecules 16, 2658–2671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svastova E. et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 577, 439–445 (2004). [DOI] [PubMed] [Google Scholar]
- Arunachalam B., Phan U. T., Geuze H. J. & Cresswell P. Enzymatic reduction of disulfide bonds in lysosomes: characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT). Proc Natl Acad Sci U S A 97, 745–750 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Wang S., Brülisauer L., Leroux J. C. & Gauthier M. A. Broad control of disulfide stability through microenvironmental effects and analysis in complex redox environments. Biomacromolecules 14, 2383–2388 (2013). [DOI] [PubMed] [Google Scholar]
- Patel P., Patel R., Wadia D. & Patel R. Dendritic macromolecules as nano-scale drug carriers: Phase solubility, in vitro drug release, hemolysis and cytotoxicity study. Asian Journal of Pharmaceutical Sciences. in press, 10.1016/j.ajps.2015.04.002. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.