

Abstract
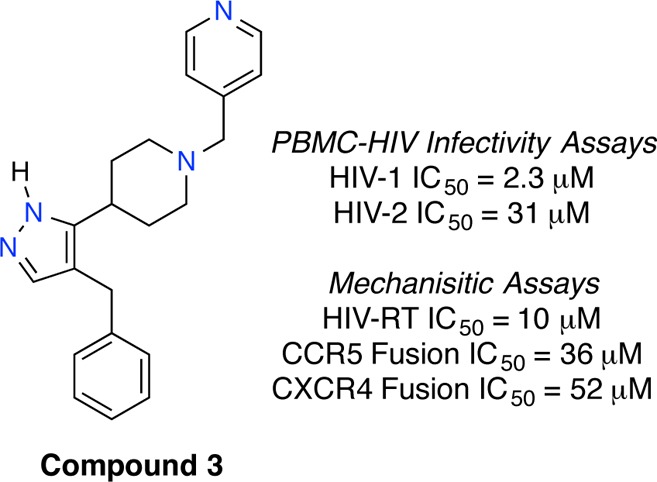
We report novel anti-HIV-1 agents with combined dual host–pathogen pharmacology. Lead compound 3, composed of a pyrazole-piperidine core, exhibits three concurrent mechanisms of action: (1) non-nucleoside reverse transcriptase inhibition, (2) CCR5-mediated M-tropic viral entry inhibition, and (3) CXCR4-based T-tropic viral entry inhibition that maintains native chemokine ligand binding. This discovery identifies important tool compounds for studying viral infectivity and prototype agents that block HIV-1 entry through dual chemokine receptor ligation.
Keywords: HIV-1, chemokine receptors, viral entry inhibitors, non-nucleoside reverse transcriptase inhibitors, multitarget drug discovery
The estimated number of people globally infected with the human immunodeficiency virus 1 (HIV-1) increased to 35 million in the last two and a half decades.1 The successful development of highly active antiretroviral therapy (HAART) delays progression to AIDS and extends life expectancy by reducing viral load within infected individuals. HAART obstructs a diverse set of viral proteins using a cocktail of drugs, which include entry and fusion inhibitors, nucleoside/non-nucleoside reverse transcriptase inhibitions (NRTIs/NNRTIs), integrase inhibitors, and protease inhibitors.2 Although effective, HAART requires coadministered single-target drugs that can result in drug–drug interactions, the development of resistance, off-target effects, high cost, and poor patient compliance. It would therefore be beneficial to identify single agent antivirals that simultaneously inhibit multiple targets, thereby potentially reducing the number of drugs required for therapy.3−5
HIV entry inhibition is a compelling antiretroviral strategy for treatment of pre-exposure prophylaxis (PrEP).6 The virus has evolved different glycoprotein variants that utilize either the CCR5 or CXCR4 coreceptor in combination with CD4 during entry: M-tropic for CCR5, T-tropic for CXCR4, and dual/mixed (D/M) tropic strains that use CCR5 and/or CXCR4. Maraviroc is an FDA-approved drug for the treatment of M-tropic HIV-1 that binds CCR5 to allosterically inhibit heterotrimer formation.7 As maraviroc targets a host protein, it is expected to exhibit a more favorable resistance profile than other approved antiretroviral agents that target viral proteins. However, individuals with T-tropic or dual/mixed phenotype variants cannot use maraviroc, which has hindered its use in combination regiments. A costly phenotypic viral tropism test (TROFILE) or a genotypic assay is required to ensure the drug’s initial and continued effectiveness further limiting the drug.8 There have been several CXCR4 antagonists in clinical trials as T-tropic HIV-1 entry inhibitors (AMD3100 and AMD11070),9 but the studies were discontinued due to off-target effects. As a consequence, dual CCR5/CXCR4 antagonists could be the next generation of chemokine based entry inhibitors,10 and previous reports indicate that such compounds are possible.11 Following this theme, we present a new class of antiviral agents with complex polypharmacology, acting on three distinct targets, the two host GPCRs (CCR5 and CXCR4) and one viral protein (HIV reverse transcriptase), to combat the evolving, promiscuous nature of HIV.
Recent successful applications of ligand-based models for multitarget GPCR ligand design prompted us to apply a similar approach for the screening of potential dual CCR5/CXCR4 antagonists.12 Bayesian statistical models for CCR5 and CXCR4 were constructed using functional chemical fingerprints to the sixth neighbor (FCFP_6) as the chemical descriptor set, and these models were trained on sets of known active and presumed inactive compounds from chemical databases (Supporting Information). The Aldrich Marketplace Select library (∼5 million compounds) was scored and ranked using the CCR5 and CXCR4 Bayesian models, and 14 compounds were selected from the top 300 to represent the total query. The initial set of compounds were purchased and screened for anti-HIV-1 activity against separate viral tropisms using CD4/CCR5/CXCR4 expressing MAGI cells (Multinuclear Activation of Galactosidase Indicator). This screen identified compound 1 with weak inhibition against both CCR5- and CXCR4-utilizing HIV-1 strains (Table 1). The compound consists of a pyrazole-piperidine core and retained anti-HIV-1 activity upon resynthesis.13 A follow-up sequential and focused screening approach aimed to improve the anti-HIV-1 activity.14 A substructure search based on 1 yielded 24 structurally similar compounds that were purchased and tested. Eleven compounds from this search exhibited antiviral activity against both M-tropic and T-tropic HIV in the MAGI assay with IC50 values below 100 μM, four below 20 μM, and two below 10 μM (Supporting Information). Compound 1–3 explored the position of the nitrogen in the pyridyl ring, and the most potent contained the 4-pyridyl group (3) and possessed single-digit micromolar anti-HIV-1 activity against both viral tropisms (Table 1). Compound 4 contained a 3-pyridyl ring and replaced the benzyl- with a phenyl-moiety. This compound was less active than 3-pyridyl/benzyl compound 2 confirming the necessity of the benzyl placement. Other compounds tested isosteres and substitutions on the pyridyl ring, but none improved anti-HIV-1 activity relative to 3. To confirm its antiviral activity, 3 was tested in peripheral blood mononuclear cells (PBMC). Agent 3 inhibited both HIV-1 tropisms in this assay with IC50 values near those obtained from the MAGI assay, while the IC90 values fell well below the toxicity threshold (Table 2).
Table 1. Anti-HIV-1 Activities of Selected Compounds from Virtual Screen.
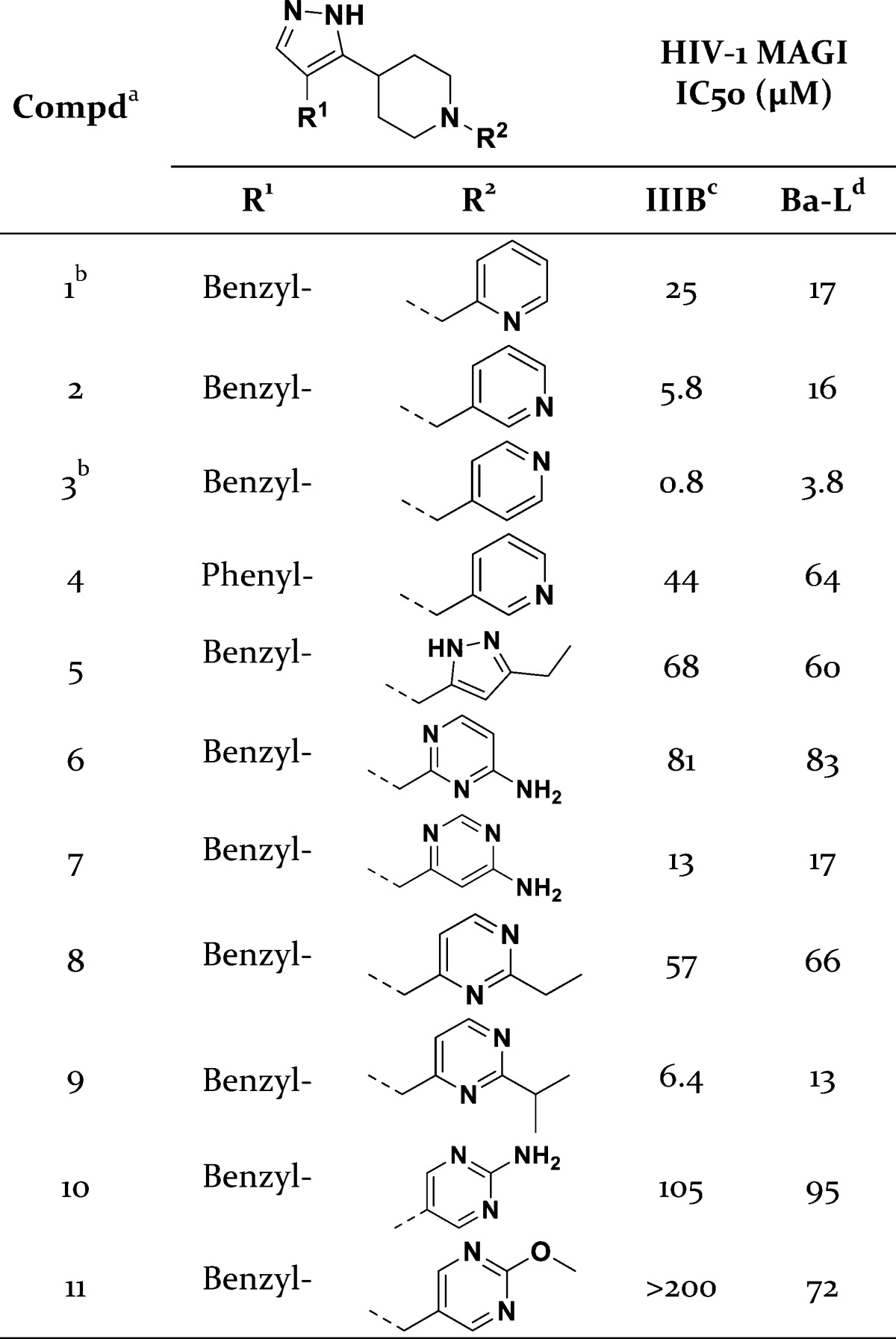
All compounds were >95% pure by HPLC.
Results from a resynthesized sample.
CXCR4-utilizing HIV-1 strain.
CCR5-utilizing HIV-1 strain.
Table 2. Bioactivities of Compound 3.
| anti-HIV-1 assay | compound 3 results |
|---|---|
| MAGI HIV-1Ba-L (CCR5) | IC50 = 3.8 μM |
| MAGI HIV-1IIIB (CXCR4) | IC50 = 0.8 μM |
| MAGI Toxicity | TC50 > 300 μM |
| PBMC HIV-1Ba-L (CCR5) | IC50 = 2.3 μM |
| PBMC HIV-1IIIB (CXCR4) | IC50 = 2.3 μM |
| PBMC toxicity | TC50 > 100 μM |
| CCR5 activity assay | compound 3 results |
|---|---|
| HeLa-C14:JRFL CD4-dependent fusion | IC50 = 36 μM |
| 125I-MIP-1β displacement | 38% at 100 μM/88% at 1 mM |
| CXCR4 activity assay | compound 3 results |
|---|---|
| Hela/67:Lai | EC50 = 70 μM |
| CD4-dependent fusion | |
| ACTOneX4:HIV-8x | EC50 = 52 μM |
| CD4-independent fusion | |
| PBMC HIV-2a | IC50 = 31 μM |
| Nef-M1:CXCR4 | |
| MMD inhibition | IC50 = 1.2 μM |
| 125I-SDF-1 displacement | 0% at 1 mM |
| NNRTI activity assay | compound 3 results |
|---|---|
| VSV-G pseudovirus | EC50 = 1.3 μM |
| isolated HIV-RT inhibition | IC50 = 9.0 μM |
| multicycle with mutant HIV-RTb | EC50 (WT) = 0.9 μM |
| EC50 (Mutb) = 19 μM (21-fold loss) |
HIV-2 type was CDC310319, a CXCR4-tropic strain.
NNRTI-resistant strain with Lys103Gln and Tyr181Cys mutations.
Since the phenotypic MAGI and PBMC assays probe antiviral activity devoid of mechanism, 3 was subjected to alternative assays to confirm coreceptor based inhibition. Cell–cell fusion assays measure specifically coreceptor mediated inhibition of gp120-induced fusion in the absence of other viral machinery, and these assays were used to explore the CCR5- and CXCR4-specific antiviral activity. A cell–cell fusion assay was constructed to simulate HIV-1 entry in the absence of other viral machinery. The assay measured fusion of effector cells expressing the JRFL envelope (CCR5-tropic/CD4-dependent) with HeLa-C14 target cells (expressing CD4, CCR5, and CXCR4) using a luciferase readout (Figure 1a). Compound 3 inhibited fusion in this assay, like the CCR5 antagonist TAK-779, establishing the CCR5-based viral entry inhibition mechanism (Figure 1b). In confirmation of CCR5 binding, 3 displaced the 125I-MIP1β chemokine ligand from the receptor in a radioligand competition assay, similar to several other compounds in this series (Table 2 and Supporting Information). Notably, compound 11 displaces the 125I-MIP1β with an apparent potency similar to the MAGI IC50 (∼70 μM) supporting that this series exhibits a CCR5-based anti-HIV mechanism of action. Molecular modeling using the CCR5 cocrystal structure suggests that 3 spans the entire extracellular binding pocket similar to maraviroc (Figure 2).15 In the top scoring pose, the 4-pyridyl ring resides in the minor subpocket composed of helices I–III, the cationic piperidine ring forms an electrostatic interaction with Glu283, and the benzyl-pyrazole moiety is buried in the major subpocket between helices IV–VII. These results support the CCR5-based anti-HIV-1 mechanism and suggest a structural model for further optimization.
Figure 1.

Chemokine-based anti-HIV-1 profile of compound 3. (a) Schematic for a CD4-dependent/CCR5-tropic envelope cell–cell fusion assay (HeLa-C14:JRFL), and (b) assay curve of 3 inhibiting fusion in this assay (blue circles) and cytotoxicity values (open circles). (c) Schematic for the CD4-independent/CXCR4-tropic cell–cell fusion assay (ACTOne-X4:HIV8x) and (d) assay curve for 3 inhibiting fusion in this assay (red diamonds) and cytotoxicity measurements (open circles). (e) Synopsis of the HIV-1 Nef-M1:CXCR4 induced mitochondrial membrane depolarization (MMD) assay with fluorescence depolarization readout. (f) Inhibition of Nef-M1-induced MMD by AMD3100 (black triangles) and 3 (red circles) with no activity observed for maraviroc or AZT (green triangles).
Figure 2.
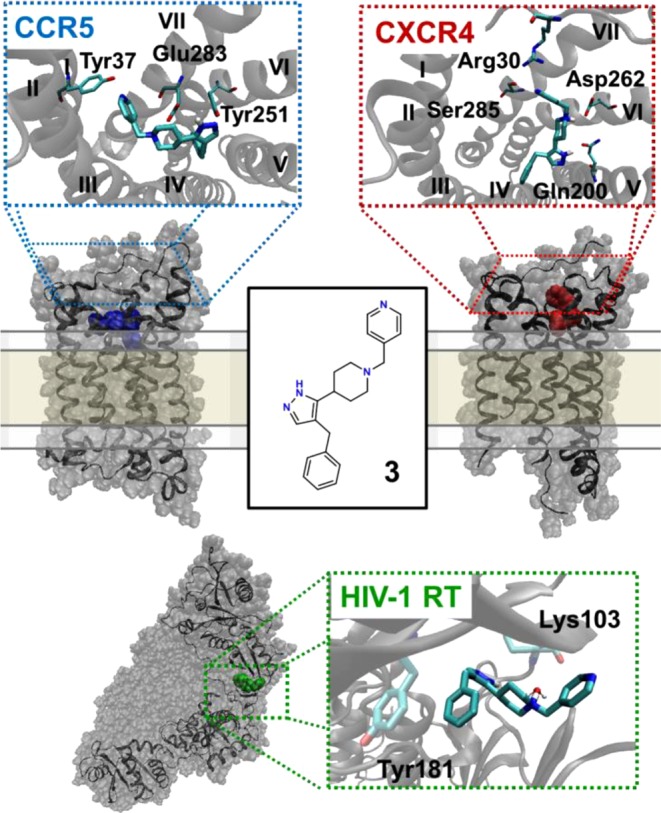
Predicted binding mode of compound 3 to the three biological targets. In all models, the piperidine nitrogen is cationic and the tautomeric state of the pyrazole positions the hydrogen nearest the piperidine ring. The model of compound 3 complexed to the extracellular binding pocket of CCR5, derived from the CCR5:Maraviroc cocrystal structure as a template, spans the entire orthosteric site (blue). The model of compound 3 binding to the extracellular binding pocket of CXCR4 using the CXCR4:CVX15 crystal structure displays interactions only in the major subpocket (helices I, V, VI, and VII) (red). Predicted U-shaped binding mode of compound 3 binding to HIV-RT p66 subunit demonstrates interactions with Tyr181 and water-mediated interactions with Lys103 (green). The chemokine receptor-bound conformations of 3 are similar to a heavy-atom RMSD of 2.0 Å, while the conformer bound to HIV-RT is distinct with an RMSD of >3.0 Å.
Cell–cell fusion assays were established to probe the CXCR4-specific anti-HIV-1 activity of 3. A CD4-dependent fusion assay used HeLa/67 as target cells (expressing CD4, CCR5, and CXCR4) and Lai envelope-expressing effector cells (CXCR4-tropic/CD4-dependent).
A separate CD4-independent fusion assay used ACTOne-X4 as target cells (expressing only CXCR4) with effector cells expressing the HIV-8x envelope (CD4-independent; requiring only CXCR4 and not CD4) (Figure 1c).16 Compound 3 blocked fusion in both CD4-dependent and CD4-independent assays (Figure 1d and Table 2) with minimal toxicity (CC50 > 200 μM) confirming CXCR4-specific viral entry inhibition. Additionally, 3 was tested for Nef-M1-induced mitochondrial membrane depolarization (MMD) inhibition, in which the viral Nef-M1 peptide induces MMD via the CXCR4 axis in Jurkat cells (Figure 1e).17 Agent 3 inhibited Nef-M1 induced MMD like the CXCR4 antagonist AMD3100, while maraviroc and AZT (an NRTI) were inactive (Figure 1f). Compound 3 was tested for antiviral activity against an HIV-2 strain that utilized CXCR4 for entry. As NNRTI compounds are inactive against HIV-2, this assay further explores the CXCR4-based anti-HIV activity.18 Compound 3 inhibited this HIV-2 strain in PBMC cells in support of the CXCR4-based viral entry inhibition mechanism (Table 2). The Nef-M1MMD inhibition assay, CD4-dependent fusion assay, CD4-independent fusion assay, and HIV-2 inhibition assay results indicate that 3 blocks HIV-1 entry and pathology by binding to the CXCR4 receptor. Compound 3 was tested for displacement of radio-labeled 125I-SDF-1 from CXCR4, but like all other compounds in the screen, did not displace the chemokine ligand at concentrations up to 1 mM (Supporting Information). Compound 3 was tested in the Ca2+ flux GPCR signaling assay with antagonist IC50 activity varying between 10 to >100 μM. The observation of activity supports direct interaction with CXCR4 suggesting 3 to be a negative allosteric modulator (NAM), but receptor internalization mechanisms cannot be ruled out. These results reveal unique pharmacology; 3 binds to CXCR4 in a pose that blocks HIV-1 binding without displacing SDF-1. To provide a rationale for this distinctive mode of action, 3 was modeled into the CXCR4 extracellular binding pocket. Previous studies concluded that the two X-ray structures of CXCR4 (cocrystallized with IT1t and CVX15)19 should be considered independently when docking novel CXCR4 antagonists.20 The CXCR4:IT1t template yielded a high-energy conformation capable of interacting with the relevant SDF-1 related residues, which was inconsistent with chemokine assay results (Supporting Information). In contrast, docking into the CXCR4:CVX15 structure placed 3 in the major binding pocket allowing the piperidine ring to adopt a low-energy conformation (Figure 2). In this pose, the protonated piperidine ring forms an electrostatic interaction with Asp262, the pyrazole ring hydrogen bonds with Gln200, and the 4-pyridyl ring acts as a hydrogen bond acceptor for Ser283 and Arg30. Previous mutagenesis demonstrates that none of these residues are required for SDF-1 binding (all critical residues are >4.5 Å away), while Asp262 is necessary for HIV-1 viral entry (Supporting Information) thereby offering a structural explanation for the unique CXCR4 pharmacology.21
The antiviral activities of 3 in chemokine and viral entry inhibition assays were up to 10-fold weaker than the potency observed from the phenotypic MAGI and PBMC assays. Although this could be due to differences in assay performance and sensitivity, the weakest activity occurred in the cell–cell fusion that specifically measures coreceptor based inhibition (as no other viral function is present). Compound 3 was tested in a VSV-G pseudovirus assay in which viral infection occurs using the vesicular stomatitis G protein pathway independent of the CCR5 or CXCR4 receptors (Supporting Information). Unlike the CXCR4 antagonist AMD3100, 3 surprisingly inhibited the VSV-G pseudovirus, indicating the potential for an additional mechanism of action (Table 2). Indeed, 3 inhibited isolated HIV-1 reverse transcriptase (HIV-RT) suggesting that this compound acts as an NNRTI in addition to its chemokine-related activities. To confirm this, 3 was tested against an NNRTI-resistant viral strain containing the K103N/Y181C mutations to probe a binding locus. Compared to wild type virus, antiviral activity for 3 decreased 20-fold against the K103N/Y181C mutant, similar to the 70-fold loss for efavirenz (an NNRTI) in this multicycle assay. The antiviral activity of 3 was the same in PBMCs for both viral tropisms (Table 2), and this value likely reflects the NNRTI-based mechanism. A model of 3 interacting with the protein was constructed using a recent crystal structure of the HIV-RT cocrystallized with a chemically similar NNRTI in the efavirenz/etravirine pocket (Figure 2).22 The model predicts that the benzyl ring of 3 π-stacks with Tyr181 and the piperidine ring forms a water-mediated hydrogen bond with the backbone of Lys103, thus furnishing a rationale for the loss of activity upon mutation. These observations also highlight the unexpected result that the viral HIV-RT protein and host CCR5 and CXCR4 receptors share association with a common ligand.
In conclusion, we report the discovery of a multitarget series of compounds that reveals distinctive anti-HIV activity. The most potent compound 3, identified from a GPCR-guided screen, blocks viral entry by binding to the host protein receptors CCR5 and CXCR4 and also displays serendipitous inhibition of viral reverse transcriptase. Based on head-to-head comparisons and control values in these assays, the priority of anti-HIV-1 effects for compound 3 is in the following order: NNRTI > CCR5 > CXCR4. Although the HIV-RT activity seems predominant, the dual chemokine activity of this series may aid in the delay of resistance compared to other NNRTIs. Interestingly, the dual chemokine activity of this series reveals common chemical space across the CCR5 and CXCR4 receptors that is silent in the CXCR4:SDF-1 signaling axis. The series provides tool compounds necessary to explore the benefits of multitarget compounds in blocking viral pathogenesis and delaying resistance acquisition. Lastly, this study provides an attractive entry point for optimization of next generation of dual host–pathogen HIV drugs to synergize with existing clinical antiretroviral agents.
Glossary
ABBREVIATIONS
- AIDS
acquired immune deficiency syndrome
- AZT
azidothymidine
- CCR5
CC chemokine receptor 5
- CXCR4
C-X-C chemokine receptor 4
- FDA
food and drug administration
- GPCR
G-protein coupled receptor
- HAART
highly activate antiretroviral therapy
- HIV
human immunodeficiency virus
- HIV-RT
HIV-1 reverse transcriptase
- MAGI
multinuclear activation of galactosidase indicator
- MIP-1β
macrophage inflammatory protein 1β
- MMD
mitochondrial membrane depolarization
- NRTI/NNRTI
(non)nucleoside reverse transcriptase inhibitor
- PBMC
peripheral blood mononuclear cells
- SDF-1
stromal cell-derived factor 1
- VSV-G
vesicular stomatitis virus-G
Supporting Information Available
Detailed methods, synthetic synopsis, activities of controls for each assay, structures and anti-HIV-1 activities of purchased compounds from the screen, and supporting figures. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00036.
Author Contributions
The manuscript was written through contributions of all authors. B.C. and L.W. conceived the project, designed approach, and selected/purchased compounds for testing. L.W. and D.L. managed and supervised the project. B.C. conducted the virtual screen, performed the molecular modeling, and is the primary author; and J.S. supervised the computational chemistry. A.P. performed synthesis and characterized the compounds. M.H. and V.B. performed the Nef-M1:CXCR4MMD assay and analyzed the data. Y.S., Z.L., S.L., and M.K. designed, performed, and analyzed coreceptor specific inhibition assays and NNRTI-related assays. B.C., A.P., L.W., J.S., M.K., and D.L. made corrections to the manuscript.
A.P. was supported by a NSF predoctoral fellowship. M.H. and V.B. were supported by NIMHD grant 8G12MD007602. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare the following competing financial interest(s): B.C., A.P., L.W., and D.L. have filed a patent application for multitarget chemokine modulators for use as anti-HIV-1 agents. M.K., Y.S., Z.L., and S.L. work for Bristol-Myers Squibb.
Supplementary Material
References
- Joint United Nations Programme on HIV/AIDS (UNAIDS). Global Report: UNAIDS Report on the Global AIDS Epidemic: 2012; UNAIDS: Geneva, Switzerland, 2012. [Google Scholar]
- Detels R.; Muñoz A.; McFarlane G.; Kingsley L. A.; Margolick J. B.; Giorgi J.; Schrager L. K.; Phair J. P. Investigators, M. A. C. S. Effectiveness of potent antiretroviral therapy on time to AIDS and death in men with known HIV infection duration. JAMA 1998, 280, 1497–1503. [DOI] [PubMed] [Google Scholar]
- Herschhorn A.; Gu C.; Espy N.; Richard J.; Finzi A.; Sodroski J. G. A broad HIV-1 inhibitor blocks envelope glycoprotein transitions critical for entry. Nat. Chem. Biol. 2014, 10, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckheit K. W.; Yang L.; Buckheit R. W. Development of dual-acting pyrimidinediones as novel and highly potent topical anti-HIV microbicides. Antimicrob. Agents Chemother. 2011, 55, 5243–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenwitheesuk E.; Horst J. A.; Rivas K. L.; Van Voorhis W. C.; Samudrala R. Novel paradigms for drug discovery: computational multitarget screening. Trends Pharmacol. Sci. 2008, 29, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A. B.; Nuttall J.; Romano J. The future of HIV microbicides: challenges and opportunities. Antiviral Chem. Chemother. 2009, 19, 143–150. [DOI] [PubMed] [Google Scholar]
- Esté J. A.; Telenti A. HIV entry inhibitors. Lancet 2007, 370, 81–88. [DOI] [PubMed] [Google Scholar]
- Poveda E.; Briz V.; Quinones-Mateu M.; Soriano V. HIV tropism: diagnostic tools and implications for disease progression and treatment with entry inhibitors. Aids 2006, 20, 1359–1367. [DOI] [PubMed] [Google Scholar]
- De Clercq E. The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil). Biochem. Pharmacol. 2009, 77, 1655–1664. [DOI] [PubMed] [Google Scholar]
- Horuk R. Promiscuous drugs as therapeutics for chemokine receptors. Expert Rev. Mol. Med. 2009, 11, e1. [DOI] [PubMed] [Google Scholar]
- Princen K.; Hatse S.; Vermeire K.; Aquaro S.; De Clercq E.; Gerlach L. O.; Rosenkilde M.; Schwartz T. W.; Skerlj R.; Bridger G.; Schols D. Inhibition of human immunodeficiency virus replication by a dual CCR5/CXCR4 antagonist. J. Virol. 2004, 78, 12996–13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X. P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser A. R.; Liotta D. C. One-pot transformation of esters to analytically pure ketones: Methodology and application in process development. Tetrahedron Lett. 2014, 10.1016/j.tetlet.2014.10.024. [DOI] [Google Scholar]
- Chen X.; Wilson L. J.; Malaviya R.; Argentieri R. L.; Yang S.-M. Virtual screening to successfully identify novel janus kinase 3 inhibitors: a sequential focused screening approach. J. Med. Chem. 2008, 51, 7015–7019. [DOI] [PubMed] [Google Scholar]
- Tan Q.; Zhu Y.; Li J.; Chen Z.; Han G. W.; Kufareva I.; Li T.; Ma L.; Fenalti G.; Li J. Structure of the CCR5 Chemokine receptor–HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Zhou N.; Sun Y.; Ray N.; Lataillade M.; Hanna G. J.; Krystal M. Activity of the HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068, against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors. Antimicrob. Agents Chemother. 20143, 57, 4172–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.-B.; Jin L. L.; James C. O.; Khan M.; Powell M. D.; Bond V. C. Characterization of Nef-CXCR4 interactions important for apoptosis induction. J. Virol. 2004, 78, 11084–11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witvrouw M.; Pannecouque C.; Switzer W. M.; Folks T. M.; De Clercq E.; Heneine W. Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antiviral Ther. 2004, 9, 57–66. [PubMed] [Google Scholar]
- Wu B.; Chien E. Y.; Mol C. D.; Fenalti G.; Liu W.; Katritch V.; Abagyan R.; Brooun A.; Wells P.; Bi F. C.; Hamel D. J.; Kuhn P.; Handel T. M.; Cherezov V.; Stevens R. C. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox B. D.; Prosser A. R.; Katzman B. M.; Alcaraz A. A.; Liotta D. C.; Wilson L. J.; Snyder J. P. Anti-HIV Small Molecule Binding in the Peptide Sub-Pocket of the CXCR4:CVX15 Crystal Structure. ChemBioChem 2014, 15, 1614–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelot A.; Heveker N.; Montes M.; Alizon M. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J. Biol. Chem. 2000, 275, 23736–23744. [DOI] [PubMed] [Google Scholar]
- Kertesz D. J.; Brotherton-Pleiss C.; Yang M.; Wang Z.; Lin X.; Qiu Z.; Hirschfeld D. R.; Gleason S.; Mirzadegan T.; Dunten P. W. Discovery of piperidin-4-yl-aminopyrimidines as HIV-1 reverse transcriptase inhibitors. N-Benzyl derivatives with broad potency against resistant mutant viruses. Bioorg. Med. Chem. Lett. 2010, 20, 4215–4218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.