

Abstract
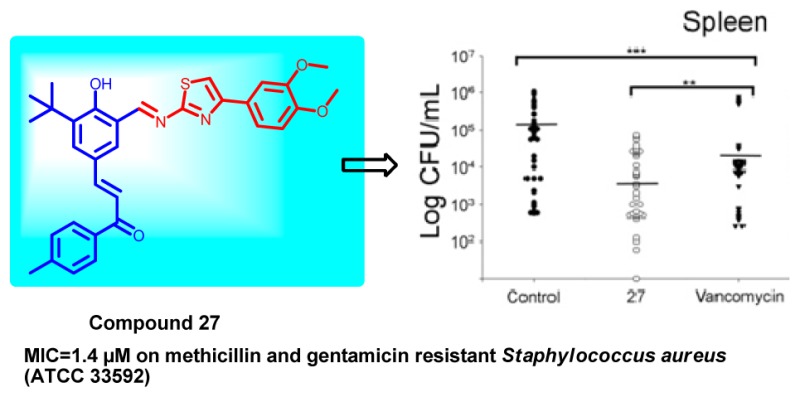
A series of novel hybrids possessing chalcone and thiazole moieties were synthesized and evaluated for their antibacterial activities. In general this class of hybrids exhibited potency against Staphylococcus aureus, and in particular, compound 27 exhibited potent inhibitory activity with respect to other synthesized hybrids. Furthermore, the hemolytic and toxicity data demonstrated that the compound 27 was nonhemolytic and nontoxic to mammalian cells. The in vivo studies utilizing a S. aureus septicemia model demonstrated that compound 27 was as potent as vancomycin. The results of antibacterial activities underscore the potential of this scaffold that can be utilized for developing a new class of novel antibiotics.
Keywords: Chalcone−thiazole hybrids, Staphylococcus aureus, antibacterial activities, MIC
Staphylococcus aureus is an opportunistic pathogen that can cause a wide spectrum of infections, ranging from severe to potentially fatal or invasive diseases. Recent data suggested that the incidence of infection due to S. aureus seems to be the highest in neonates, immune compromised, and/or hospitalized individuals with a mortality rate as high as 50%. In USA alone, S. aureus has accounted for 80,461 infections and 11,285 deaths per year, indicating the seriousness of the problem.1 Currently to treat S. aureus infections, a wide range of FDA approved drugs are available ranging from ciprofloxacin to oritavancin. Unfortunately, acquiring resistance to the antibiotics by S. aureus has recently increased the number of untreatable infections.2 Recent strategies to develop a vaccine for S. aureus have not been very successful as data suggested that vaccination with bacterial cell-surface antigens increases disease severity instead of offering protection.3 Given this, Centers for Disease Control, USA have listed S. aureus as a microorganism of serious threat level.1
Regrettably, many pharmaceutical companies have abolished their programs on new antibiotic discovery and development long ago4,5 and recent search for novel targets by genomic and proteomic based approaches also have yielded only limited success.6,7 Given the alarming situation of drug-resistant S. aureus, the search for new antimicrobial agents against S. aureus, especially strains resistant to multiple antibiotics, is an urgent and unmet medical need.
Natural and synthetic chalcones have been reported to exhibit a broad spectrum of pharmacological activity.8 They are of special interest due to their pronounced antimicrobial activities.9,10 However, thiazole ring is found to be a common structural component in a large number of biological agents.11 Thiazoles have also been extensively studied for their antibacterial activities.12−15 Though in some cases combining two pharmacophores may lead to inactive and molecular obese compounds, the design of hybrid molecules has generated immense interest in medicinal chemists as it can lead to compounds with superior efficacy while circumventing the problem of resistance.16,17 Increasingly in literature examples of hybrid molecules, incorporating chalcone and thiazoles are reported as being active as antimicrobials.18 Thus, keeping in view the antimicrobial potential of chalcones and thiazoles, it was envisaged that the synthesis and antibacterial evaluation of hitherto novel chalcone–thiazole hybrids is worth the attempt. Figure 1 shows the representative structures of potent antibacterial compounds that contain either chalcone or thiazoles and form the basis of our designed prototype.
Figure 1.
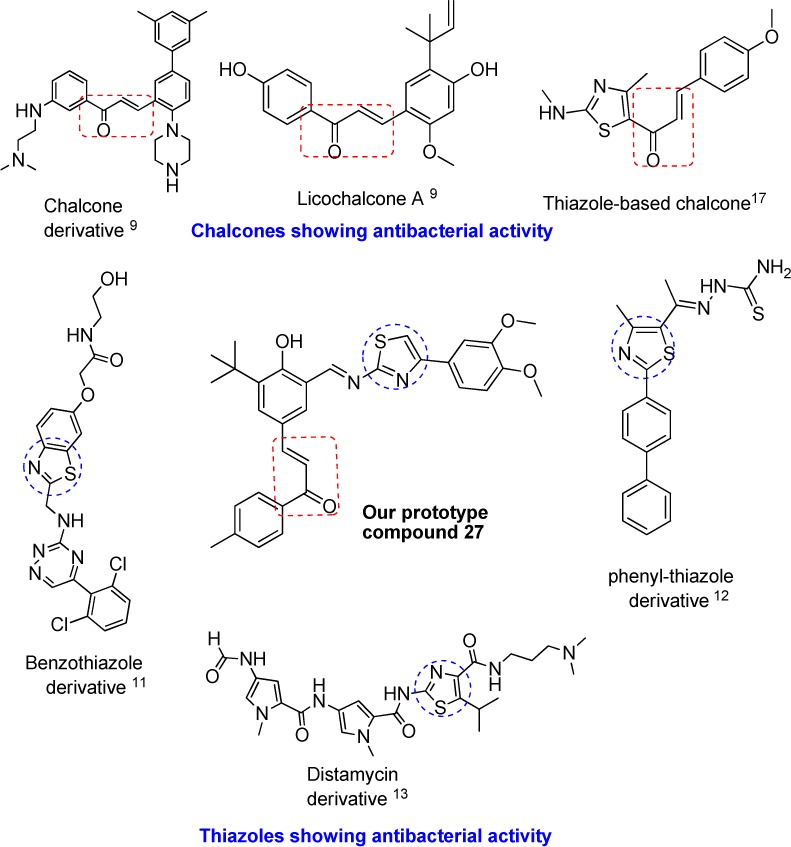
Designing chalcone–thiazole hybrids by pharmacophore hybridization.
The synthetic routes for the preparation of the target chalcone–thiazole hybrids are depicted in Scheme 1. The synthesis of aromatic dicarbaldehydes (4–6) were achieved by the Duff formylation reaction on 2-substituted phenols with hexamethylenetetramine (HMTA) and trifluoroacetic acid (TFA) at 120 °C followed by hydrolysis using 10% aqueous H2SO4.19 The regioselective synthesis of chalcones (7–15) was achieved using acid catalyzed condensation between aromatic dicarbaldehydes and substituted acetophenones.20 The 2-aminothiazole derivatives (19–21) were synthesized by the reaction between thiourea and phenacyl bromide derivatives in the presence of ethanol as solvent. Finally, the target compounds were synthesized via formation of the Schiff’s bases between chalcones (7–15) and 2-aminothiazole derivatives (19–21) in the presence of ethanol. All the new synthesized compounds were characterized using NMR, IR, and mass spectrometry (Supporting Information).
Scheme 1. Synthesis of Chalcone–Thiazole Hybrids.

Reagents and conditions: (i) HMTA, TFA, 120 °C, 4 h; (ii) aq. H2SO4, 100 °C, 2 h; (iii) conc. HCl, p-R1C6H4COCH3, 1,4-dioxane, 80–90 °C, 2.5–3.5 h; (iv,v) ethanol, reflux, 3 h.
Initially, we screened all the synthesized hybrids for their antibacterial activity in vitro with norfloxacin and gentamycin as reference drugs. The in vitro antibacterial results confirmed that some of the chalcone–thiazole hybrids exhibited antibacterial activity against various strains of Staphylococcus aureus as shown in Table 1 with compounds 22, 25, and 27 being the most potent in the series. A pictorial representation of the preliminary structure–activity relationships (SARs) could be summarized as depicted in Figure 2. The presence of bulky tert butyl side chain (22, 25, 27) is preferred over isopropyl and n-propyl groups at ortho position to hydroxyl group. In the chalcone part, though both aromatic and heteroaromatic groups are tolerated, the presence of 4-methylphenyl group is optimum. While in the thiazole core phenyl with dimethoxy substitution (27) is preferred over monomethoxy (26) and ethyl ester (23, 24, 31).
Table 1. Antibacterial Activity of Chalcone–Thiazole Hybridsa.
| minimum inhibitory conc (MIC) in μM |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| bacteria |
fungi |
||||||||||||
| compound | 1 | 2 | 3 | 3a | 3b | 3c | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 22 | >104.9 | >104.9 | 6.5 | 13.1 | 6.5 | 3.3 | >104.9 | >104.9 | >104.9 | >104.9 | >104.9 | >104.9 | >104.9 |
| 23 | >101.5 | >101.5 | 12.7 | 12.7 | 12.7 | 3.2 | >101.5 | >101.5 | >101.5 | >101.5 | >101.5 | >101.5 | >101.5 |
| 24 | >110.5 | >110.5 | 13.8 | 13.8 | 13.8 | 13.8 | >110.5 | >110.5 | >110.5 | >110.5 | >110.5 | >110.5 | >110.5 |
| 25 | >106.7 | >106.7 | 13.3 | 13.3 | 6.7 | 3.3 | >106.7 | >106.7 | >106.7 | >106.7 | >106.7 | >106.7 | >106.7 |
| 26 | >97.9 | >97.9 | 12.2 | 12.2 | 12.2 | 12.2 | >97.9 | >97.9 | >97.9 | >97.9 | >97.9 | >97.9 | >97.9 |
| 27 | >92.5 | >92.5 | 5.8 | 11.6 | 5.8 | 1.4 | >92.5 | >92.5 | >92.5 | >92.5 | >92.5 | >92.5 | >92.5 |
| 28 | >89.8 | >89.8 | 11.2 | 11.2 | 11.2 | 5.6 | >89.8 | >89.8 | >89.8 | >89.8 | >89.8 | >89.8 | >89.8 |
| 29 | >93.9 | >93.9 | 23.5 | 11.7 | 11.7 | 11.7 | >93.9 | >93.9 | >93.9 | >93.9 | >93.9 | >93.9 | >93.9 |
| 30 | >94.9 | >94.9 | 11.9 | 11.9 | 11.9 | 3.0 | >94.9 | >94.9 | >94.9 | >94.9 | >94.9 | >94.9 | >94.9 |
| 31 | >108.1 | >108.1 | 27.0 | 13.5 | 13.5 | 13.5 | >108.1 | >108.1 | >108.1 | >108.1 | >108.1 | >108.1 | >108.1 |
| 32 | >104.5 | >104.5 | 26.1 | 26.1 | 26.1 | 26.1 | >104.5 | >104.5 | >104.5 | >104.5 | >104.5 | >104.5 | >104.5 |
| 33 | >102.4 | >102.4 | 51.2 | 51.2 | 51.2 | 51.2 | >102.4 | >102.4 | >102.4 | >102.4 | >102.4 | >102.4 | >102.4 |
| 34 | >110.0 | >110.0 | 27.5 | 27.5 | 27.5 | 13.7 | >110.0 | >110.0 | >110.0 | >110.0 | >110.0 | >110.0 | >110.0 |
| Gen | 6.5 | 1.6 | 1.6 | 104.9 | 3.3 | 104.9 | 3.3 | >104.9 | 104.9 | 104.9 | 104.9 | 104.9 | 104.9 |
| Nor | 0.0 | 1.0 | 0.5 | 33.6 | 1.0 | 0.5 | 0.1 | >33.6 | 33.6 | 33.6 | 33.6 | 33.6 | 33.6 |
| Van | >156.6 | >156.6 | 2.4 | 9.8 | 1.2 | 2.4 | >156.6 | >156.6 | 156.6 | 156.6 | 156.6 | 156.6 | 156.6 |
| Flu | >34.5 | >34.5 | >34.5 | >34.5 | >34.5 | >34.5 | >34.5 | 0.7 | 1.4 | 1.4 | 22.1 | 22.1 | 1.4 |
| AmB | >54.1 | >54.1 | >54.1 | >54.1 | >54.1 | >54.1 | >54.1 | 0.0 | 0.1 | 0.3 | 0.3 | 0.5 | 0.0 |
1. E. coli (ATCC 9637); 2. P. aeruginosa (ATCC BAA-427); 3. S. aureus (ATCC 25923); 3a. S. aureus (ATCC 700699 Methicillin Resistant); 3b. S. aureus (ATCC 29213); 3c. S. aureus (ATCC 33592 Methicillin and Gentamicin Resistant); 4. K. pneumoniae (ATCC 27736); 5. C. albicans; 6. C. neoformans; 7. S. schenckii; 8. T. mentagrophytes; 9. A. fumigates; 10. C. parapsilosis (ATCC 22019); Gen, Gentamicin; Nor, Norfloxacin; Van, Vancomycin; Flu, Fluconazole; AmB, Amphotericin-B.
Figure 2.
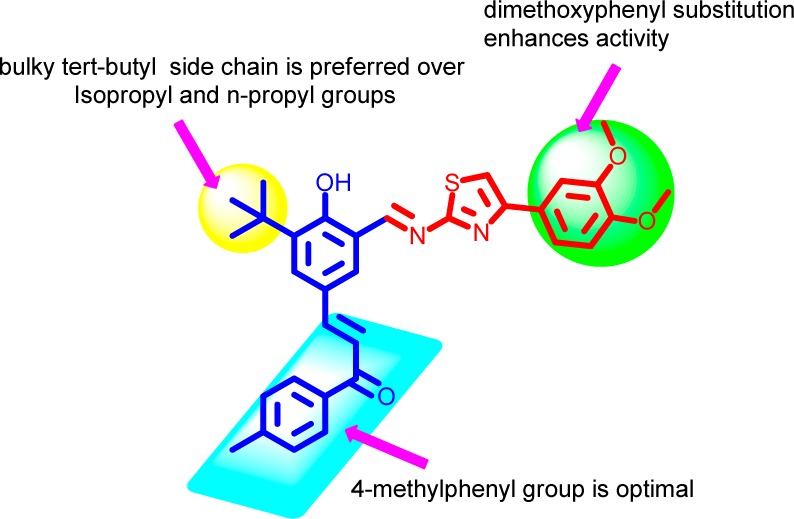
SAR of synthesized hybrids.
By using human RBCs, hemolytic activity for all the test compounds was evaluated, and the results showed that all the compounds were nonhemolytic (Figure 3A). Among all the compounds, 22 and 27 were selected to screen in the MTT assay as these molecules showed lower MIC values and no hemolytic activity (Figure 3A).
Figure 3.

Effect of chalcone–thiazole hybrids on eukaryotic cells. (A) Influence of test compounds (22–36) on erythrocytes in the absence of human serum. Hemolytic effects of the test compounds were investigated, and the corresponding data for vancomysin and polymyxin B-sulfate are included for comparison. RBC was incubated at 100 μg/mL and 2% Trition X-100 was used as positive control. Hemoglobin released was measured at 540 nm and is expressed as a percentage of Trition X-100 induced hemolysis. (B) The MTT assay (right top panel) was used to measure the viability of Mammalian fibroblast cell line, L929 in the presence of compounds 22 and 27. In the assay, enzymes associated with metabolic activity will modify the MTT into a dye, blue formazan, which is measured at 550 nm. (C) Morphological changes on the L929 were investigated by using phase contrast microscopy. Cells were incubated for 24 h at 1× MIC for 24 h and stained with Giemsa stain before observation under the phase contrast microscope for the morphological changes.
These results clearly demonstrated that the tested molecules do not have any toxicity issue at tested (10× MIC) concentrations. In order to prove that the test molecules are indeed nontoxic, MTT assay was carried out to measure the toxicity of 22 and 27 against mammalian fibroblast cell line, L929 (Figure 3B). Results showed that the tested compounds did not show any toxicity against L929 cells at MIC and lower concentrations. Even though the test compounds did not have any immediate toxic effects, we were interested to investigate whether the molecules induced any morphological changes over 24 h (Figure 3C). L929 cells were exposed to test compounds at 1× MIC, followed by staining and observation under the phase contrast microscope to see any changes in the morphology of the cells. Results demonstrated that the test compounds have no immediate toxicity nor induced any morphological changes. In comparison to control, compound treated L929 cells were normal in appearance, transparent, and attached to the surface of the wells of tissue culture plate (Figure 3C).
Next, in order to determine whether the compounds exhibited time-dependent or concentration-dependent antimicrobial activity, compound 27 was tested at 1× and 10× MIC against S. aureus ATCC 29213, and aliquots were plated at regular intervals. As can be seen in Figure 4A, the compound exhibits concentration-dependent activity with a 10 log10 reduction in cfu in comparison to no-drug control at 24 h, which is comparable to levofloxacin. Based upon the above results, we were of the opinion that the developed molecule could be tested in in vivo conditions.
Figure 4.
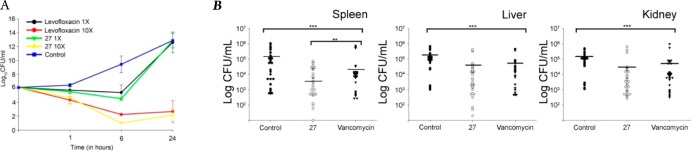
Static and cidal curve with different concentrations of compound 27. (A) Compound 27 (1× and 10× MIC concentration) were incubated with S. aureus for 24 h, and at regular time points, aliquot was taken and CFU was calculated. In vivo antimicrobial activity of the compound 27. (B) BALB/C mice were initially made neutropenic by injecting cyclophosphamide (150 mg/kg of body weight), followed by S. aureus ATCC 29213 injection. After 3 h of post infection, compound 27 and vancomycin at 15 mg/kg of body weight was injected into mice twice at an interval of 3 h between injection. After 24 h, the mice were sacrificed and the organs (spleen, kidney, liver) were crushed before plating them on the TH plates for CFU determination. The data from three independent experiments were pooled, and the P-value was determined using the ANOVA on ranks by using Sigma stat software (***P < 0.001 and **P < 0.05).
In order to investigate the possible in vivo efficacy of compound 27, we injected the compound into mice infected intraperitoneally with S. aureus ATCC 29213. In the control group, mice injected with bacteria developed severe signs of sepsis, whereas the treatment group mice showed symptoms of mild to average sick behavior, but later recovered upon a second dose of injection. Compared to the controls, treatments with compound 27 yielded an increase in survival and significantly lower bacterial numbers in the spleen and liver of the animals as compared to kidney (Figure 4B). We believe that the lower activity of the drug in the kidney may be due to excretion of the drug through urine or drug metabolism.21
In conclusion, we have designed and synthesized a new class of hybrids by using a pharmacophore hybridization approach. The majority of these hybrid compounds, especially, 27, exhibited promising in vitro and in vivo antibacterial activity against S. aureus. Furthermore, these compounds were not showing the hemolytic activity against human erythrocytes at 50 μg/mL concentration. Further studies to unravel the mechanism of action and structural optimization of this lead are currently underway in our laboratory. It is intended that this study will help to discover and develop a new class of next generation antibacterial agents.
Acknowledgments
We thank to SAIF division, CSIR-CDRI for the analytical facilities and A.Lal for technical assistance. This is CSIR-CDRI communication number 8996.
Supporting Information Available
Experimental procedures, characterization of all compounds together with protocols for biological assays. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00169.
K.B.R. and R.K.M. are thankful to CSIR, New Delhi, P.K. for UGC, and I.S. for ICMR fellowships.
The authors declare no competing financial interest.
Supplementary Material
References
- Antibiotic resistance threats in the United States. In Centers for Disease Control and Prevention (CDC); CDC: Atlanta, GA, 2013; available from http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. [Google Scholar]
- Chugh T. D. Emerging and re-emerging bacterial diseases in India. J. Biosci. 2008, 33, 549–555. [DOI] [PubMed] [Google Scholar]
- Spaulding A. R.; Salgado-Pabon W.; Merriman J. A.; Stach C. S.; Ji Y.; Gillman A. N.; Peterson M. L.; Schlievert P. M. Vaccination against Staphylococcus aureus pneumonia. J. Infect. Dis. 2013, 209, 1955–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadio S.; Maffioli S.; Monciardini P.; Sosio M.; Jabes D. Antibiotic discovery in the twenty-first century: current trends and future perspectives. J. Antibiot. 2010, 63, 423–430. [DOI] [PubMed] [Google Scholar]
- Livermore D. M. The need for new antibiotics. Clin. Microbiol. Infect. 2004, 10 (Suppl 4), 1–9. [DOI] [PubMed] [Google Scholar]
- Brotz-Oesterhelt H.; Sass P. Postgenomic strategies in antibacterial drug discovery. Future Microbiol. 2010, 5, 1553–1579. [DOI] [PubMed] [Google Scholar]
- Fournier P. E.; Raoult D. Prospects for the future using genomics and proteomics in clinical microbiology. Annu. Rev. Microbiol. 2011, 65, 169–188. [DOI] [PubMed] [Google Scholar]
- Nowakowska Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007, 42, 125–137. [DOI] [PubMed] [Google Scholar]
- de Carvalho Tavares L.; Johann S.; Maria de Almeida Alves T.; Guerra J. C.; Maria de Souza-Fagundes E.; Cisalpino P. S.; Bortoluzzi A. J.; Caramori G. F.; de Mattos Piccoli R.; Braibante H. T.; Braibante M. E.; Pizzolatti M. G. Quinolinyl and quinolinyl N-oxide chalcones: synthesis, antifungal and cytotoxic activities. Eur. J. Med. Chem. 2011, 46, 4448–4456. [DOI] [PubMed] [Google Scholar]
- Nielsen S. F.; Larsen M.; Boesen T.; Schonning K.; Kromann H. Cationic chalcone antibiotics. Design, synthesis, and mechanism of action. J. Med. Chem. 2005, 48, 2667–2677. [DOI] [PubMed] [Google Scholar]
- Paget C. J.; Kisner K.; Stone R. L.; DeLong D. C. Heterocyclic substituted ureas. II. Immunosuppressive and antiviral activity of benzothiazole- and benzoxazoleureas. J. Med. Chem. 1969, 12, 1016–1018. [DOI] [PubMed] [Google Scholar]
- Desroy N.; Denis A.; Oliveira C.; Atamanyuk D.; Briet S.; Faivre F.; LeFralliec G.; Bonvin Y.; Oxoby M.; Escaich S.; Floquet S.; Drocourt E.; Vongsouthi V.; Durant L.; Moreau F.; Verhey T. B.; Lee T. W.; Junop M. S.; Gerusz V. Novel HldE-K inhibitors leading to attenuated Gram negative bacterial virulence. J. Med. Chem. 2013, 56, 1418–1430. [DOI] [PubMed] [Google Scholar]
- Mohammad H.; Mayhoub A. S.; Ghafoor A.; Soofi M.; Alajlouni R. A.; Cushman M.; Seleem M. N. Discovery and characterization of potent thiazoles versus methicillin- and vancomycin-resistant Staphylococcus aureus. J. Med. Chem. 2014, 57, 1609–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalaf A. I.; Waigh R. D.; Drummond A. J.; Pringle B.; McGroarty I.; Skellern G. G.; Suckling C. J. Distamycin analogues with enhanced lipophilicity: Synthesis and antimicrobial activity. J. Med. Chem. 2004, 47, 2133–2156. [DOI] [PubMed] [Google Scholar]
- LaMarche M. J.; Leeds J. A.; Amaral A.; Brewer J. T.; Bushell S. M.; Deng G. J.; Dewhurst J. M.; Ding J.; Dzink-Fox J.; Gamber G.; Jain A.; Lee K.; Lee L.; Lister T.; McKenney D.; Mullin S.; Osborne C.; Palestrant D.; Patane M. A.; Rann E. M.; Sachdeva M.; Shao J.; Tiamfook S.; Trzasko A.; Whitehead L.; Yifru A.; Yu D. H.; Yan W. L.; Zhu Q. M. Discovery of LFF571: An Investigational Agent for Clostridium difficile Infection. J. Med. Chem. 2012, 55, 2376–2387. [DOI] [PubMed] [Google Scholar]
- Marco-Contelles J.; Soriano E. The Medicinal Chemistry of Hybrid-Based Drugs Targeting Multiple Sites of Action. Curr. Top. Med. Chem. 2011, 11, 2714–2715. [DOI] [PubMed] [Google Scholar]
- Viegas-Junior C.; Danuello A.; Bolzani V. D.; Barreir E. J.; Fraga C. A. M. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [DOI] [PubMed] [Google Scholar]
- Liaras K.; Geronikaki A.; Glamoclija J.; Ciric A.; Sokovic M. Thiazole-based chalcones as potent antimicrobial agents. Synthesis and biological evaluation. Bioorg. Med. Chem. 2011, 19, 3135–3140. [DOI] [PubMed] [Google Scholar]
- Sashidhara K. V.; Kumar M.; Khedgikar V.; Kushwaha P.; Modukuri R. K.; Kumar A.; Gautam J.; Singh D.; Sridhar B.; Trivedi R. Discovery of coumarin-dihydropyridine hybrids as bone anabolic agents. J. Med. Chem. 2013, 56, 109–122.23214410 [Google Scholar]
- Sashidhara K. V.; Kumar A.; Kumar M.; Sarkar J.; Sinha S. Synthesis and in vitro evaluation of novel coumarin-chalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 7205–7211. [DOI] [PubMed] [Google Scholar]
- Lohr J. W.; Willsky G. R.; Acara M. A. Renal drug metabolism. Pharmacol. Rev. 1998, 50, 107–141. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.