

Abstract

A focused library of analogues of the dual PLK1 kinase/BRD4 bromodomain inhibitor BI-2536 was prepared and then analyzed for BRD4 and PLK1 inhibitory activities. Particularly, replacement of the cyclopentyl group with a 3-bromobenzyl moiety afforded the most potent BRD4 inhibitor of the series (39j) with a Ki = 8.7 nM, which was equipotent against PLK1. The superior affinity of 39j over the parental compound to BRD4 possibly derives from improved interactions with the WPF shelf. Meanwhile, substitution of the pyrimidine NH with an oxygen atom reversed the PLK1/BRD4 selectivity to convert BI-2536 into a BRD4-selective inhibitor, likely owing to the loss of a critical hydrogen bond in PLK1. We believe further fine-tuning will furnish a BRD4 “magic bullet” or an even more potent PLK1/BRD4 dual inhibitor toward the expansion and improved efficacy of the chemotherapy arsenal.
Keywords: Bromodomain, BRD, BET, protein−protein interaction, kinase, BI-2536
Cancer remains one of the most challenging pathologies of our time owing to its manifestation through the aberrant regulation of multiple signaling pathways; for example, the upregulation of the antiapoptotic Bcl-2 proteins and the dysregulation of various kinases.1 A conventional strategy to block cancer cell growth is to inhibit at least one of the proteins involved in these pathways. However, while target-selective inhibitors may function well in vitro, the upregulation of compensatory signaling pathways often compromises their efficacies in cells. Rationally designed polypharmacology, in which a single synthetic agent is fashioned to recognize several key biological targets, carries the promise to deliver drugs that are more efficacious in cells, as well as overcome the drawbacks associated with multidrug regimens.2 Imatinib, like most of the other FDA-approved kinase inhibitors, owes much of its success to targeting multiple kinases involved in tumor development and progression.3 Given the efficacy of imatinib, a notion that is gaining much traction is that the next generation of anticancer agents should go one stage further and target multiple protein targets of different families that are involved in tumorigenesis.4
Polo-like kinase 1 (PLK1) and bromodomain 4 (BRD4) are both drivers in acute myeloid leukemia (AML).5,6 They are also intricately involved in mitosis.7,8 Their dual inhibition by a single drug molecule might provide the platform for a new strategy to treat AML as well as various other cancers. Recently, the laboratories of Knapp and Schönbrunn independently discovered that the PLK1 inhibitor BI-2536 (1) is also a potent inhibitor of BRD4, which was confirmed through an Alpha Screen (IC50 = 25 nM),9 isothermal titration calorimetry (Kd = 37 nM),10 and cocrystal structures.9,10 The BRD proteins recognize ε-N-acetylated lysines of histones and have been dubbed epigenetic “readers”.11 The bromo and extra-terminal domain (BET) proteins of the BRD family (BRD2, BRD3, BRD4, and BRDT) have recently emerged as druggable targets for the development of new anticancer agents owing to their roles in the transcriptional regulation of genes involved in tumor development (e.g., c-MYC) and survival (e.g., BCL2).12−14 Particularly, since the BET proteins regulate the transcription of c-Myc, their inhibition provides an alternative and indirect strategy to counter tumorigenesis,15 which is urgently needed given the difficulties associated through targeting c-Myc with synthetic agents.16−22
Herein, we describe a focused structure–activity relationship (SAR) study of BI-2536 (1) against BRD4. Our findings hint at possible strategies to enhance the dual inhibitory activity of BI-2536 as well as to render the ligand more selective for BRD4 over PLK1. The ability to fine-tune a ligand in this way may help in the expansion of personalized medicines and, more generally, may assist in the delineation of biochemical pathways targeted by other drugs with polypharmacological profiles.
The crystal structure of BI-2536 bound to the first bromodomain of BRD4, BRD4(1), is shown in Figure 1 (PDB ID: 4OGI(10)). The methylated amide of BI-2536 functions as a mimetic of ε-N-acetylated lysine wherein the carbonyl forms a water-mediated hydrogen bond with the side chain amide of N140, while the methyl group is directed into a hydrophobic subpocket formed from F83, M132, and C136. The aniline NH and one of the pyrimidine nitrogen atoms bind the backbone amide of Q85 also through an intermediary water molecule. The ethyl group is projected into a small hydrophobic subpocket (V87/L92/L94/Y97), while the cyclopentyl moiety and the N-methylpiperidine point to the solvent. Close inspection of the binding site indicates that replacement of the amide methyl group with a slightly larger ethyl or isopropyl might enhance binding affinity to BRD4; large groups, such as benzyl, conversely, are not expected to be accommodated here. Introduction of a kink into the cyclopentyl group might allow it to interact better with the WPF shelf (W81, P82, F83). Finally, elaboration of the 3-methoxy group is predicted to improve binding affinity through interactions with L92 and/or L94. One of the pyrimidine nitrogens and the aniline NH form critical hydrogen bonds with C133 in PLK1 (PDB ID = 2RKU). As suggested by Knapp et al., substitution of this NH with NMe will result in the loss of a hydrogen bond donor, specifically the interaction with the backbone carbonyl of Cys133, and a concomitant reduction in PLK1 binding affinity.10 Therefore, toward the optimization of the BRD4 inhibitory activity of BI-2536, we elected to vary the methyl, ethyl, and cyclopentyl moieties, as well as the 3-methoxy group. Substitution of the aniline NH with an oxygen atom should render this fragment incapable of functioning as a hydrogen bond donor yet still permit a water-mediated hydrogen bond, and was, therefore, predicted to furnish BRD4 selectivity. It was decided to retain the aminopiperidine moiety to assist with compound solubility.
Figure 1.
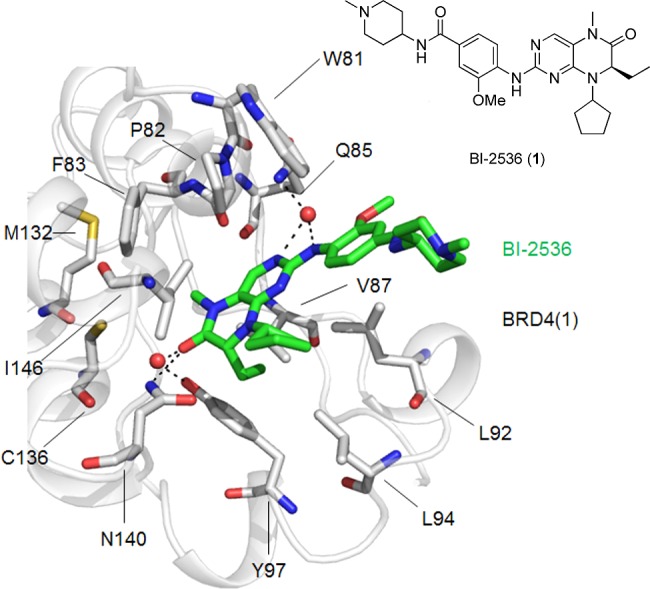
Crystal structure of BI-2536 bound to BRD4(1) (PDB ID = 4OGI).10 Key side chains of BRD4(1) are shown in stick format and colored by atom type (gray = carbon; blue = nitrogen; red = oxygen; yellow = sulfur). BI-2536 is shown in stick format and colored by atom type (green = carbon; blue = nitrogen; red = oxygen). Red sphere = water molecule; dashed line = hydrogen bond.
Accordingly, we prepared a focused library of analogues of BI-2536 by adapting a previously described synthesis of BI-2536.23 Briefly, the appropriate amino acids 2–5 were first esterified and then N-alkylated (R2 group) by reductive amination to deliver compounds 6–11 (Scheme 1). Regioselective nucleophilic aromatic substitutions (SNAr) of 2,6-dichloro-3-nitropyrimidine furnished tertiary anilines 12–17, wherein the nitro group directed ortho attack of amines 6–11 through intramolecular, hydrogen-bonded, six-membered transition states.24,25 The dihydropteridinone scaffold was next constructed by reductive heterocyclizations of 12–17 into 18–23, respectively, with iron powder in hot AcOH. Introduction of the R3 group was achieved by deprotonation of the anilide NH followed by quenching with various alkyl iodides and bromides to yield the 2-chloropyrimidines 24–33. Meanwhile, to complete the synthesis of BI-2536 and its analogues, the requisite anilines were prepared as in Scheme 2. In short, various 4-nitrobenzoic acids (34a–g) were activated by HBTU and then coupled to 4-amino-1-methylpiperidine to deliver amides 35a–g, which were subsequently reduced with tin(II) chloride (SnCl2) to yield anilines 36a–g. Phenol 38 was prepared by the condensation of vanillic acid (37) with 4-amino-1-methylpiperidine. Finally, HCl-assisted SNAr of the 2-chloropyrimidines 24–33 with anilines 36a–g and phenol 38 furnished the lead compound BI-2536 (1) and its analogues 39a–q, whose structures are given in full in Table 1.
Scheme 1.

Reagents and conditions: (a) SOCl2, MeOH, 0 °C to reflux, 16 h; (b) cyclopentanone, isobutyraldehyde or 3-bromobenzaldehyde, NaBH(OAc)3, NaOAc, CH2ClCH2Cl, RT, 16 h; (c) 2,4-dichloro-5-nitropyrimidine, K2CO3, acetone, RT, 16 h; (d) Fe, AcOH, 70 °C, 1 h, 100 °C, 4–5 h; (e) alkyl iodide or benzyl bromide, NaH, DMF, 0 °C to RT, 3−16 h. Cp = cyclopentyl.
Scheme 2.

Reagents and conditions: (a) 4-amino-1-methylpiperidine, HBTU, Et3N, DMF, RT, 16 h; (b) SnCl2·2H2O, EtOAc/ethanol, 10:3, 50 °C, 16 h; (c) 24–33, c. HCl, EtOH/dioxane/H2O, 1:1:1, 100 °C, 24–48 h; (d) 4-amino-1-methylpiperidine, EDCI·HCl, HOBt·H2O, Et3N, CH3CN, RT, 16 h.
Table 1. Structure–Activity Relationships of the R1, R2, R3, R4, and X Groups; Cp = Cyclopentyl.
| compd | R1 | R2 | R3 | R4 | X | BRD4 Ki (nM)a | PLK1 Ki (nM)a |
|---|---|---|---|---|---|---|---|
| BI-2536 | (R)-Et | Cp | Me | 3-OMe | NH | 56 ± 9 | 0.22 ± 0.01 |
| 39a | (R)-Et | Cp | H | 3-OMe | NH | 7950 ± 1550 | ND |
| 39b | (R)-Et | Cp | Et | 3-OMe | NH | 470 ± 10 | 0.91 ± 0.13 |
| 39c | (R)-Et | Cp | iPr | 3-OMe | NH | 4500 ± 300 | ND |
| 39d | (R)-Et | Cp | iBu | 3-OMe | NH | >10000 | ND |
| 39e | (R)-Et | Cp | Bn | 3-OMe | NH | >10000 | ND |
| 39f | H | Cp | Me | 3-OMe | NH | 1400 ± 400 | ND |
| 39g | (S)-Et | Cp | Me | 3-OMe | NH | 54 ± 4 | 0.42 ± 0.04 |
| 39h | (R)-Bn | Cp | Me | 3-OMe | NH | >10000 | 4.20 ± 0.99 |
| 39i | (R)-Et | iBu | Me | 3-OMe | NH | 970 ± 30 | 6.95 ± 1.63 |
| 39j | (R)-Et | 3-Br-C6H4CH2 | Me | 3-OMe | NH | 8.7 ± 1.3 | 5.80 ± 0.99 |
| 39k | (R)-Et | Cp | Me | H | NH | 60 ± 7 | ND |
| 39l | (R)-Et | Cp | Me | 2-OMe | NH | 37 ± 5 | 0.52 ± 0.01 |
| 39m | (R)-Et | Cp | Me | 3-F | NH | 46 ± 2 | ND |
| 39n | (R)-Et | Cp | Me | 3-OiBu | NH | 29 ± 9 | ND |
| 39o | (R)-Et | Cp | Me | 3-OBn | NH | 100 ± 0 | ND |
| 39p | (R)-Et | Cp | Me | 3-OCp | NH | 27 ± 8 | 0.84 ± 0.01 |
| 39q | (R)-Et | Cp | Me | 3-OMe | O | 99 ± 11 | 240 ± 14 |
Data represent the mean and standard deviation of two independent experiments. ND = not determined.
Table 1 shows the derivatives with variations of substituents on the dihydropteridinone ring along with associated BRD4 and PLK1 inhibitory activities. The data in Table 1 shows BRD4 and PLK1 inhibitory activities, and is presented as Ki values, which were obtained with BROMOscan and KINOMEscan, DiscoveRx’s proprietary ligand binding competition assays that measure interactions between test compounds and bromodomains or kinases, respectively. We obtained similar binding data for the parent compound BI-2536 to that reported previously for BRD49,10 and PLK1.26 It is unsurprising that BI-2536 is >250-fold more selective for PLK1 given the extensive optimization that led to its discovery.26 Replacement of the CONMe group with a CONH group reduced the inhibitory activity by almost 150-fold, supporting its functional role as a mimetic of ε-N-acetylated lysine moieties. Interestingly, although the binding pocket that accommodates this methyl group appears large enough to allow occupancy by slightly larger groups such as ethyl and isopropyl, all groups bigger than methyl resulted in weaker inhibitors. For example, the Ki values for compounds with R3 = Me, Et, iPr, and Bn were 56, 470, 4500, and >10 000 nM, respectively. Marginally increasing the size of the R3 group was not tolerated by PLK1, either.
In the cocrystal structure of BRD4(1)–BI-2536, the ethyl group at the R1 position points into the pocket formed by V87, L92, L94, and Y97. Changing the stereochemistry from R to S did not affect the binding affinity to BRD4 (compare 1 with 39g) but resulted in a 2-fold drop in potency against PLK1. With respect to BRD4 inhibition, the S-Et group can point into a pocket on the other side of V87/L92/L94/Y97 formed by P82, F83, and I146, and this may account for the subtle effect of the chirality change. Deleting the ethyl group afforded a reduction in BRD4 inhibitory activity of 26-fold (39f: Ki = 1400 nM), while replacing it with a bulky R-benzyl group (compound 39h) completely abolished binding. Together, these data indicate that a small hydrophobic group is optimal here, consistent with the crystal structure. In PLK1 (PDB ID = 2RKU), the R-Et group is surrounded by L59, C67, A80, K82, and L130. Similar to BRD4, this pocket is largely tolerant of the chirality change but cannot accommodate a larger benzyl group.
While modifying the R1 and R3 groups exhibited only detrimental impacts on BRD4 binding, changing the R2 group from cyclopentyl to 3-bromobenzyl increased the BRD4 binding affinity by 7-fold (39j: Ki = 8.7 nM) although a bulkier isobutyl group (39i) was not tolerated. The potent binding affinity of 39j places it among the most active BRD4 inhibitors known14 and its increased affinity over BI-2536 might be explained by hydrophobic interactions with the WPF shelf. Notably, compound 39j bound PLK1 25-fold weaker than BI-2536, such that affinities for BRD4 and PLK1 had become effectively equipotent. Taken together, these data indicate that the R2 group appears to be a dictator of selectivity, and further optimization here might furnish a selective, subnanomolar BRD4 inhibitor.
Shifting the 3-methoxy group to the 2-position (compound 39l) did not have a significant impact on the binding to BRD4. Moreover, its deletion (39k) or replacement with a fluorine atom (39m) also had little effect. However, substitution of the 3-methoxy group with bulkier isobutoxy (39n) and cyclopentyloxy (39p) moieties afforded a 2-fold enhancement in binding affinity to BRD4, which may be due to improved contacts with L92 and/or L94. Concomitantly, a 4-fold reduction in binding affinity to PLK1 was observed, as predicted.10 Akin to our findings with the R2 group, careful modification of the R4 group might provide further improvements to BRD4 inhibitory activity. One of the most striking observations of our SAR work is seen when comparing the in vitro data for BI-2536 with 39q. Substitution of the pyrimidine NH with an isosteric O afforded more than a 1000-fold drop in activity against PLK1, yet resulted in an only 2-fold reduction in activity against BRD4, effectively transforming the PLK1 selective inhibitor into a BRD4 selective inhibitor. This result is consistent with the NH, as a hydrogen bond donor, engaging in a critical hinge interaction with C133 in the active site of PLK1, an interaction that is lost upon its replacement with an oxygen atom; this group engages in a water-mediated hydrogen bond in BRD4 that can presumably still form with 39q and so a limited effect on binding affinity was observed. This dramatic impact on binding profiles indicates that the nature of the functional group here is a key determinant of ligand specificity and may be exploited further (e.g. NMe) toward achieving even greater BRD4 selectivity.
To gain a better appreciation of how our most potent compound 39j likely binds BRD4(1), we employed molecular modeling with GOLD.27 Figure 2 shows a GOLD high scoring docking pose of 39j bound to BRD4(1). Upon comparison of Figures 1 and 2, it will be noticed that 39j and BI-2536 bind to BRD4 in very similar patterns, and form largely identical interactions with BRD4. Structurally, the only difference between these two molecules is the replacement of the cyclopentyl group in BI-2536 with a 3-bromobenzyl moiety. In BI-2536, the cyclopentyl group does not appear to be involved in any notable interactions with BRD4 as it is directed out into the solvent. In contrast, the installation of the benzyl CH2 in 39j provides the extra flexibility to reach out to the WPF shell (W81, P82, and F83), possibly accounting for the 7-fold improvement in activity. Furthermore, the bromine atom potentially forms a hydrophobic interaction with I146, which might also explain the greater binding affinity of 39j. Therefore, future investigation into the R2 position with flexible and hydrophobic arylmethyl groups may lead to more potent BRD4 inhibitors.
Figure 2.
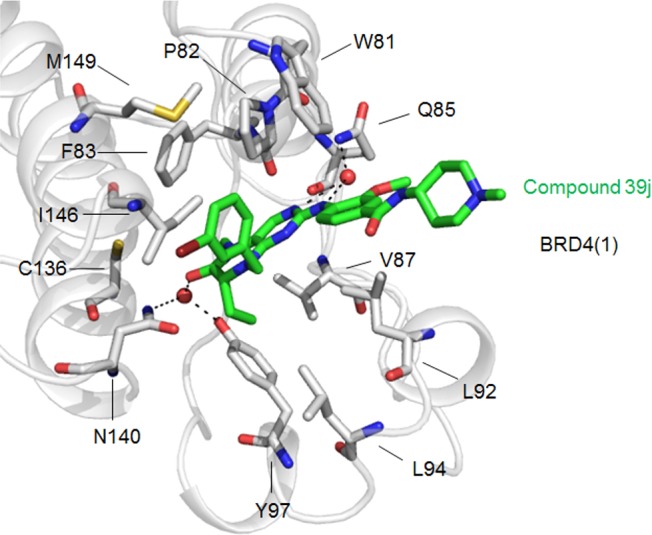
High scoring, GOLD-predicted docking mode of compound 39j in BRD4(1). Compound 39j shown in stick format and colored by atom type (green = carbon; blue = nitrogen; red = oxygen). Red sphere = water molecule; dashed line = hydrogen bond.
Compounds were also analyzed in a CellTiter-Blue cell viability assay with the MV4-11 cell line, which is an acute myeloid leukemia cell line that expresses high levels of BRD4 and PLK1. As positive controls, the BRD4-selective inhibitor (+)-JQ-1 and the PLK1-selective inhibitor GSK-461364 were selected. The growth inhibition (GI50) data is shown in Table 2, which represents the concentration of the small molecule that elicits 50% growth inhibition of the MV4-11 cells. The overall trend of in vitro data is mirrored by the cell data, which supports the conclusion that cell death is through the inhibition of BRD4 and PLK1. There are a few anomalous results that require discussion. The cell data for 39j and 39p are less potent, while that for 39f and 39i are more potent, than would be anticipated given their in vitro data. Considering its polypharmacological profile, it is possible that BI-2536, and its analogues, bind other targets in addition to BRD4 and PLK1, such as PLK2 and PLK3,26 which may account for the imperfect correlation between the in vitro and cell data.
Table 2. MV4-11 Cell Viability Exposed to BI-2536 Analogues.
| compd | MV4-11, GI50 (μM)a | compd | MV4-11, GI50 (μM)a |
|---|---|---|---|
| BI-2536 | 0.0152 | 39h | 1.87 |
| (+)-JQ-1 | 0.08 | 39i | 0.04 |
| GSK-461364 | 0.679 | 39j | 0.675 |
| 39a | 0.219 | 39k | 0.003 |
| 39b | 0.392 | 39l | 0.015 |
| 39c | 2.67 | 39m | 0.004 |
| 39d | 2.53 | 39n | 0.154 |
| 39e | 7.28 | 39o | 0.356 |
| 39f | 0.219 | 39p | 0.240 |
| 39g | 0.019 | 39q | 1.34 |
Compound concentration that elicits 50% growth inhibition of MV4-11 cells.
In summary, building on the initial discoveries by Knapp and Schönbrunn, an SAR campaign of the PLK1 inhibitor BI-2536 has revealed opportunities for its continued optimization as a dual inhibitor of PLK1 and BRD4, as well as a selective inhibitor of the latter. The impacts of structural modifications were largely in agreement with predictions made based on the BRD4(1)–BI-2536 cocrystal structure. We believe that combining further modifications to the R2 and R4 groups, as well as switching the pyrimidine NH to O, could provide selective, subnanomolar BRD4 inhibitors. At the same time, we anticipate it will be feasible to enhance BRD4 inhibitory activity and yet maintain dual PLK1/BRD4 inhibitory activity through careful selection of the R4 side chain while retaining, most critically, the pyrimidine NH, as well as the cyclopentyl group and the R-stereochemistry of the ethyl group. More generally, it is considered that drugs with polypharmacological profiles may likewise be optimized for inhibitory activities against specific deconvoluted targets, which may assist in a better understanding of the biochemical pathways targeted within cells.
Glossary
ABBREVIATIONS
- BRD4
bromodomain 4
- BET
bromodomain and extra-terminal domain
- PLK1
polo-like kinase 1
Supporting Information Available
Synthetic procedures and assay protocols. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00084.
Author Contributions
All authors have given approval to the final version of the manuscript.
We thank the University of Maryland School of Pharmacy (start-up funds (to S.F.); fellowships (to L.C. and J.L.Y.)), ConverGene and the American Foundation for Pharmaceutical Education (fellowship to J.L.Y.) for financial support of this research. Chemietek is acknowledged for the generous donation of GSK-461364. PyMOL was used for rendering Figures 1 and 2.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang J.; Yang P. L.; Gray N. S. Targeting Cancer with Small Molecule Kinase Inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [DOI] [PubMed] [Google Scholar]
- Peters J.-U. Polypharmacology - Foe or Friend?. J. Med. Chem. 2013, 56, 8955–8971. [DOI] [PubMed] [Google Scholar]
- Capdeville R.; Buchdunger E.; Zimmermann J.; Matter A. Glivec (STI571, Imatinib), a Rationally Developed, Targeted Anticancer Drug. Nat. Rev. Drug Discovery 2002, 1, 493–502. [DOI] [PubMed] [Google Scholar]
- Petrelli A.; Giordano S. From Single- to Multi-Target Drugs in Cancer Therapy: When Aspecificity Becomes an Advantage. Curr. Med. Chem. 2008, 15, 422–432. [DOI] [PubMed] [Google Scholar]
- Renner A. G.; Dos Santos C.; Recher C.; Bailly C.; Créancier L.; Kruczynski A.; Payrastre B.; Manenti S. Polo-like Kinase 1 Is Overexpressed in Acute Myeloid Leukemia and Its Inhibition Preferentially Targets the Proliferation of Leukemic Cells. Blood 2009, 114, 659–662. [DOI] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature 2011, 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gutierrez P.; Mundi M.; Garcia-Dominguez M. Association of Bromodomain BET Proteins with Chromatin Requires Dimerization through the Conserved Motif B. J. Cell Sci. 2012, 125, 3671–3680. [DOI] [PubMed] [Google Scholar]
- Van Vugt M. A. T. M.; Medema R. H. Getting in and out of Mitosis with Polo-like Kinase-1. Oncogene 2005, 24, 2844–2859. [DOI] [PubMed] [Google Scholar]
- Ember S. W. J.; Zhu J.-Y.; Olesen S. H.; Martin M. P.; Becker A.; Berndt N.; Georg G. I.; Schönbrunn E. Acetyl-Lysine Binding Site of Bromodomain-Containing Protein 4 (BRD4) Interacts with Diverse Kinase Inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciceri P.; Müller S.; O’Mahony A.; Fedorov O.; Filippakopoulos P.; Hunt J. P.; Lasater E. A.; Pallares G.; Picaud S.; Wells C.; Martin S.; Wodicka L. M.; Shah N. P.; Treiber D. K.; Knapp S. Dual Kinase-Bromodomain Inhibitors for Rationally Designed Polypharmacology. Nat. Chem. Biol. 2014, 10, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J.-P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A.-C.; Arrowsmith C. H.; Knapp S. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyce A.; Ganji G.; Smitheman K. N.; Chung C.-W.; Korenchuk S.; Bai Y.; Barbash O.; Le B.; Craggs P. D.; McCabe M. T.; Kennedy-Wilson K. M.; Sanchez L. V.; Gosmini R. L.; Parr N.; McHugh C. F.; Dhanak D.; Prinjha R. K.; Auger K. R.; Tummino P. J. BET Inhibition Silences Expression of MYCN and BCL2 and Induces Cytotoxicity in Neuroblastoma Tumor Models. PLoS One 2013, 8, e72967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M.; Measures A. M.; Wilson B. G.; Cortopassi W. A.; Alexander R.; Höss M.; Hewings D. S.; Rooney T. P.; Paton R. S.; Conway S. J. Small Molecule Inhibitors of Bromodomain–Acetyl-lysine Interactions. ACS Chem. Biol. 2015, 10, 22–39. [DOI] [PubMed] [Google Scholar]
- Mertz J. A.; Conery A. R.; Bryant B. M.; Sandy P.; Balasubramanian S.; Mele D. A.; Bergeron L.; Sims R. J. Targeting MYC Dependence in Cancer by Inhibiting BET Bromodomains. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S.; Prochownik E. V. Small-Molecule Inhibitors of the Myc Oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albihn A.; Johnsen J. I.; Henriksson M. A. MYC in Oncogenesis and as a Target for Cancer Therapies. Adv. Cancer Res. 2010, 107, 163–224. [DOI] [PubMed] [Google Scholar]
- Yap J. L.; Chauhan J.; Jung K.-Y.; Chen L.; Prochownik E. V.; Fletcher S. Small-Molecule Inhibitors of Dimeric Transcription Factors: Antagonism of Protein–Protein and Protein–DNA Interactions. MedChemComm 2012, 3, 541. [Google Scholar]
- Chauhan J.; Wang H.; Yap J. L.; Sabato P. E.; Hu A.; Prochownik E. V.; Fletcher S. Discovery of Methyl 4′-Methyl-5-(7-nitrobenzo[c][1,2,5]oxadiazol-4-Yl)-[1,1′-Biphenyl]-3-Carboxylate, an Improved Small-Molecule Inhibitor of c-Myc-Max Dimerization. ChemMedChem 2014, 9, 2274–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Chauhan J.; Hu A.; Pendleton K.; Yap J. L.; Sabato P. E.; Jones J. W.; Perri M.; Yu J.; Cione E.; Kane M. A.; Fletcher S.; Prochownik E. V. Disruption of Myc-Max Heterodimerization with Improved Cell-Penetrating Analogs of the Small Molecule 10074-G5. Oncotarget 2013, 4, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap J. L.; Wang H.; Hu A.; Chauhan J.; Jung K.-Y.; Gharavi R. B.; Prochownik E. V.; Fletcher S. Pharmacophore Identification of c-Myc Inhibitor 10074-G5. Bioorg. Med. Chem. Lett. 2013, 23, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K. Y.; Wang H.; Teriete P.; Yap J. L.; Chen L.; Lanning M. E.; Hu A.; Lambert L. J.; Holien T.; Sundan A.; Cosford N. D.; Prochownik E. V.; Fletcher S. Perturbation of the c-Myc-Max Protein-Protein Interaction via Synthetic α-Helix Mimetics. J. Med. Chem. 2015, 58, 3002–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budin G.; Yang K. S.; Reiner T.; Weissleder R. Bioorthogonal Probes for Polo-like Kinase 1 Imaging and Quantification. Angew. Chem., Int. Ed. 2011, 50, 9378–9381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap J. L.; Hom K.; Fletcher S. Ortho-Selectivity in the Nucleophilic Aromatic Substitution (SNAr) Reactions of 3-Substituted, 2,6-Dichloropyridines with Alkali Metal Alkoxides. Tetrahedron Lett. 2011, 52, 4172–4176. [Google Scholar]
- Wang X.; Salaski E. J.; Berger D. M.; Powell D. Dramatic Effect of Solvent Hydrogen Bond Basicity on the Regiochemistry of S(N)Ar Reactions of Electron-Deficient Polyfluoroarenes. Org. Lett. 2009, 11, 5662–5664. [DOI] [PubMed] [Google Scholar]
- Steegmaier M.; Hoffman M.; Baum A.; Lénárt P.; Petronczki M.; Krssák M.; Gürtler U.; Garin-Chesa P.; Lieb S.; Quant J.; Grauert M.; Adolf G. R.; Kraut N.; Peters J. M.; Rettig W. J. BI 2536, a Potent and Selective Inhibitor of Polo-Like Kinase 1, Inhibits Tumor Growth In vivo. Curr. Biol. 2007, 17, 316–322. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.