

Abstract
The principles of molecular property optimization in drug design have been understood for decades, yet much drug discovery activity today is conducted at the periphery of historical druglike property space. Lead optimization trajectories aimed at reducing physicochemical risk, assisted by ligand efficiency metrics, could help to reduce clinical attrition rates.
The biological, metabolic, and toxicological effects of a test compound or a marketed drug are direct consequences of its physicochemical profile, which determines the frequency and strength of molecular interactions with a vast pool of macromolecules, and in vivo access to cells and tissues. The physicochemical properties of approved oral drugs result from the synthetic, design, and testing principles that led to their invention, and the subsequent attrition pressures applied by successful drug development. While the underlying science and processes used have evolved radically over the past 60 years, the human body has not.
Molecular properties including size, shape, lipophilicity, hydrogen bonding capability, and polarity are surrogates for compound quality. The principle of minimal hydrophobicity, proposed by Hansch and colleagues in 1987,1 states that “without convincing evidence to the contrary, drugs should be made as hydrophilic as possible without loss of efficacy.” This hypothesis is surviving the test of time and has been quantified as lipophilic ligand efficiency (LLE or LipE).2,3 Given the long established relevance of lipophilicity and the development of druglike and leadlike concepts, it may be surprising that molecules patented by pharmaceutical companies during 2000–103,4 have higher mean lipophilicities and sizes than marketed oral drugs (Figure 1). Some oral drug properties are increasing over time, with molecular weight increasing more rapidly than lipophilicity since the 1950s.3 The mean oral molecular weight and lipophilicity of drugs invented post-1990 are similar to optimized compounds from patents4 (Figure 1). Among all oral drugs invented post-1950, 44% have molecular weight < 400 and cLogP < 3, compared with only 6.6% of mean 2000–10 patent targets. Is an opportunity being missed by conducting so few drug discovery projects in historical druglike space?
Figure 1.
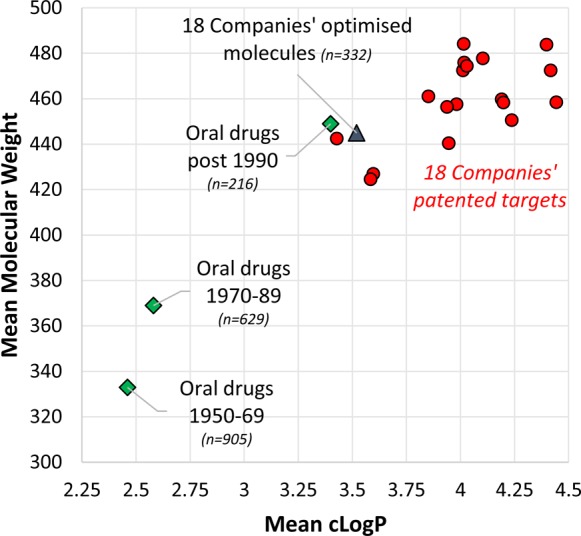
Mean molecular weight and cLogP values of oral drugs according to time of publication (from ref (100), updated with drugs launched up to 2014), and patented targets and optimized compounds from 18 companies during 2000–10 (from ref (4)). Post-1990 oral drugs show increased physical properties versus earlier drugs and are further examined in Table 2. cLogP is the calculated 1-octanol–water partition coefficient (from Biobyte (http://www.biobyte.com/bb/prod/clogp40.html).
Relationships between molecular properties of various compound sets and pharmacokinetic, metabolic, and toxicological data consistently display benefits of optimal lipophilicity and can be used to assess the probability of development risk. For example, several developability measures of GlaxoSmithKline compounds were analyzed using the property forecast index, a hydrophobicity measure combining chromatographically determined lipophilicity and aromatic ring count (Figure 2a). The effect of PFI is assay dependent, but in lowering PFI, the fraction of compounds meeting defined assay criteria tends to increase, as does the population of marketed drugs (Figure 2b). The likelihood of meeting multiple criteria, a typical requirement for a candidate drug, increases substantially with ‘low fat, low flat’ molecules where PFI is <7, versus >7. In considering a portfolio of drug candidates, the probabilistic argument hypothesizes that successful outcomes will increase as the portfolio’s balance of biological and physicochemical properties becomes more similar to that of marketed drugs.
Figure 2.
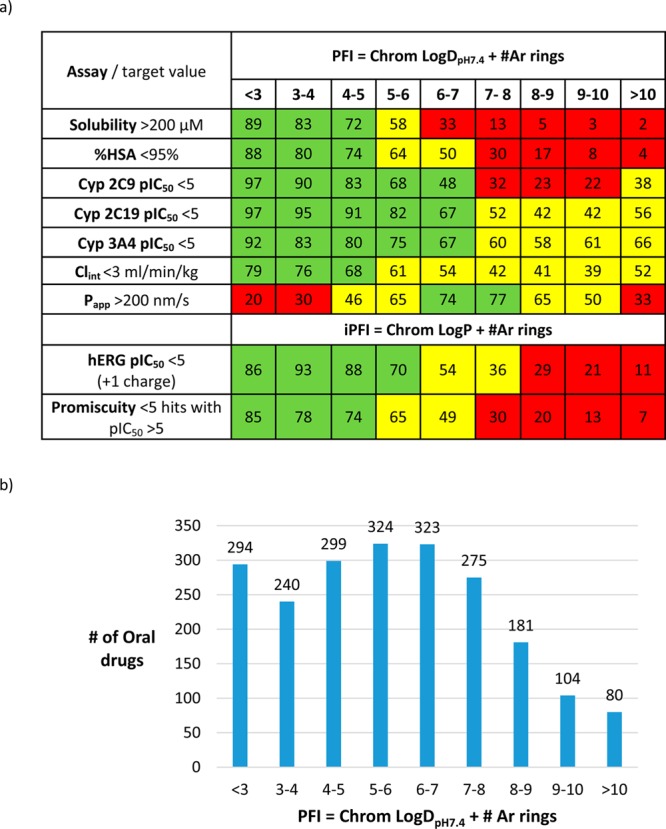
Impact of hydrophobicity on developability assays and the profile of marketed oral drugs. (a) Percentages of GlaxoSmithKline compounds meeting developability criteria according to their property forecast indices (PFI and iPFI (intrinsic PFI based on LogP)), composite measures of hydrophobicity using the combination of chromatographically determined lipophilicities, and aromatic ring count. Some data, e.g., Cyp inhibition and passive permeability (Papp), display nonlinear behavior. Green, ≥67%; Yellow, 34–66%; Red, <33%. All data from Young et al,101 reproduced with permission. (b). Calculated PFI (from ref (101)) distribution of marketed oral drugs (from ref (100), updated with drugs launched up to 2014) shows 70% are in the lower risk zone (PFI < 7).
Is property design being used regularly today? Among 261 publications from January to July 2014 disclosing hit or lead optimization, 33% specifically addressed lipophilic optimization: 40% in industry and 15% in academia (the “Yes” group in Table 1). In both “Yes” and “No” groups, ligand efficiency (LE) did not change and LLE increased, mirroring changes seen in lead to drug optimizations.5 However, property-based thinking paid off because the mean cLogP and LLE of values of optimized compounds from the “Yes” group are significantly improved versus the “No” group (Table 1). The 33% “Yes” is probably a minimum figure because property design in the “No” group could still have been used, specifically or intuitively, or was not disclosed because it was unsuccessful.
Table 1. Summary of Mean in Vitro activity (pX50) and Property Changes in Optimizations Reported during January to July 2014 in the Journal of Medicinal Chemistry, Bioorganic and Medicinal Chemistry Letters, and ACS Medicinal Chemistry Letters.
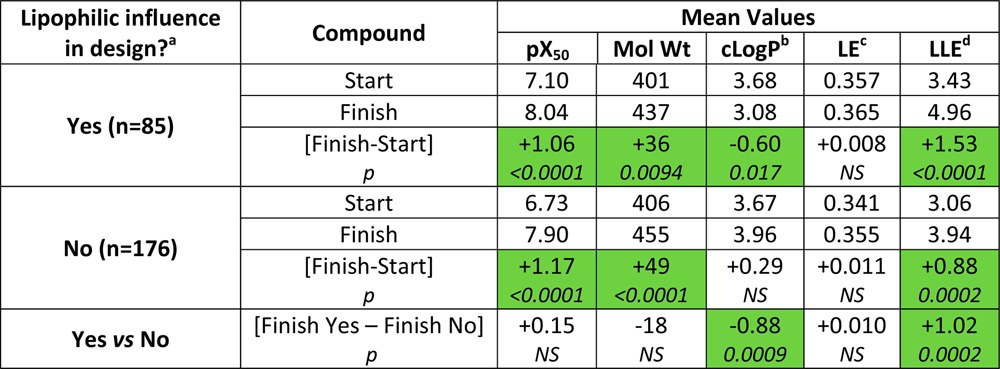
Lipophilic influence in design was assessed as Yes or No based on tactics employed, tabulation of data, and discussion. Optimizations (n = 261) cover lead optimization (n = 169), high throughput screening hit optimization (n = 75) and other lead generation (n = 17). Industrial publications Yes/No = 73:108 (40% Yes); academic publications Yes/No = 12:68 (15% Yes).
cLogP is the calculated 1-octanol–water partition coefficient (from Biobyte (http://www.biobyte.com/bb/prod/clogp40.html).
LE = pX50 × 1.37/# heavy atoms (kcal/mol/atom).
LLE (or LipE) = pX50 – cLogP (unitless). p values are from t tests assuming equal variances; shaded entries show statistically significant changes. NS = not significant (p > 0.05).
Some companies lowered mean lipophilicity in patented compounds during 2000–10, but most did not.4 Views obtained from senior medicinal chemistry leaders in three large pharmaceutical companies recognize the issues and indicate progress is being made, but varying challenges remain: “the quality of molecules is getting better and we’re becoming smarter at design, but strive to further improve” and “despite championing the monitoring and controlling of compound properties, some chemists in my group have not routinely adopted the approach” and “I still see chemists making compounds that they perhaps should not, by just looking at the structure.” A former head of drug discovery at a major contract research organization noted: “it is shocking how many companies value numbers of compounds over quality.”
The stakes are high. Only ∼4% of candidate drugs reach the market, with the success rate in phase II the lowest, at 23%. Recent disclosures acknowledge that suboptimal compounds were progressed to costly clinical trials in the period 2005–10. In Pfizer, low confidence in candidate drug exposure meant that the biological mechanism could not be tested adequately in 43% of phase II failures.6 In AstraZeneca, 38% of projects that advanced to the clinic had low confidence in safety, and 78% of these eventually failed due to toxicity.7 Medicinal chemists’ accountability is obvious: to ensure that compounds reaching the clinic will be able to unambiguously test the disease hypotheses, while minimizing the possibility of failure due to inadequate exposure, off-target toxicity, or poor solubility. Compound quality is fixed at the point of design and is controllable during lead optimization.
Why are medicinal chemists synthesizing so many compounds closer to the periphery than the center of historical druglike space (Figure 1)?
-
•
Misinterpreting the rule-of-5? A molecule possessing cLogP 4.5–5 and molecular weight 450–500 is “rule-of-5 compliant” on these properties (cLogP < 5 and molecular weight < 500), but because these cut-offs are defined by 90th percentile values, only 1% of oral drugs are in this range. Molecules only just meeting the rule-of-five risk having poor developability properties.
-
•
Precedent exists beyond the rule-of-5. Some 8.2% of oral drugs published since 1950 break the rule-of-5 by failing two or more rules (see Table 2). This is incompletely understood territory but possible “compensating” features include natural product origin, complexity such as high chirality and sp3 carbon fraction (e.g., antiviral agents, Table 2), low aromatic ring count, macrocyclic structures, intramolecular hydrogen bonding, and transporter-mediated permeation. Pursuing “exception space” becomes more justifiable if the target is of high therapeutic value, there are no alternative leads, and the portfolio is not heavily weighted by such projects. However, even highly challenging “lipophilic” targets, for example, cholesteryl ester transfer protein, can succumb to property-based inhibitor optimization.2,8
-
•
Increase in less “druggable” targets. Some say the “low-hanging fruit” has been picked, though one wonders what the pioneering innovators from the past might think of that. Antiviral drugs, acting on targets requiring high molecular weight ligands (Table 2), mainly break the rule-of-five and have impressively addressed severe medical need. Protein–protein interactions also present a tough, but not always insurmountable, druggability challenge.
-
•
Disease risk/benefit ratio. In 2012–14, 36% of FDA approvals were orphan drugs, many for treatment of cancer and severe low incidence diseases, where greater safety risk and dosing inconvenience than other diseases can be tolerated. Kinase inhibitors for cancer, along with antivirals, account in part for the molecular weight and lipophilicity increases in recently marketed drugs (Table 2). Ligand efficiency metrics of approved kinase inhibitors are less optimized versus other oral drugs.2
-
•
Hit selection and ‘potency hunting’. Hit selection is a key decision, kicking off investment in design-make-test optimization cycles. Molecular weight often increases in optimization (see Table 1 and ref (5)) and low molecular weight “leadlike” starting points are ideal, allowing molecular growth into druglike space. However, it appears that high affinity often outweighs leadlike considerations in hit selection. Influencing medicinal chemists’ design decisions by using peer review and input from computational and drug metabolism scientists can help curb overemphasis on increasing potency. Multiparameter optimization of predicted human dose is more important than optimizing activity alone, not least, because low doses and exposures reduce in vivo toxicity risks.
-
•
Synthetic feasibility versus design desirability. If high synthetic productivity is sought, there is a risk that hit molecules with good properties may not be prioritized if library synthesis is not readily available. Library profiling in advance is important because parallel synthesis often increases bulk properties. Time spent optimizing and expanding synthesis is worthwhile because it can create opportunities and allow carefully designed molecules to be made.
-
•
Divergent design practices. The physical properties of patented molecules vary markedly between originating Companies (Figure 1), and the differences are not dependent on targets pursued.4 An example is the 2 log unit variance in mean lipophilicity across four Companies pursuing an identical CCR5 receptor antagonist pharmacophore.3 While the impact on design of culture, history, experience, local expertise, and strategy is clearly very powerful, improved physical properties can be found if they are deliberately targeted. For example, AstraZeneca improved solubility by setting specific goals and using predictive methods prior to synthesis.9
Table 2. Properties of 216 Oral Drugs Published since 1990.
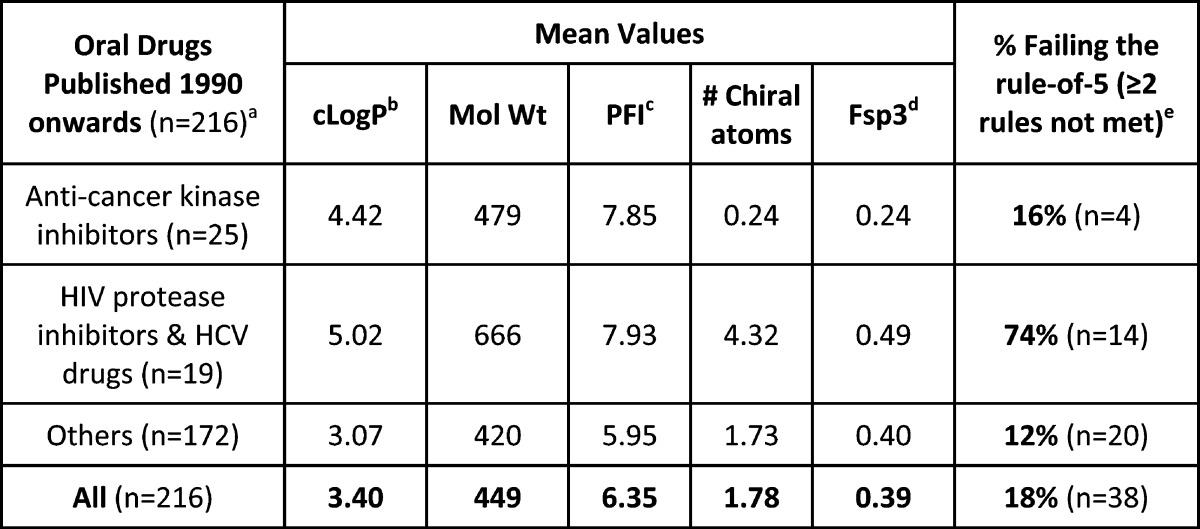
From ref (5), updated with drugs launched up to 2014. Antivirals and anticancer kinase inhibitors contribute to increased cLogP and molecular weight versus earlier drugs (see Figure 1).
cLogP is the calculated 1-octanol−water partition coefficient from Biobyte (http://www.biobyte.com/bb/prod/clogp40.html).
Calculated property forecast index (from ref (6)) = Chrom LogDpH7.4 + #Ar rings.
Fraction of carbon sp3 atoms/total C atoms.
What else can be done? Many oral drugs occupy optimal combined ligand efficiency space (LE and LLE, Table 1) for their specific targets,2 showing the importance of balancing biological activity, size, and lipophilicity in lead optimization. Increasing LLE is likely to be associated with better selectivity,3 efficient polar atom ligand–protein contacts,10 and specific hydrophobic binding. An example is the CCR5 antagonist maraviroc, a drug with optimal LE and LLE,2 where seven available polar atoms make six polar interactions and four lipophilic groups fill nonpolar pockets on the CCR5 receptor.11 Application of LE is useful in hit triage to identify leadlike starting points, and in fragment optimization where its control can provide improved druglike outcomes.4 There is debate about the thermodynamic and mathematical basis of some ligand efficiency measures, and they may be less relevant when lead molecules are highly polar or very small. However, metrics do not have to be perfect to be useful. Application of LE, LLE, their variants, and potency versus property analysis is needed because it promotes awareness of physical property impact.
Every compound or library that is synthesized costs effort, time, and money and must be designed to provide beneficial information. Innovation is vital, but so too is efficient decision-making in avoiding predictably poor compounds9 and making too many compounds.12 Effective optimization is not about meeting idealized druglike principles or rules because targets, disease requirements, and routes of administration will differ. Instead, the focus should be on hypothesis-driven design, guiding optimization trajectories continually toward less risky property space. Drug discovery is highly challenging, but you can improve compound quality if you aim for it. This is not really an option, but an obligation that lies at the heart of medicinal chemistry. Is there any other viable strategy for medicinal chemists to improve the output of marketed drugs?
Views expressed in this editorial are those of the authors and not necessarily the views of the ACS.
The authors declare no competing financial interest.
References
- Hansch C.; Björkroth J. P.; Leo A. Hydrophobicity and central nervous system agents: on the principle of minimal hydrophobicity in drug design. J. Pharm. Sci. 1987, 76, 663–687. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; St-Gallay S. A. The influence of the ‘organizational factor’ on compound quality in drug discovery. Nat. Rev. Drug Discovery 2011, 10, 749–765. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; St-Gallay S. A.; Wenlock M. C. Impact of ion class and time on oral drug molecular properties. MedChemComm 2011, 2, 91–105. [Google Scholar]
- Young R. J.; Green D. V.; Luscombe C. N.; Hill A. P. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discovery Today 2011, 16, 822–830. [DOI] [PubMed] [Google Scholar]
- Perola E. An analysis of the binding efficiencies of drugs and their leads in successful drug discovery programs. J. Med. Chem. 2010, 53, 2986–2997. [DOI] [PubMed] [Google Scholar]
- Morgan P.; Van Der Graaf P. H.; Arrowsmith J.; Feltner D. E.; Drummond K. S.; Wegner C. D.; Street S. D. A. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discovery Today 2012, 17, 419–424. [DOI] [PubMed] [Google Scholar]
- Cook D.; Brown D.; Alexander R.; March R.; Morgan P.; Satterthwaite G.; Pangalos M. N. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat. Rev. Drug Discovery 2014, 13, 419–431. [DOI] [PubMed] [Google Scholar]
- Treiselmann T.; Wagner H.; Fuchs K.; Hamprecht D. W.; Berta D. G.; Cremonesi P.; Streicher R.; Luippold G.; Volz A.; Markert M.; Nar H. Potent cholesteryl ester transfer protein inhibitors of reduced lipophilicity: 1,1’-spiro-substituted hexahydrofuroquinoline derivatives. J. Med. Chem. 2014, 57, 8766–76. [DOI] [PubMed] [Google Scholar]
- Cumming J. G.; Davis A. M.; Muresan S.; Haeberlein M.; Chen H. Chemical predictive modelling to improve compound quality. Nat. Rev. Drug Discovery 2013, 12, 948–962. [DOI] [PubMed] [Google Scholar]
- Lipinksi C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- Higueruelo A. P.; Schreyer A. G.; Bickerton G. R. J.; Blundell T. L.; Pitt W. R. What can we learn from the evolution of protein-ligand interactions to aid the design of new therapeutics?. PLoS One 2012, 7 (12), e51742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q.; Zhu Y.; Li J.; Chen Z.; Han G. W.; Kufareva I.; Li T.; Ma L.; Fenalti G.; Li J.; Zhang W.; Xie X.; Yang H.; Jiang H.; Cherezov V.; Liu H.; Stevens R. C.; Zhao Q.; Wu B. Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshire D. R. How well do medicinal chemists learn from experience?. Drug Discovery Today 2011, 16, 817–821. [DOI] [PubMed] [Google Scholar]