

Abstract
The DDR1 and DDR2 receptor tyrosine kinases are activated by extracellular collagen and have been implicated in a number of human diseases including cancer. We performed a fragment-based screen against DDR1 and identified fragments that bound either at the hinge or in the back pocket associated with the DFG-out conformation of the kinase. Modeling based on crystal structures of potent kinase inhibitors facilitated the “back-to-front” design of potent DDR1/2 inhibitors that incorporated one of the DFG-out fragments. Further optimization led to low nanomolar, orally bioavailable inhibitors that were selective for DDR1 and DDR2. The inhibitors were shown to potently inhibit DDR2 activity in cells but in contrast to unselective inhibitors such as dasatinib, they did not inhibit proliferation of mutant DDR2 lung SCC cell lines.
Keywords: discoidin domain receptor, fragment-based drug design, back to front kinase design
Discoidin domain receptors, DDR1 and DDR2, are transmembrane receptor tyrosine kinases that are activated by collagen binding to their extracellular domain.1,2 DDR1 and DDR2 have been associated with extracellular remodeling, cell adhesion, proliferation and migration, and they have been linked to a number of human diseases, including fibrotic disorders, atherosclerosis and cancer.3−5 There has recently been evidence suggesting that DDR2 inhibitors would be useful for the treatment of lung squamous cell cancer6,7 although the role of DDR2 may be more complex than first realized.8−10 Hammerman et al.6,7 have shown that DDR2 is mutated in approximately 4% of lung squamous cell cancer and have presented data to suggest that these are gain of function mutations. Hammerman et al.6 have also shown that cell lines harboring these mutations are selectively sensitized through knockdown of DDR2 by RNA interference or by treatment of the multitargeted kinase inhibitor dasatinib.11 Selective DDR2 inhibitors would therefore be useful to probe the role of DDR2 and may have utility for the treatment of lung cancer.
To date, most DDR1/2 inhibitors have been derived from cross-screening of existing kinase inhibitors.12 Initial compounds often lacked selectivity over homologous enzymes such as Bcr-Abl, but a number of recent papers have described more selective inhibitors which all bind to the DFG-out form of the enzyme.13−15 Gray and co-workers have identified DDR1/2 inhibitors by screening a library of compounds that had previously been designed by mixing and matching motifs from known Type II kinase inhibitors.14 The resulting compounds were reported to be selective with IC50 values versus DDR1 and DDR2 of approximately 100 nM, and experimental binding modes in DDR1 were determined.14,16 Gao et al. have published a series of potent Type II DDR1 inhibitors that are selective over other kinases, including DDR2.13
In this Letter we describe a fragment based approach to the discovery of potent and selective DDR1/2 inhibitors. We first obtained a soakable crystal form of DDR1 suitable for high-throughput crystallography and fragment screening (see Supporting Information). Approximately 1500 compounds from the Astex fragment library were screened against DDR1 using a protein thermal shift assay (TM). Hits from this assay were progressed into X-ray structure determination, and around 50 fragment–protein complexes were determined. Additionally protein–ligand complexes were also determined for 10–15 nonselective kinase inhibitors, including dasatinib. Most fragments bound at the hinge region, but intriguingly, about 10 fragments bound in the back pocket of DDR1, which is only accessible to the inactive DFG-out form of the protein. In our experience of many fragment screens against kinases,17−22 it is somewhat unusual to obtain X-ray structures of fragments binding in this region because only a small proportion of kinases support fragment binding here,23 and we have found such fragments tend to have relatively low solubility.
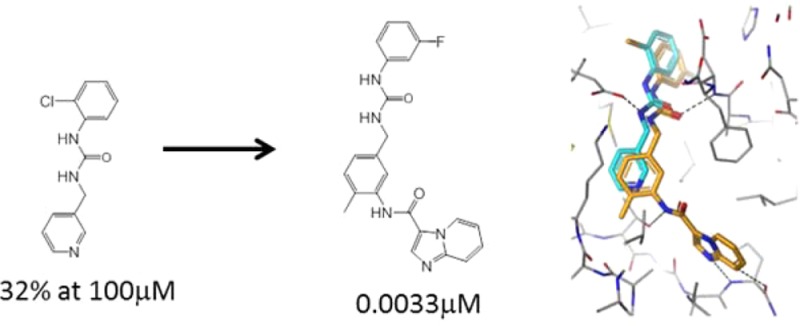
We were particularly interested in fragment 1 (see Figure 1(a) and Figure 2), which places a chlorophenyl in the back pocket region and a pyridyl in the selectivity pocket proximal to the small gatekeeper residue (Thr701 in DDR1/2). The linker region of fragment 1 contains a urea moiety which hydrogen bonds to the side chain of Glu672 and to the backbone N–H of Asp784. The molecule is similar in structure and binding mode to fragments of a number of Type II kinase inhibitors (e.g., sorafenib) but is unusual in containing a benzylic methylene in the linker region (Figure 2). Fragment 1 is larger and more lipophilic than the vast majority of compounds in our current fragment library and is a relatively weak inhibitor (>100 μM) with poor ligand efficiency (LE <0.3).24 However, we believed that fragment 1 could be improved rapidly because (i) it did not optimally fill the selectivity pocket and (ii) it lacked a hinge binding moiety. Additionally we believed that the incorporation of the unusual benzylic methylene could potentially lead to novel and selective DDR inhibitors. The approach we adopted is known as “back-to-front” kinase design, reflecting the fact that design begins in the back pocket of the kinase and works its way to the hinge binding region at the “front” of the ATP binding cleft (Figure 1(a)). This approach has only rarely been attempted,25−28 although the discovery of one of the earliest and most influential DFG-out inhibitors, BIRB796, started from an HTS hit that occupied the back pocket of p38 MAP kinase.27
Figure 1.

Crystal structures of compounds bound to DDR1 (PDB Codes: 5bvk, 5bvw, 5bvn and 5bvo). (a) Fragment 1 in DDR1 with pockets annotated. (b) Superimposition of the DDR1–dasatinib cocomplex and the DDR1–fragment 1 cocomplex. (c) Compound 4 superimposed on the crystallographic positioning of fragment 1. The four hydrogen bonding residues are marked. (d) Compound 9 in DDR1.
Figure 2.
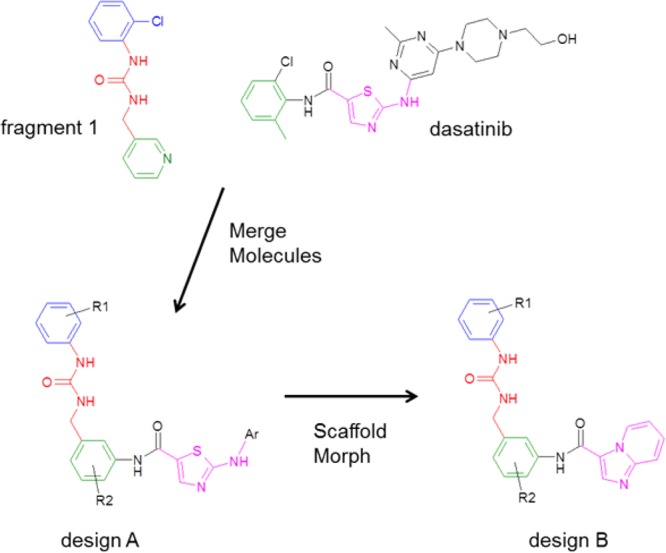
Schematic of the design process. Molecules are colored according to the binding mode in DDR1: blue = back pocket region, red = linker region, green = selectivity pocket, magenta = hinge binding group.
The first stage of the design process was to identify potential hinge binding motifs that could be grafted onto fragment 1 without significantly perturbing its binding mode. Given the large number of internal DDR1 structures and internal/public domain kinase structures, we chose to employ a computational tool, AstexMerge, to suggest potential designs. AstexMerge is an internal implementation of the program, BREED, described by Pierce et al.29 The method starts from a pre-existing web page of proteins in complex with small molecules superimposed in the same frame of reference; this is often the same web page used by medicinal chemists and modelers to visualize all the relevant crystal structures associated with a particular project. The user then selects a starting molecule/fragment, and AstexMerge looks for bonds in all other molecules that superimpose well with the starting molecule. When a suitable hit molecule is located, two merged molecules are displayed, each containing part of the starting fragment and part of the hit molecule. The merged molecules are scored according to the quality of bond and neighboring atom superimposition. The user then visualizes a selection of the best scoring merged molecules. Fragment 1 was used as the starting fragment in AstexMerge, and a number of promising designs were suggested, including design A (Figure 2), based on the superimposition with dasatinib (Figure 1(b)). This design was further modified based on our previous knowledge of FGFR inhibitors,21 suggesting that the thiazole hinge binder of dasatinib could be replaced by an imidazopyridine (design B in Figure 2).
Compounds 2 and 3 were the first compounds that we synthesized in the imidazopyridine series (Table 1). Gratifyingly, addition of the hinge binding element in compound 2 led to a >300-fold increase in affinity for DDR2, and inclusion of the methyl group in 3 gave a further 30-fold improvement in affinity. Limited further SAR exploration led to compound 4, which, after further characterization, became the initial lead molecule in this series. Compound 4 showed promising in vitro PK and in vivo PK in mice (Table 1 and Table S5). The selectivity profile of compound 4 was also encouraging (Table 1 and Table S1) because, in contrast to dasatinib, it showed selectivity over c-src. Figure 1(c) shows the experimental binding mode of 4 in DDR1, superimposed on the starting fragment 1. The binding mode is in agreement with the original design hypothesis, and the imidazopyridine forms the anticipated hydrogen bonds with the hinge.
Table 1. Assaya, Pharmacokineticb and Property Data for Compounds from the Text.
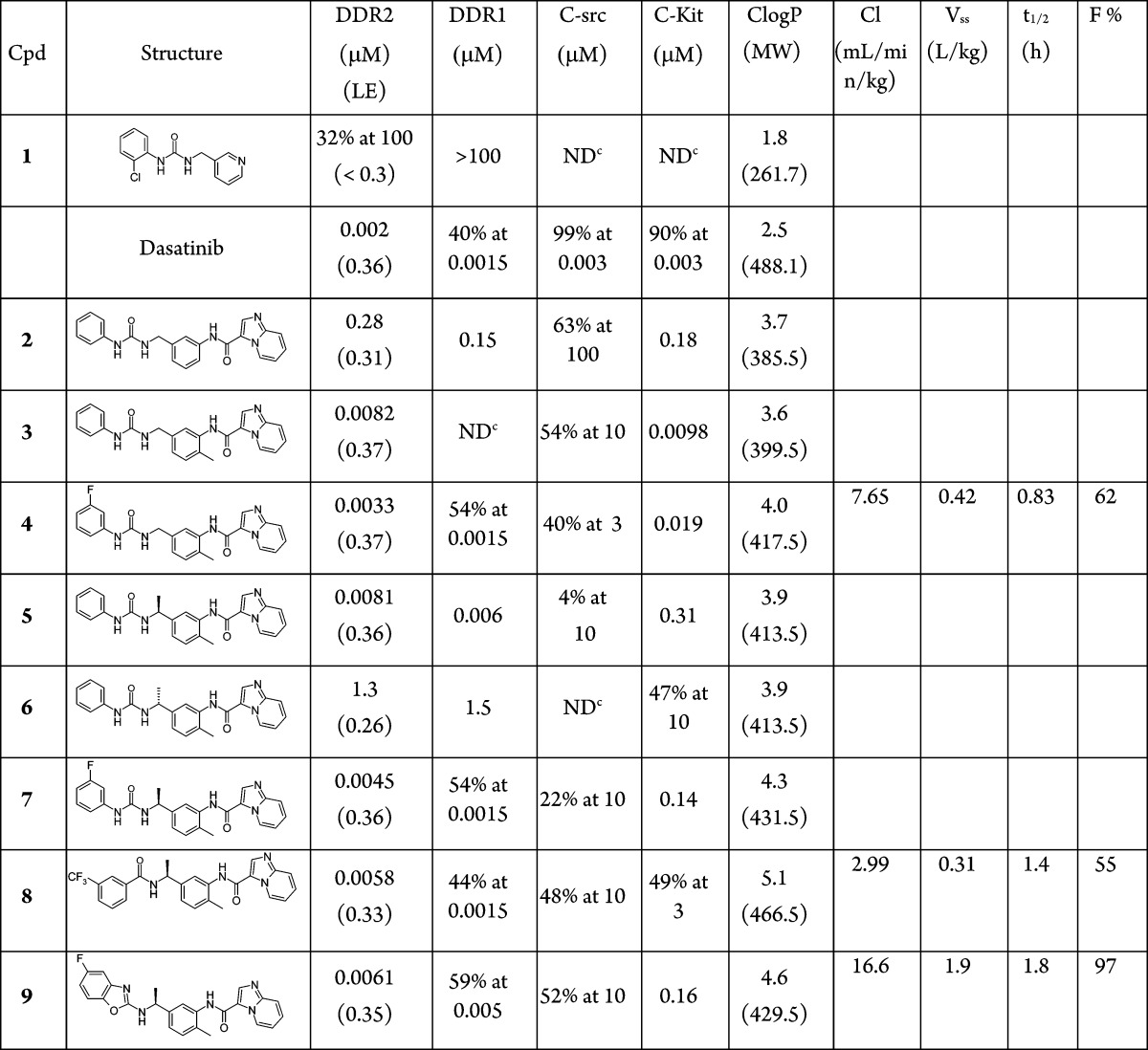
Assay data given in IC50 or percentage inhibition at specified concentration. More details on assays including error estimates are provided in the Supporting Information. Where quoted LE values are given in kcal/mol/(heavy atom).
IV dosing in mouse at 1 mg/kg and oral dosing in mouse at 5 mg/kg. More details on pharmacokinetic data including additional in vitro data are provided in the Supporting Information.
No inhibition value was determined in this case.
Compound 4 showed off-target activity against a number of tyrosine kinases and in particular was a potent c-kit inhibitor (19 nM). Inspection of the crystal structure for compound 4 indicated that there was space to grow off the benzylic methylene. It was hypothesized that this change might improve selectivity because our analysis of kinase literature indicated that branched sp3 centers were poorly precedented in this region for type II kinase inhibitors. The α-methyl compounds, 5, 6 and 7 were synthesized and the assay data (Table 1) showed the S-isomer was tolerated whereas, in agreement with the structure, the R-isomer was considerably less active. The compounds showed improved selectivity profiles (see Table 1 and Table S1).
Further design assessed the scope to replace the urea linker. It has been suggested that 3-trifluorobenzamide is a privileged group for Type II inhibitors,30 and so we incorporated this idea into the branched series resulting in the potent amide, 8 (Table 1). Benzoxazoles have been employed as urea replacements in Type II inhibitors of VEGFR2,31 and so it was also interesting to explore this change within the branched DDR series. Compound 9 showed good activity in the assay and exhibited the expected experimental binding mode in DDR1 (Table 1 and Figure 1(d)). Compounds 8 and 9 show promising pharmacokinetics in mouse (Table 1 and Table S5) and good selectivity (Table 1 and Table S1)
The properties of the lead compound 4 were assessed in cells. Attempts to confirm inhibition of DDR2 phosphorylation (pDDR2) in mutant-DDR2 lung squamous cell cancer cell lines (NCI-H2286 and HCC-366) were unsuccessful because DDR2 expression could not be detected in these cell lines (Figure S2). HEK293 cells were therefore stably transfected with either wild type DDR2 or mutant DDR2 (I638F), leading to overexpression of DDR2 in these cells. Compound 4 significantly reduces basal and collagen I-stimulated DDR2 phosphorylation in both wild type DDR2- and mutant DDR2-expressing HEK293 cells (Figure 3(a)). The multikinase inhibitors, dasatinib and nilotinib, also reduce pDDR2 in this experiment; however, the relatively selective c-src inhibitor, saracatinib, only inhibits pDDR2 at the highest concentration. The inhibition of DDR2 activity conferred by compound 4 was quantified using a MesoScale Discovery (MSD) assay, and an EC50 of 8.7 nM was determined (Figure S1).
Figure 3.
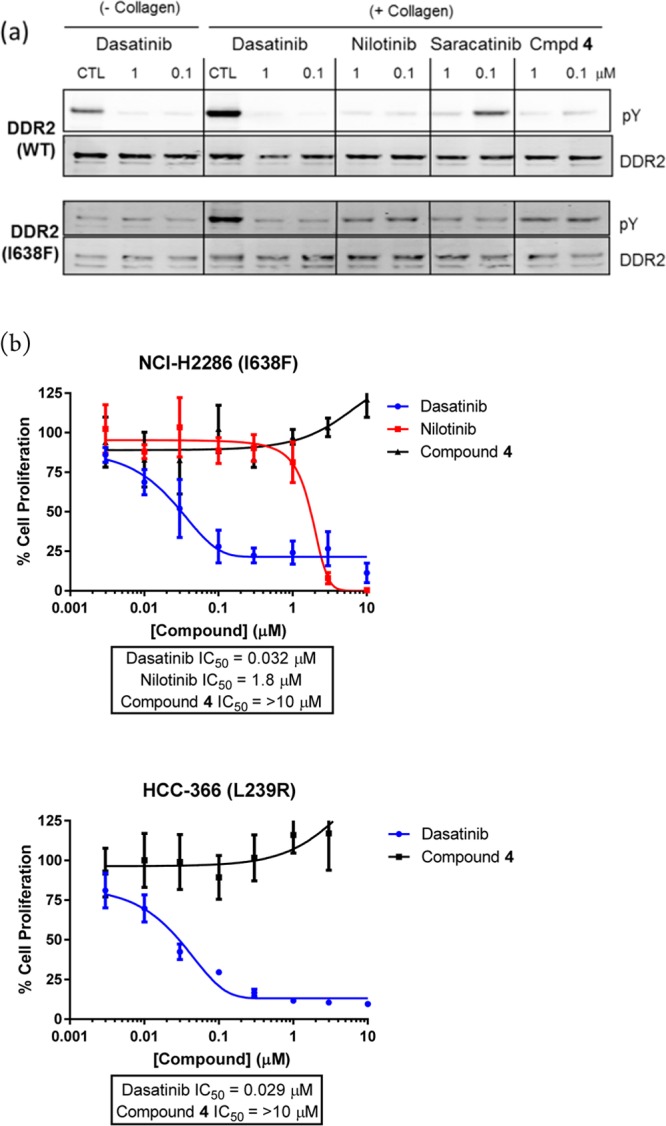
(a) Knockdown of DDR2 phosphorylation (pY) in HEK293 cells stably transfected with wild type (upper blots) or mutant DDR2 (I638F) (lower blots). Knockdown is reported with specified compounds for collagen I stimulated cells (right) or unstimulated cells (left). (b) Effect of compounds on proliferation of two mutant DDR2 cell lines. More experimental details are provided in the Supporting Information.
As previously shown by Hammerman et al.,6 dasatinib and nilotinib inhibit proliferation in DDR2 mutant cell lines (Figure 3(b)). Despite potently inhibiting pDDR2 in cells, compound 4 has no effect on the cell proliferation (Figure 3(b)), thus suggesting that the antiproliferative effects conferred by dasatinib and nilotinib are not attributable to DDR2 inhibition. Taken together with the observation that the DDR2 mutant cell lines NCI-H2286 and HCC-366 lack detectable DDR2 expression or activity, we conclude that the DDR2 mutations in these cell lines are unlikely to be activating. Recent papers in the literature have also questioned whether DDR2 mutants in lung cancer are activating.8−10 We note that DDR1 and/or DDR2 inhibitors may be useful as anti-invasive or antimetastatic agents,13 or indeed that combination with other kinase inhibitors may be beneficial.14 However, the lack of a direct antiproliferative phenotype in mutant-DDR2 squamous cell lines meant that the project was halted in favor of more attractive targets.
In conclusion, we have described novel inhibitors of DDR1 and DDR2 that contain a substituted sp3 center in the linker region of Type II kinase inhibitors. The inhibitors were generated using fragment based drug design starting from fragments binding in the back pocket of the kinase (so-called back-to-front design). Structural biology and computational modeling tools were employed to guide the design of the inhibitors. We have demonstrated the sp3 center in the linker region can be used in conjunction with a variety of linker groups, including ureas, amides and benzoxazoles. The resulting compounds not only are potent and selective but also display promising pharmacokinetic properties. Biological data on our DDR2 inhibitors were not supportive of an antiproliferative role for DDR2 inhibitors in mutant-DDR2 squamous cell lung cancer. However, the compounds should be useful chemical probes for assessing the potential role of DDR1 or DDR2 inhibitors in other therapeutic indications.
Acknowledgments
The authors would like to acknowledge Chris Johnson and David Rees for useful comments on the manuscript, Brent Graham for help with the cell assays, and Torren Peakman and Sharna Rich for help with in vitro pharmacokinetics data.
Glossary
Abbreviations
- DDR
discoidin domain receptor
- pDDR2
phosphorylated DDR2
- LE
ligand efficiency
- MW
molecular weight
- HTS
high throughput screening
Supporting Information Available
Assay conditions and error estimates; more extensive selectivity data; additional data and methods on in vivo and in vitro pharmacokinetics; experimental synthetic procedures and characterization data for novel compounds and intermediates; additional cell data and associated methods; crystallographic details. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00143.
The authors declare no competing financial interest.
Supplementary Material
References
- Shrivastava A.; Radziejewski C.; Campbell E.; Kovac L.; McGlynn M.; Ryan T. E.; Davis S.; Goldfarb M. P.; Glass D. J.; Lemke G.; Yancopoulos G. D. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol. Cell 1997, 1, 25–34. [DOI] [PubMed] [Google Scholar]
- Leitinger B. Transmembrane collagen receptors. Annu. Rev. Cell Dev. Biol. 2011, 27, 265–290. [DOI] [PubMed] [Google Scholar]
- Vogel W. F.; Abdulhussein R.; Ford C. E. Sensing extracellular matrix: an update on discoidin domain receptor function. Cell. Signal. 2006, 18, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Valiathan R. R.; Marco M.; Leitinger B.; Kleer C. G.; Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012, 31, 295–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Corsa C. A.; Ponik S. M.; Prior J. L.; Piwnica-Worms D.; Eliceiri K. W.; Keely P. J.; Longmore G. D. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 2013, 15, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerman P. S.; Sos M. L.; Ramos A. H.; Xu C.; Dutt A.; Zhou W.; Brace L. E.; Woods B. A.; Lin W.; Zhang J.; Deng X.; Lim S. M.; Heynck S.; Peifer M.; Simard J. R.; Lawrence M. S.; Onofrio R. C.; Salvesen H. B.; Seidel D.; Zander T.; Heuckmann J. M.; Soltermann A.; Moch H.; Koker M.; Leenders F.; Gabler F.; Querings S.; Ansen S.; Brambilla E.; Brambilla C.; Lorimier P.; Brustugun O. T.; Helland A.; Petersen I.; Clement J. H.; Groen H.; Timens W.; Sietsma H.; Stoelben E.; Wolf J.; Beer D. G.; Tsao M. S.; Hanna M.; Hatton C.; Eck M. J.; Janne P. A.; Johnson B. E.; Winckler W.; Greulich H.; Bass A. J.; Cho J.; Rauh D.; Gray N. S.; Wong K. K.; Haura E. B.; Thomas R. K.; Meyerson M. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011, 1, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp E. M.; Woods B. A.; Dulak A. M.; Tan L.; Xu C.; Gray N. S.; Bass A. J.; Wong K. K.; Meyerson M.; Hammerman P. S. Acquired resistance to dasatinib in lung cancer cell lines conferred by DDR2 gatekeeper mutation and NF1 loss. Mol. Cancer Ther. 2014, 13, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai L. K.; Payne L. S.; Luczynski M. T.; Chang F.; Xu H.; Clinton R. W.; Paul A.; Esposito E. A.; Gridley S.; Leitinger B.; Naegle K. M.; Huang P. H. Phosphoproteomics of collagen receptor networks reveals SHP-2 phosphorylation downstream of wild-type DDR2 and its lung cancer mutants. Biochem. J. 2013, 454, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y.; Kim J. Y.; Watters J. M.; Fang B.; Kinose F.; Song L.; Koomen J. M.; Teer J. K.; Fisher K.; Chen Y. A.; Rix U.; Haura E. B. Adaptive Responses to Dasatinib-Treated Lung Squamous Cell Cancer Cells Harboring DDR2 Mutations. Cancer Res. 2014, 74, 7217–7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne L. S.; Huang P. H. Discoidin domain receptor 2 signaling networks and therapy in lung cancer. J. Thorac. Oncol. 2014, 9, 900–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J.; Chen P.; Norris D.; Padmanabha R.; Lin J.; Moquin R. V.; Shen Z.; Cook L. S.; Doweyko A. M.; Pitt S.; Pang S.; Shen D. R.; Fang Q.; de Fex H. F.; McIntyre K. W.; Shuster D. J.; Gillooly K. M.; Behnia K.; Schieven G. L.; Wityak J.; Barrish J. C. 2-Aminothiazole as a Novel Kinase Inhibitor Template. Structure–Activity Relationship Studies toward the Discovery of N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1- piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (Dasatinib, BMS-354825) as a Potent pan-Src Kinase Inhibitor. J. Med. Chem. 2006, 49, 6819–6832. [DOI] [PubMed] [Google Scholar]
- Li Y.; Lu X.; Ren X.; Ding K. Small Molecule Discoidin Domain Receptor Kinase Inhibitors and Potential Medical Applications. J. Med. Chem. 2015, 58, 3287–3301. [DOI] [PubMed] [Google Scholar]
- Gao M.; Duan L.; Luo J.; Zhang L.; Lu X.; Zhang Y.; Zhang Z.; Tu Z.; Xu Y.; Ren X.; Ding K. Discovery and optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 2013, 56, 3281–3295. [DOI] [PubMed] [Google Scholar]
- Kim H. G.; Tan L.; Weisberg E. L.; Liu F.; Canning P.; Choi H. G.; Ezell S. A.; Wu H.; Zhao Z.; Wang J.; Mandinova A.; Griffin J. D.; Bullock A. N.; Liu Q.; Lee S. W.; Gray N. S. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem. Biol. 2013, 8, 2145–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richters A.; Nguyen H. D.; Phan T.; Simard J. R.; Grutter C.; Engel J.; Rauh D. Identification of type II and III DDR2 inhibitors. J. Med. Chem. 2014, 57, 4252–4262. [DOI] [PubMed] [Google Scholar]
- Canning P.; Tan L.; Chu K.; Lee S. W.; Gray N. S.; Bullock A. N. Structural mechanisms determining inhibition of the collagen receptor DDR1 by selective and multi-targeted type II kinase inhibitors. J. Mol. Biol. 2014, 426, 2457–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill A. L.; Frederickson M.; Cleasby A.; Woodhead S. J.; Carr M. G.; Woodhead A. J.; Walker M. T.; Congreve M. S.; Devine L. A.; Tisi D.; O’Reilly M.; Seavers L. C.; Davis D. J.; Curry J.; Anthony R.; Padova A.; Murray C. W.; Carr R. A.; Jhoti H. Identification of novel p38alpha MAP kinase inhibitors using fragment-based lead generation. J. Med. Chem. 2005, 48, 414–426. [DOI] [PubMed] [Google Scholar]
- Howard S.; Berdini V.; Boulstridge J. A.; Carr M. G.; Cross D. M.; Curry J.; Devine L. A.; Early T. R.; Fazal L.; Gill A. L.; Heathcote M.; Maman S.; Matthews J. E.; McMenamin R. L.; Navarro E. F.; O’Brien M. A.; O’Reilly M.; Rees D. C.; Reule M.; Tisi D.; Williams G.; Vinkovic M.; Wyatt P. G. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J. Med. Chem. 2009, 52, 379–388. [DOI] [PubMed] [Google Scholar]
- Johnson C. N.; Berdini V.; Beke L.; Bonnet P.; Brehmer D.; Coyle J. E.; Day P. J.; Frederickson M.; Freyne E. J.; Gilissen R. A.; Hamlett C. C.; Howard S.; Meerpoel L.; McMenamin R.; Patel S.; Rees D. C.; Sharff A.; Sommen F.; Wu T.; Linders J. T. Fragment-based discovery of type I inhibitors of maternal embryonic leucine zipper kinase. ACS Med. Chem. Lett. 2015, 6, 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxty G.; Woodhead S. J.; Berdini V.; Davies T. G.; Verdonk M. L.; Wyatt P. G.; Boyle R. G.; Barford D.; Downham R.; Garrett M. D.; Carr R. A. Identification of inhibitors of protein kinase B using fragment-based lead discovery. J. Med. Chem. 2007, 50, 2293–2296. [DOI] [PubMed] [Google Scholar]
- Squires M.; Ward G.; Saxty G.; Berdini V.; Cleasby A.; King P.; Angibaud P.; Perera T.; Fazal L.; Ross D.; Jones C. G.; Madin A.; Benning R. K.; Vickerstaffe E.; O’Brien A.; Frederickson M.; Reader M.; Hamlett C.; Batey M. A.; Rich S.; Carr M.; Miller D.; Feltell R.; Thiru A.; Bethell S.; Devine L. A.; Graham B. L.; Pike A.; Cosme J.; Lewis E. J.; Freyne E.; Lyons J.; Irving J.; Murray C.; Newell D. R.; Thompson N. T. Potent, selective inhibitors of fibroblast growth factor receptor define fibroblast growth factor dependence in preclinical cancer models. Mol.Cancer Ther. 2011, 10, 1542–1552. [DOI] [PubMed] [Google Scholar]
- Wyatt P. G.; Woodhead A. J.; Berdini V.; Boulstridge J. A.; Carr M. G.; Cross D. M.; Davis D. J.; Devine L. A.; Early T. R.; Feltell R. E.; Lewis E. J.; McMenamin R. L.; Navarro E. F.; O’Brien M. A.; O’Reilly M.; Reule M.; Saxty G.; Seavers L. C.; Smith D. M.; Squires M. S.; Trewartha G.; Walker M. T.; Woolford A. J. Identification of N-(4-piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a novel cyclin dependent kinase inhibitor using fragment-based X-ray crystallography and structure based drug design. J. Med. Chem. 2008, 51, 4986–4999. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Brown M. J.; Christopher J. A.; Chung C. W.; Mellor G. W. Selectivity of kinase inhibitor fragments. J. Med. Chem. 2011, 54, 5131–5143. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Baldwin I.; Bamborough P.; Haslam C. G.; Hunjan S. S.; Longstaff T.; Mooney C. J.; Patel S.; Quinn J.; Somers D. O. Kinase array design, back to front: biaryl amides. Bioorg. Med. Chem. Lett. 2008, 18, 5285–5289. [DOI] [PubMed] [Google Scholar]
- Iwata H.; Oki H.; Okada K.; Takagi T.; Tawada M.; Miyazaki Y.; Imamura S.; Hori A.; Lawson J. D.; Hixon M. S.; Kimura H.; Miki H. A Back-to-Front Fragment-Based Drug Design Search Strategy Targeting the DFG-Out Pocket of Protein Tyrosine Kinases. ACS Med. Chem. Lett. 2012, 3, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan J.; Breitfelder S.; Cirillo P.; Gilmore T.; Graham A. G.; Hickey E.; Klaus B.; Madwed J.; Moriak M.; Moss N.; Pargellis C.; Pav S.; Proto A.; Swinamer A.; Tong L.; Torcellini C. Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate. J. Med. Chem. 2002, 45, 2994–3008. [DOI] [PubMed] [Google Scholar]
- Sanphanya K.; Wattanapitayakul S. K.; Phowichit S.; Fokin V. V.; Vajragupta O. Novel VEGFR-2 kinase inhibitors identified by the back-to-front approach. Bioorg. Med. Chem. Lett. 2013, 23, 2962–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce A. C.; Rao G.; Bemis G. W. BREED: Generating novel inhibitors through hybridization of known ligands. Application to CDK2, p38, and HIV protease. J. Med. Chem. 2004, 47, 2768–2775. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Gray N. S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [DOI] [PubMed] [Google Scholar]
- Potashman M. H.; Bready J.; Coxon A.; DeMelfi T. M. Jr.; DiPietro L.; Doerr N.; Elbaum D.; Estrada J.; Gallant P.; Germain J.; Gu Y.; Harmange J. C.; Kaufman S. A.; Kendall R.; Kim J. L.; Kumar G. N.; Long A. M.; Neervannan S.; Patel V. F.; Polverino A.; Rose P.; Plas S.; Whittington D.; Zanon R.; Zhao H. Design, synthesis, and evaluation of orally active benzimidazoles and benzoxazoles as vascular endothelial growth factor-2 receptor tyrosine kinase inhibitors. J. Med. Chem. 2007, 50, 4351–4373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.