

Abstract
Substitution of rigidified A3 adenosine receptor (AR) agonists with a 2-((5-chlorothiophen-2-yl)ethynyl) or a 2-(4-(5-chlorothiophen-2-yl)-1H-1,2,3-triazol-1-yl) group provides prolonged protection in a model of chronic neuropathic pain. These agonists contain a bicyclo[3.1.0]hexane ((N)-methanocarba) ring system in place of ribose, which adopts a receptor-preferred conformation. N6-Small alkyl derivatives were newly optimized for A3AR affinity and the effects of a 1-deaza-adenine modification probed. 1-Deaza-N6-ethyl alkyne 20 (MRS7144, Ki 1.7 nM) and 1-aza N6-propyl alkyne 12 (MRS7154, Ki 1.1 nM) were highly efficacious in vivo. Thus, the presence of N1 is not required for nanomolar binding affinity or potent, long-lasting functional activity. Docking of 1-deaza compounds to a receptor homology model confirmed a similar binding mode as previously reported 1-aza derivatives. This is the first demonstration in nonribose adenosine analogues that the 1-deaza modification can maintain high A3AR affinity, selectivity, and efficacy.
Keywords: G protein-coupled receptor, purines, chronic neuropathic pain, molecular modeling, adenosine receptor, crystallographic structure
Agonists of the A3 adenosine receptor (AR) have been shown to have anticancer and anti-inflammatory properties in vivo, and two nucleosides that are selective for this subtype have progressed to Phase III clinical trials for psoriasis and rheumatoid arthritis.1−3 We recently developed a series of rigidified nucleoside full agonists that are thousands-fold selective for the A3AR and display prolonged protection in models of chronic neuropathic pain.4−6 The rigidity of these analogues, which seems to maintain a preferred conformation at the A3AR, results from a bicyclic substitution of the ribose moiety, e.g., an appropriately positioned bicyclo[3.1.0]hexane ring system, termed North (N)-methanocarba. A combination with a rigid extension at the C2 position of the adenine in the form of a 2-(5-chlorothiophen-2-yl)ethynyl) or a 2-(4-(5-chlorothiophen-2-yl)-1H-1,2,3-triazol-1-yl) group further enhances A3AR selectivity.5,6 This modification suggested structural plasticity of the second transmembrane helix (TM2) of the A3AR to accommodate the highly rigidified analogues.
Previous studies of modified ribonucleosides as AR ligands have demonstrated the importance of the adenine N3 and N7 positions; however, several 1-deaza-adenine analogues 1–3 of commonly used AR agonists were reported to display activity at various AR subtypes but with lower affinity (Chart 1).7−9 Among these three analogues, the average loss of binding affinity at rat (r) ARs upon 1-deaza modification was: A1, 12-fold; A2A, 35-fold; A3, 3.6-fold.8,9 This suggested that the 1-deaza modification could be a means of enhancing A3AR selectivity, possibly with a loss of affinity, but it had not been applied to nucleosides optimized for that AR subtype.
Chart 1. Previous Examples of 1-Deaza Nucleosides as AR Ligands, with Binding Ki Values (nM) at the rA3AR.

We first extended the structure–activity relationship (SAR) of the previous alkynyl and triazolyl series with analogues containing various N6 groups synthesized by reported methods (Schemes S1 and S2, Supporting Information),5,6 and then selected compounds were prepared in a 1-deaza form. The N6 substituent was modified as small alkyl or cycloalkyl groups because the earlier studies emphasized N6-methyl or N6-(3-chlorobenzyl),5,6 i.e., leaving a knowledge gap. The terminal aryl group was 5-chlorothiophen-2-yl, which was previously found to prolong the duration of the protective response in pain models.5,6 The preparation of the 1-deaza-(N)-methanocarba derivatives containing an extended C2 alkynyl group is shown in Scheme 1. A 2′,3′-isopropylidene [3.1.0]bicyclohexanol derivative 4(5,15) was converted to nucleoside precursor 6 through a Mitsunobu reaction with 1-deaza-2-iodo-6-chloropurine 5 (Scheme S3, Supporting Information).16,17 Ester aminolysis followed by nucleophilic substitution of the 6-chloro with an alkyl or cycloalkyl amine gave the N6-substituted intermediates 8. The C2 alkynyl group was installed by a Sonogashira reaction,19 followed by isopropylidene deprotection in aqueous acid to provide 1-deaza nucleosides 19–22. The 1-deaza C2-triazolyl derivative 29 was prepared by a modification of this route to include a click [3 + 2] cycloaddition step as shown in Scheme 2.
Scheme 1. Synthesis of 1-Deaza-(N)-methanocarba-adenosine Alkynyl Analogues.

Ar = 5-chlorothiophen-2-yl. N6 groups of 8, 9: a, Me; b, Et; c, n-Pr; d, c-Pr. Reagents: (i) 1-deaza-6-chloro purine, Ph3P, DIAD, THF, 51%; (ii) 40% MeNH2, MeOH, rt, 72%; (iii) RNH2, DIPEA, i-PrOH, 150 °C, microwave, 74-81%; (iv) Ar–C≡CH, PdCl2(Ph3P)2, CuI, Et3N, DMF, rt, 84-95%; (v) 10% TFA, MeOH, 70 °C, 86-91%.
Scheme 2. Synthesis of a 1-Deaza-(N)-methanocarba-2-triazolyladenosine Analogue 29.

Ar = 5-chlorothiophen-2-yl. Reagents: (i) NaN3, sodium ascorbate, CuSO4.5H2O, l-proline, Na2CO3, tBuOH-H2O, 100 °C, microwave, 1.5 h, 92%; (ii) Ar–C≡CH, sodium ascorbate, CuSO4·5H2O, TBTA, tBuOH-H2O, rt, 89%; (iii) 10%TFA-MeOH, 70 °C, 78%.
The nucleosides were assayed by radioligand binding at three human (h) AR subtypes by standard methods (Table 1).5 Only two compounds, 12 and 20, were evaluated and found to be inactive at the hA2BAR; it was shown previously that (N)-methanocarba adenosine derivatives have greatly reduced hA2BAR affinity.5 Reference alkyne 10 and triazoles 23, 24, 27, and 28 were included for comparison.5,6
Table 1. Structures and Binding Affinitiesa of A3AR Agonists, Including Reference Compounds 10, 23, 24, 27, and 28.
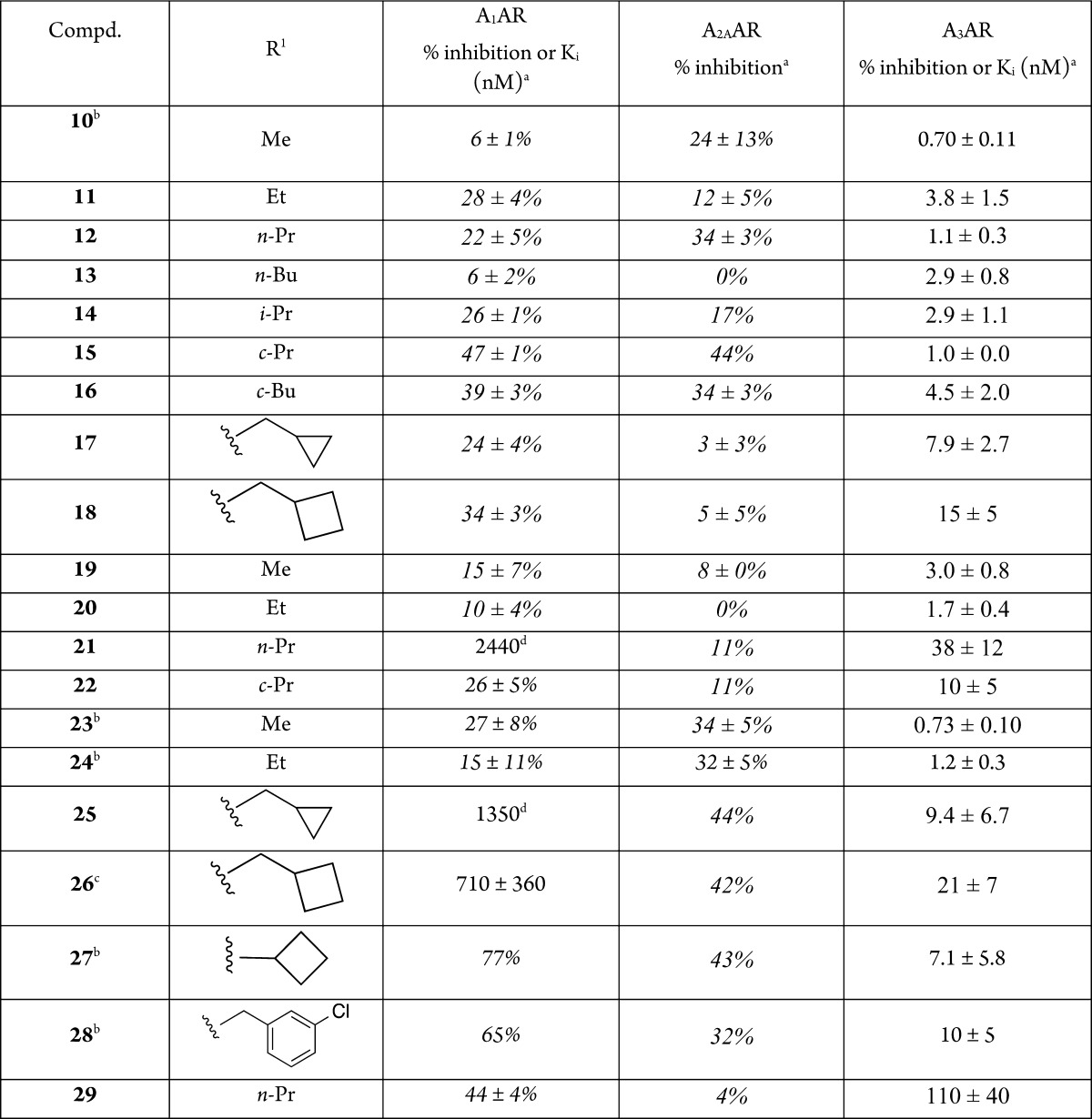
Binding in membranes of CHO or HEK293 (A2A only) cells stably expressing one of three hAR subtypes. The binding affinity for hA1, A2A, and A3ARs was expressed as Ki values using agonists [3H]N6-R-phenylisopropyladenosine 41, [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine 42, or [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide 43, respectively. A percent in italics refers to inhibition of binding at 10 μM. Nonspecific binding was determined using adenosine 5′-N-ethyluronamide 44 (10 μM). Values are expressed as the mean ± SEM (n = 3, unless noted). Ki values were calculated as reported.14 Compounds 12 and 20 were shown to be inactive at the hA2BAR (Supporting Information).
Low aqueous solubility observed for 26.
N = 1.
We first optimized affinity in the 1-aza series and then proceeded to introduce the 1-deaza modification. Except for 21, the nucleoside alkyne derivatives only weakly inhibited binding (<50% at 10 μM) at the hA1AR and hA2AAR, and many bound at the hA3AR with nearly nanomolar affinity. N6-Cyclopropyl derivative 15 displayed close to 50% inhibition at the hA1AR and hA2AAR. In the 1-aza ethynyl series 10–18, homologation to N6-ethyl 11 diminished A3AR affinity, but the N6-propyl analogue 12 was more potent with a Ki value of 1.1 nM at hA3AR. The effects on affinity of branching of the N6 group at the α-carbon were mixed (cf. 15, 16), and a cycloalkyl group on the β-carbon (17, 18) lowered the hA3AR affinity. In the 1-deaza ethynyl series 19–22, homologation to N6-ethyl 20 increased A3AR affinity to a Ki value of 1.7 nM, but the N6-propyl analogue 21 was less selective and 23-fold less potent than 20. Direct comparison of N6-ethyl analogues 11 and 20 indicated slightly higher affinity with a 1-deaza modification. Compounds 19 and 20 were completely inactive in binding to the A1AR and A2AAR. In the 1-aza triazolyl series 23–28, homologation beyond N6-ethyl diminished A3AR affinity and somewhat increased the micromolar affinity at A1AR. Thus, the N6-methyl triazole analogue 23 was favored in this in vitro A3AR assay. Like the corresponding alkyne 21, the 1-deaza N6-propyl triazolyl analogue 29 displayed only moderate A3AR affinity. Analogues 21 and 25–28 displayed significant micromolar A1AR affinity; 26 displayed an A3AR Ki value of 21 nM (only 33-fold A3AR-selective). The A3AR selectivities of the highest affinity nucleosides in the present study are much greater than 2000-fold.
Selected alkyne derivatives were assayed in binding to the mouse (m) and canine (c) A3ARs in membranes of A3AR-expressing HEK293 cells.5 The affinities (nM; m, c; mean ± SEM, n = 3) were: 10, (36 ± 5,5 8.5 ± 0.7); 11, (27 ± 2, 1.5 ± 0.2); 12, (6.8 ± 0.3, 5.8 ± 0.2); 19 (31 ± 2, 75 ± 7); 20 (16 ± 3, 49 ± 4). Thus, 12 was consistently potent in binding at h and mA3ARs. Off-target activity at various receptors (Psychoactive Drug Screening Program,20Supporting Information) indicated only an occasional interaction in the micromolar range. Analogue 12 showed none, and 20 showed only one such interaction (Ki at 5HT2BR = 2.5 μM).
The most potent 1-deaza compound 20 was shown to be a full agonist with EC50 of 1.5 ± 0.3 nM (cf. 38 ± 16 nM for 44) in hA3AR-induced inhibition of the production of cyclic AMP in membranes of hA3AR-expressing CHO cells (Supporting Information).5
Potent in vivo efficacy of the A3AR-selective nucleosides was demonstrated in a mouse chronic constriction injury (CCI) model of neuropathic pain,10 as described in our previous studies.5,6 The compounds were administered by oral gavage at the point of peak pain (day 7) to assess bioavailability and duration of action (Table 2). Unlike many of the compounds we reported earlier that lost most of the protection by 3 h post-administration, in this phenotypic screen most of the compounds examined maintained at least 80% of full protection at the 3 h time-point. Homologation of the N6 group to ethyl in 11 in the alkynyl 1-aza series prolonged the duration of action in vivo with only a small increase in the ED50 value to 0.43 mg/kg (Figure S1, Supporting Information). By comparison, the reference agonist 10 displayed an ED50 value of 0.55 mg/kg.4 For comparison with an established treatment of neuropathic pain, the ED50 of gabapentin (i.p.) at maximal reversal (1 h) in the CCI model was 140 μmol/kg,18 i.e., much less potent than 10 and 11 (footnote b, Table 1). N6-Propyl analogue 12 also displayed a long duration of action in vivo. Other small N6 groups in 13–15 resulted in a reduction of peak protective effect (Figure 1). However, the N6-cyclobutyl analogue 16 displayed 95% protection at 3 h. Comparable high efficacies were observed in the 1-deaza series, indicating that the lack of N1 does not detract from in vivo activity. The most potent 1-deaza analogue in A3AR binding 20 achieved 93% and 84% reduction of chronic neuropathic pain at 1 and 3 h, respectively. The full time course for the in vivo action of 1-deaza N6 ethyl analogue 20 indicated a very long duration of action of at least 5 h (Figure 1). The cLog P values were in a favorable range for drug-like molecules, e.g., 3.49 and 2.71 for N6-ethyl analogues 20 and 11, respectively (Supporting Information).11
Table 2. Activity of Orally Administered AR Agonists (3 μmol/kg) in CCI Model of Neuropathic Pain in the Mousea.
| compd | max. effect Emax (% ± SEM)a | effect at 3 h (% ± SEM) |
|---|---|---|
| 10b | 93 ± 5 | 46 ± 9 |
| 11b | 98 ± 2 | 87 ± 6 |
| 12 | 95 ± 2 | 93 ± 4 |
| 13 | 68 ± 11 | 24 ± 6 |
| 14 | 88 ± 6 | 39 ± 9 |
| 15 | 76 ± 1 | 36 ± 7 |
| 16 | 97 ± 3 | 95 ± 5 |
| 17c | 94 ± 11 | 85 ± 18 |
| 18 | 98 ± 2 | 90 ± 2 |
| 19 | 91 ± 6 | 80 ± 8 |
| 20 | 93 ± 4 | 84 ± 7 |
| 27d | 91 ± 6 | 85 ± 9 |
| 28d | 95 ± 3 | 91 ± 5 |
Effect shown for ipsilateral hind paw; there is no effect on the contralateral side. Time of peak protection (corresponding to Emax) was 1 h for each compound in Table 2. n = 4, unless noted.
ED50 values at peak effect were 0.34 mg/kg (0.73 μmol/kg; n = 5)4 for 10 and 0.43 mg/kg (0.9 μmol/kg; n = 5) for 11 (Supporting Information).
n = 3.
Figure 1.

Time course of protection hind paw mechanoallodynia of the sciatic nerve in the CCI mouse model (p.o. administration on day 7, 3 μmol/kg, p.o.; n = 4). Data are the mean ± SEM analyzed by two-tailed, two-way ANOVA with Bonferroni comparisons: *P < 0.05 vs D0; †P < 0.05 vs D7. Additional plots are shown in the Supporting Information.
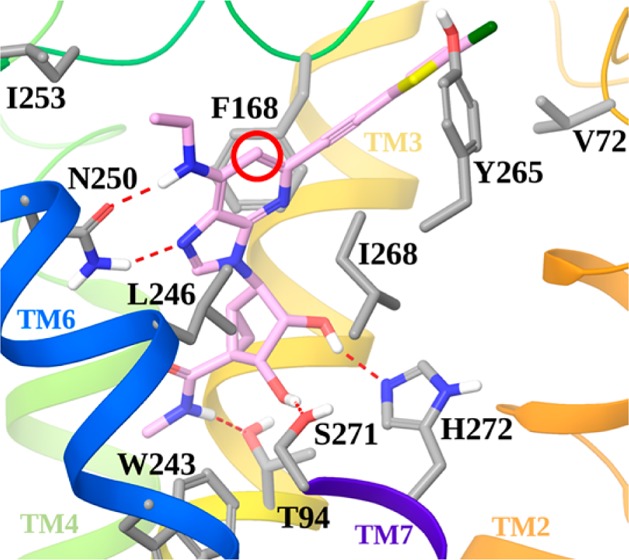
The observation that nanomolar A3AR affinity could be maintained in the 1-deaza series was explored structurally through molecular modeling using a hybrid homology model5,6 of the receptor (methodological details have been previously reported).12 Figure 2 shows the docking pose of compound 20 at the hA3AR model.
Figure 2.
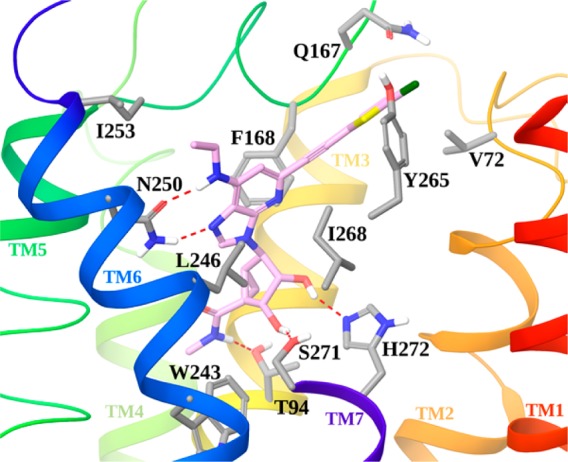
Docking pose of compound 20 (pink carbon sticks) at the hA3AR model. Side chains of residues important for ligand recognition are shown in sticks (gray carbons), and H-bonding interactions are indicated by red, dashed lines. Nonpolar hydrogen atoms are not displayed. The view of TM7 is partially omitted.
As with the 1-aza analogues,5,6 conserved H-bonds for adenosine derivatives were maintained between the 3′- and 2′-hydroxyl groups of docked 20 and Ser271 (7.42) and His272 (7.43), respectively. As expected, the 5′-N-methyluronamide of 20 formed a H-bond with the side chain of Thr94 (3.36). The necessity of having an N7 reflected its association as H-bond acceptor with Asn250 (6.55); the same residue accepts a hydrogen bond from the 6-amino group. As with the 1-aza analogues, the adenine ring formed a π–π stacking with Phe168 (EL2) and strong hydrophobic interactions with Leu246 (6.51) and Ile268 (7.39). The C2 group of 20 was accommodated in an exofacial interface region generated by the outward movement of TM2 in the hybrid A3AR model, as previously proposed for derivatives bearing rigid and extended C2 substituents.12,13 Thus, the major conserved recognition points for A3AR agonists were preserved in the 1-deaza analogues, and as expected, the N1 of adenosine is not required for binding. Several different N6 substitutions can be tolerated in this series, slightly modulating the affinity depending on their accommodation in a region delimited by TM6 and EL2 and exposed toward the extracellular environment. Consistent with our previous report,6 there was a correspondence in A3AR affinity between 1-aza alkynes and 1-aza triazoles. We have no structural explanation for the lack of correlation of A3AR affinity between 1-aza and 1-deaza variants of the alkynes containing N6-Pr or c-Pr substituents.
In conclusion, we have found that the 1-deaza modification may promote affinity and selectivity in (N)-methanocarba nucleosides that are optimized for activation of the A3AR subtype, but not consistently in all cases. A3AR docking suggests no major difference in the binding mode of the 1-aza and 1-deaza nucleosides. The preferred N6 substituents, i.e., small alkyl groups, provided high A3AR affinity and selectivity and maintained the in vivo efficacy. 1-Deaza N6-ethyl 20 and 1-aza N6-propyl 12 analogues were particularly potent in vitro and in vivo and displayed a long duration of action in reducing chronic neuropathic pain.
Glossary
ABBREVIATIONS
- AR
adenosine receptor
- CHO
Chinese hamster ovary
- EL
extracellular loop
- GPCR
G protein-coupled receptor
- HEK
human embryonic kidney
- TM
transmembrane helix
Supporting Information Available
Synthetic procedures, physicochemical properties, mass spectra, NMR and mass spectra, HPLC, biological assay procedures and results, and off-target activity. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00150.
Author Contributions
All authors contributed to this manuscript and have given approval to its final version.
We thank the NIH Intramural Research Program (NIDDK); National Cancer Institute (R01CA169519); and National Heart Lung Institute (R01HL077707) for support and John Lloyd and Noel Whittaker (NIDDK) for mass spectral determinations.
The authors declare no competing financial interest.
Supplementary Material
References
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Linden J.; Müller C. Nomenclature and classification of adenosine receptors – An update. Pharmacol. Rev. 2011, 63, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman P.; Bar-Yehuda S.; Liang B. T.; Jacobson K. A. Pharmacological and therapeutic effects of A3 adenosine receptor (A3AR) agonists. Drug Discovery Today 2012, 17, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borea P. A.; Varani K.; Vincenzi F.; Baraldi P. G.; Tabrizi M. A.; Merighi S.; Gessi S. The A3 adenosine receptor: History and perspectives. Pharmacol. Rev. 2015, 67, 74–102. [DOI] [PubMed] [Google Scholar]
- Little J. W.; Ford A.; Symons-Liguori A. M.; Chen Z.; Janes K.; Doyle T.; Xie J.; Luongo L.; Tosh D. K.; Maione S.; Bannister K.; Dickenson A.; Vanderah T. W.; Porreca F.; Jacobson K. A.; Salvemini D. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 2015, 138, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Finley A.; Paoletta S.; Moss S. M.; Gao Z. G.; Gizewski E.; Auchampach J.; Salvemini D.; Jacobson K. A. In vivo phenotypic screening for treating chronic neuropathic pain: Modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J. Med. Chem. 2014, 57, 9901–9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Paoletta S.; Chen Z.; Crane S.; Lloyd J.; Gao Z. G.; Gizewski E.; Auchampach J. A.; Salvemini D.; Jacobson K. A. Structure-based design, synthesis by click chemistry and in vivo activity of highly selective A3 adenosine receptor agonists. MedChemComm 2015, 6, 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristalli G.; Grifantini M.; Vittori S. Adenosine and 2-chloroadenosine de-aza analogues as adenosine receptor agonists. Nucleosides Nucleotides 1985, 4, 625–639. [Google Scholar]
- van Galen P. J. M.; van Bergen A. H.; Gallo-Rodriguez C.; Melman N.; Olah M. E.; IJzerman A. P.; Stiles G. L.; Jacobson K. A. A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol. Pharmacol. 1994, 45, 1101–1111. [PMC free article] [PubMed] [Google Scholar]
- Siddiqi S. M.; Jacobson K. A.; Esker J. L.; Melman N.; Tiwari K. N.; Secrist J. A.; Schneller S. W.; Cristalli G.; Johnson C. R.; IJzerman A. P. Search for new purine- and ribose-modified adenosine analogues as selective agonists and antagonists at adenosine receptors. J. Med. Chem. 1995, 38, 1174–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett G. J.; Xie Y. K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [DOI] [PubMed] [Google Scholar]
- Di L.; Rong H.; Feng B. Demystifying brain penetration in central nervous system drug discovery. Miniperspective. J. Med. Chem. 2013, 56, 2–12. [DOI] [PubMed] [Google Scholar]
- Paoletta S.; Tosh D. K.; Finley A.; Gizewski E. T.; Moss S. M.; Gao Z. G.; Auchampach J. A.; Salvemini D.; Jacobson K. A. Rational design of sulfonated A3 adenosine receptor-selective nucleosides as pharmacological tools to study chronic neuropathic pain. J. Med. Chem. 2013, 56, 5949–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Deflorian F.; Phan K.; Gao Z. G.; Wan T. C.; Gizewski E.; Auchampach J. A.; Jacobson K. A. Structure-guided design of A3 adenosine receptor-selective nucleosides: combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J. Med. Chem. 2012, 55, 4847–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y. C.; Prusoff W. H. Relationship between inhibition constant (K1) and concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic-reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Choi W. J.; Moon H. R.; Kim H. O.; Yoo B. N.; Lee J. A.; Shin D. H.; Jeong L. S. Preparative and stereoselective synthesis of the versatile intermediate for carbocyclic nucleosides: Effects of the bulky protecting groups to enforce facial selectivity. J. Org. Chem. 2004, 69, 2634–2636. [DOI] [PubMed] [Google Scholar]
- Kumara Swamy K. C.; Bhuvan Kumar N. N.; Balaraman E.; Pavan Kumar K. V. P. Mitsunobu and related reactions: Advances and applications. Chem. Rev. 2009, 109, 2551–2651. [DOI] [PubMed] [Google Scholar]
- Yang M.; Zhou J.; Schneller S. W. The Mitsunobu reaction in preparing 3-deazapurine carbocyclic nucleosides. Tetrahedron 2006, 62, 1295–1300. [Google Scholar]
- Chen Z.; Janes K.; Chen C.; Doyle T.; Tosh D. K.; Jacobson K. A.; Salvemini D. Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 2012, 26, 1855–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchilla R.; Nájera C. Recent advances in Sonogashira reactions. Chem. Soc. Rev. 2011, 40, 5084–5121. [DOI] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X. P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.