

Abstract
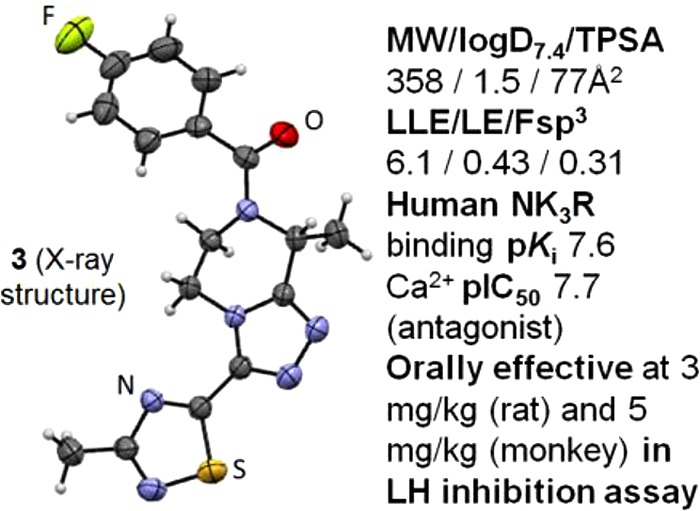
Further lead optimization on N-acyl-triazolopiperazine antagonists to the neurokinin-3 receptor (NK3R) based on the concurrent improvement in bioactivity and ligand lipophilic efficiency (LLE) is reported. Overall, compound 3 (LLE > 6) emerged as the most efficacious in castrated rat and monkey to lower plasma LH, and it displayed the best off-target safety profile that led to its clinical candidate nomination for the treatment of sex-hormone disorders.
Keywords: NK3 antagonist, triazolopiperazine, neurokinin B, LH, FSH, GnRH
The neurokinin-3 receptor (NK3R) is a class A GPCR with neurokinin B (NKB) as its endogenous agonist. We present here the sequel on the lead optimization of N-acyl-triazolopiperazine NK3R antagonists (1 and 2, Figure 1).1
Figure 1.
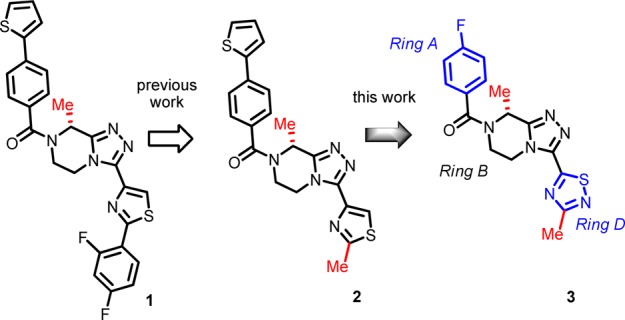
Lead progression: iv POC (1), oral POC (2), and clinical candidate (3). (The “magic methyl” groups are shown in red.)
NK3R antagonists were speculated as therapeutically relevant for CNS dysfunctions, e.g., schizophrenia, predicated on the “hyperdopaminergic hypothesis”, which repeatedly met with clinical failures in over a decade of efforts.2 Meanwhile, emerging biology has unambiguously established the role of NK3R/NKB signaling in reproductive neuroendocrinology. Importantly, recent studies have revealed NK3R as a key regulatory component of the hypothalamic–pituitary–gonadal (HPG) axis wherein its tonic activation positively regulates the gonadotropin-releasing hormone (GnRH) pulse frequency.3 In turn, the GnRH pulse frequency is known to differentially control the circulating levels of luteinizing hormone (LH) versus follicle-stimulating hormone (FSH). Thus, high frequency pulses stimulate LH release, whereas low frequency pulses favor FSH induction.4 These gonadotropins ultimately act on the ovary and testis to promote production of gametes and sex-hormone release. In 2011, it was reported that patients with a loss of function mutation in NK3R display a phenotype of normosomic congenital hypogonadotropic hypogonadism, low plasma LH, and attendant low LH/FSH ratios that could be restored through exogenous administration of GnRH.5 We have demonstrated elsewhere6 that the foregoing NK3R antagonists slow the LH pulse and decrease circulating LH levels without affecting FSH, consistent with the literature reports.4 As such, these antagonists are subtle modulators of gonadotropin secretion unlike GnRH ligands that abrogate both LH and FSH with the consequent decline in plasma estrogen to castration levels thereby triggering menopausal-like adverse events such as bone mineral density loss and incidences of hot flashes.7 Hence, NK3R antagonists offer a potentially safer therapeutic approach due to a decreased rather than abrogated GnRH pulse frequency. Collectively, these findings offer a strong rationale for repositioning NK3R antagonists to address sex-hormones disorders such as polycystic ovary syndrome (PCOS) and uterine fibroids (UF), among others.8
The synthetic approach to the analogues herein was previously described.1 With minor modifications, Scheme 1 was used for the GMP scale-up of 3 in overall 42% yield (2.7 kg) with 99.3% purity and >99.9% enantiomeric excess.
Scheme 1.

Reagents and conditions: (a) Et3OBF4, Na2CO3, CH2Cl2, 45 min, 68%; (b) MeOH, 70 °C, 8 h, 80%; (c) TFA, 2 h, >99% conversion; (d) 4-fluorobenzoyl chloride, NaHCO3, 15 min, 97%; (e) recrystallization (EtOH/H2O), 97% (DMB = 2,4-dimethoxybenzyl).
The in vitro bioactivity structure–activity relationship (SAR) was established through radioligand binding (pKi) and aequorin functional assays (pIC50) data from recombinant human NK3R in CHO cells. The lead optimization strategy1 of combined improvement in both bioactivity and ligand lipophilic efficiency (LLE = pKi – logD7.4)9 was maintained. An initial emphasis was placed on LLE as a predictive marker of improved safety profiles.10 Other efficiency metrics such as LE11 and Fsp3 12 (Table 1) were also tracked, though not as a primary focus. The previously discovered “magic methyl” groups in Rings B and D (Figure 1),1 so-called due to their significant impact in improving potency and LLE, remained crucial and rendered feasible improvements in Fsp3 and LE as well (see below).
Table 1. Human NK3R In Vitro Bioactivity, LogD7.4, Ligand Efficiency Metrics,10−12 and Off-Target Safety SAR.
N = 3, %RSD ≤ 5.
CYP 3A4, 2D6, 2C9, 2C19, and 1A2, respectively (N = 2, <10% variability).
N = 3, coefficient of variation < 6%.
To recall, replacing 2-methylthiazole (2) at Ring D with 3-methyl-1,2,4-thiadiazole (8) was reported earlier to markedly improve bioactivity and LLE (Table 1).1 Moreover, 1,2,4-thiadiazole is regarded as a means of circumventing bioactivation liabilities potentially relevant to the thiazole ring.13 These considerations overall prompted us to retain the 1,2,4-thiadiazole, but to modify the 4-(thiophen-2-yl)phenyl in 8 to the 4-fluorophenyl Ring A (i.e., 3) present in the earlier lead structures.1 Progression from 8 to 3 helped reduce lipophilicity (ΔlogD7.4 = −1.5), which not surprisingly right-shifted bioactivity by nearly one log. However, in evaluating 3 against 8, the improved LLE and absence of the thiophene ring (a potential structural safety alert)14 was considered of greater importance due to the reduced toxicological risk. In addition, 3 was nearly equipotent to the oral POC lead 2, while >1-log superior in LLE. Narrowing our focus on the thiadiazole Ring D and related variants, we observed the following descending trend in bioactivity (Table 1): 1,2,4-thiadiazole (3) > 1,2,4-oxadiazole (10) ≫ 1,3,4-thiadiazole (9). As with the Ring A cases, increased lipophilicity in Ring D helped improve bioactivity albeit offset by a loss in LLE, e.g., 11 vs 10 (Table 1). The impact of Rings B and D magic methyl groups on SAR trends was quite pronounced, as expected based on previous results,1 since the corresponding des-Me analogues of 3, whether at Ring B or D (12 and 13, respectively), were decidedly inferior in both bioactivity and LLE (Table 1). Gem dimethyl Ring B substitution substantially eroded the bioactivity (14 vs 3) in keeping with the unfavorable impact of (S)-Me at this Ring B position (data not shown).1 Once again, improved bioactivity followed increased lipophilicity whether at Ring A (15) or at Ring D (16) positions, but this gain was negated by a deteriorated LLE against 3. Conversely, replacing 4-fluorophenyl with phenyl at Ring A, i.e., 17, helped diminish lipophilicity, but it also deteriorated bioactivity. Interestingly, a hydroxyethyl substitution at Ring B (18) afforded an alternative means of reducing lipophilicity (ΔlogD7.4 = −0.5 vs 3) with minimal impact on bioactivity, thus resulting in a 0.4-log superior LLE vs 3. Despite this, 18 proved inferior to 3 due to Pgp efflux that in turn markedly diminished its brain exposure level (Table 2 and discussion further below).
Table 2. Permeability, Plasma and Brain Fraction Unbound, Brain Exposure, and PK Dataa.
| Caco-2 Papp (nm/sec) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cpd | AB | BA | ER | species | plasma fu | brain fu | Cplasma,u (nM) | Cbrain,u (nM) | (B/P)u | iv ClT (min/mL/kg) | iv Vss (L/kg) | iv T1/2 (min) | %F |
| 2b | 339 | 224 | 0.7 | rat | 0.055 | 0.028 | 54.4 | 7.79 | 0.14 | 7.4 | 1.4 | 126 | 119 |
| 2c | − | − | − | monkey | 0.043 | − | 17.9 | − | − | 16.5 | 1.52 | 210 | 12 |
| 3b | 487 | 465 | 1.0 | rat | 0.639 | 0.525 | 1507 | 343 | 0.23 | 1.5 | 0.60 | 279 | 62.5 |
| 3c | − | − | − | monkey | 0.532 | − | 5040 | − | − | 3.17 | 1.15 | 324 | 107 |
| 8d | 360 | 221 | 0.6 | rat | 0.065 | 0.031 | 58.6 | 39.3 | 0.67 | 7.3 | 2.8 | 267 | − |
| 12d | 477 | 528 | 1.1 | rat | 0.674 | 0.436 | 1767 | 212 | 0.12 | 1.94 | 0.65 | 239 | 126 |
| 15d | 467 | 345 | 0.7 | rat | 0.408 | − | − | − | − | 2.2 | 0.91 | 294 | 73.6 |
| 16d | 419 | 368 | 0.9 | rat | 0.291 | 0.109 | 348.0 | 85.4 | 0.25 | 1.07 | 2.25 | 1479 | 98.2 |
| 17d | 437 | 519 | 1.2 | rat | 0.592 | 0.604 | 406.7 | 68.6 | 0.17 | 13.7 | 0.68 | 35 | 55 |
| 18d | 91 | 345 | 3.8 | rat | 0.662 | 0.995 | 897.4 | 45.6 | 0.05 | 5.07 | 1.59 | 215 | 46.6 |
PK doses: iv, 1 mg/kg (rat), 10 mg/kg (monkey); oral, 3 mg/kg (rat), 5 mg/kg (monkey). Brain exposure dose: 1 mg/kg. Mean values for N = 3–4 rats, or 4 monkeys, per group. All rat data at 60 min: (B/P)u = Cbrain,u/Cplasma, u with Cbrain,u = Cbrain, total × bfu and Cplasma,u = Cplasma,total × fu. Monkey Cplasma,u data at 90 min (oral).
PK formulation: HPβCD.
PK formulation: iv HPβCD; oral 0.5% MC/water.
PK formulation: 1% DMSO, HPβCD in 0.9% NaCl.
The hERG SAR herein (Table 1) was governed by the interplay between lipophilicity and the hydrogen-bond acceptor (HBA) count in the heteroaryl Ring D, as previously reported.1 For instance, in progressing from 8 to 3 (ΔlogD7.4 = 1.5), the hERG IC50 was improved by over 12-fold. However, the poor hERG IC50 = 1.6 μM in 11 (logD7.4 = 1.7) was in keeping with the increased HBA count in the Ring D oxadiazole (N + N + O) in stark contrast to the thiadiazole (N + N) Ring D variations (3 and 15–18), all of which displayed a superior hERG, IC50 ≥ 39 μM (logD7.4 = 1.3–2.4). Interestingly, the Ring B hydroxyl group in 18 did not adversely impact hERG (IC50 = 50 μM) suggesting that the HBA effect on hERG SAR is primarily a Ring D related effect. Finally, the Ring B magic methyl also reduced hERG efficacy, i.e., 3 (IC50 > 100 μM) vs 12 (IC50 = 50 μM). Compound 3 was the best overall in the hERG and CYP safety profile evaluation.
Based on the free drug hypothesis, the unbound fraction rather than total drug is relevant for PKPD analysis.15 The NK3R is mainly expressed on KNDy neurons in the ARC region of the hypothalamus3 that is part of the circumventricular organs lacking blood–brain barrier and are therefore exposed to blood solutes.16 As such, both the unbound plasma (fu) and the unbound brain levels (bfu) must be considered here (Table 2). While lipophilicity alone does not correlate well to albumin binding, this trend is often apparent in a congeneric series.17 Hence, a compound with balanced lipophilicity such as 3 (logD7.4 = 1.5) displayed high fu and bfu levels (>50%) in contrast to the more lipophilic congeners, e.g., 16 (Table 2). It is noteworthy that despite an increase in unbound plasma concentration, the systemic clearance levels (CLT) remained low (e.g., 3, Table 2). The comparatively lower CLT in para substituted phenyl Ring A (3, 15) against the unsubstituted congener 17 is likely due to the metabolic blocking effect. All analogues except 18 displayed high Caco-2 permeability with no evidence of appreciable Pgp efflux (ER = 0.6–1.2), consistent with the high oral availability (%F) and brain-to-plasma ratios observed. The so-called Pgp rule-of-4 suggests that increasing the number of HBA atoms to (N + O) ≥ 8 tends to confer an increasing likelihood of Pgp efflux.18 This is in keeping with the Pgp efflux in 18 (ER = 3.8) given its HBA atom count (N + O = 8). As with 2,1 a complete oral absorption (%F > 100) was also observed in rat with compound 12 and in monkey with 3. This phenomenon is well-known and various underlying causes have been reported.19 No drug accumulation was observed in 5-day once-daily oral dosing studies in rats (3 and 12) or monkeys (3), despite administration of elevated doses (e.g., up to 1 g/kg in rats with 12), in step with the relatively short half-life values and the previous related observations with 2.1 Moreover, no adverse hepatotoxicity (AST, ALT, and bilirubin levels normal) was detected in these subchronic studies. Furthermore, 3 displayed the highest bfu = 0.525 and brain unbound concentration (Cbrain,u = 343 nM) herein (Table 2). In contrast, 18 although nearly completely unbound in the brain displayed a comparatively low Cbrain,u = 45.6 nM consistent with its elevated Pgp efflux ratio.
The key PKPD parameters for interpreting the LH inhibition data are the unbound plasma and brain levels normalized with respect to the bioactivity, i.e., Cplasma,u/Ki and Cbrain u/Ki (Table 3).1 The plasma and brain levels were determined at the Tmax for the minimum effective dose (MED). As noted before for 2 and 8,1 a statistically significant effect was attained at Cplasma,u/Ki ≥ 7.6 and Cbrain,u/Ki > 1 in rat oral LH inhibition studies. This was also the case here, i.e., for analogues 3, 12, and 16–18, with MED values ranging from 3 mg/kg (3) to 30 mg/kg (12 and 17). For example, in rats, 3 was 20-fold more efficacious in vivo against the initial POC lead 2 despite being 3-fold right-shifted in Ki. This ameliorated efficacy is reflected in their respective MED-normalized plasma and brain PKPD parameters (Table 3, the last two columns). Otherwise stated, the >1-log LLE superiority of 3 vs 2 underscores the greater unbound exposure levels and consequently the greater in vivo efficacy of 3. Likewise, the monkey LH data (Figure 2) mirrored these trends with 3 4-fold more efficacious (MED levels) although nearly equipotent to 2 in monkey Ki values (Table 3) in keeping with the significantly better MED-normalized plasma PKPD parameter for 3 vs 2.
Table 3. PKPD Analysis of the Oral LH Inhibition Studiesa.
| Cpd | species | Ki (nM) | LLE | MED (mg/kg) | Tmax (min) | Cplasma,u/Ki | Cbrain,u/Ki | (Cplasma,u/Ki)/MED | (Cbrain,u/Ki)/MED |
|---|---|---|---|---|---|---|---|---|---|
| 2 | rat | 76 | 4.0 | 60 | 150 | 16.4 | 2.32 | 0.273 | 0.039 |
| 2 | monkey | 20 | 4.6 | 20 | 60 | 13.3 | − | 0.665 | − |
| 3 | rat | 219 | 5.2 | 3 | 150 | 22.0 | 5.03 | 7.33 | 1.68 |
| 3 | monkey | 25 | 6.1 | 5 | 90 | 192 | − | 38.4 | − |
| 8 | rat | 22 | 4.6 | 10 | 150 | 15.0 | 12.4 | 1.5 | 1.24 |
| 12 | rat | 2033 | 4.5 | 30 | 150 | 22.5 | 2.77 | 0.75 | 0.09 |
| 16 | rat | 85 | 4.7 | 10 | 150 | 23.8 | 5.87 | 2.38 | 0.59 |
| 17 | rat | 573 | 4.9 | 30 | 45 | 47.3 | 7.71 | 1.58 | 0.26 |
| 18 | rat | 244 | 5.6 | 10 | 45 | 38.9 | 1.82 | 3.89 | 0.18 |
Plasma concentrations coincident with LH measurements. MED determined by a significant decrease (p < 0.05) in LH vs baseline with a lower nonsignificant dose established in all cases.
Figure 2.
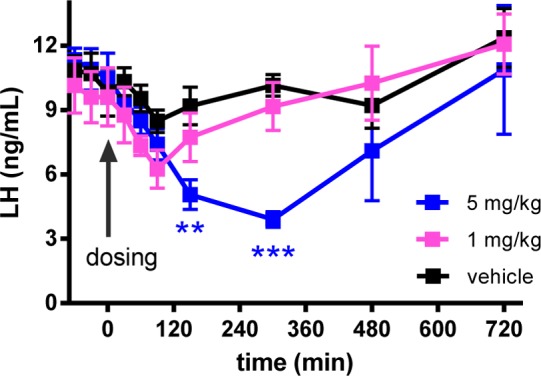
Oral LH inhibition with 3 (0.5% MC/water) in castrated cynomolgus monkey (2-way ANOVA and Dunnet’s comparison to the vehicle; ***p < 0.001, **p < 0.01).
In summary, 3 proved a superior lead candidate based on bioactivity, LLE, LE, and Fsp3 (Table 1) criteria. Apart from its excellent hERG and CYP safety profile, 3 was highly efficacious in LH inhibition, showed >2.5-log selectivity against NK1R and NK2R subtypes, proved >300-fold selective against related HPG axis receptors1 (KOR, GnRH, GnIH-R, GPR54), and was highly selective in the broad CEREP off-target screen (<25% inhib at 10 μM). Finally, 3 showed no effect either in Langendorff cardiac safety in rabbits (up to 30 μM) or in AMES genotoxicity test (up to 100 μM). Compound 3 (ESN364) is currently in phase 2 clinical trials for the treatment of PCOS and UF.
Acknowledgments
We thank Prof. Tony Plant and Dr. Suresh Ramaswamy (University of Pittsburgh) for the monkey LH assays.
Glossary
ABBREVIATIONS
- ARC
arcuate nucleus
- AST
aspartate transaminase
- ALT
alanine aminotransferase
- bfu
brain fraction unbound
- (B/P)u
unbound brain-to-plasma
- CYP
cytochrome P-450
- FSH
follicle-stimulating hormone
- Fsp3
fraction sp3 carbon content
- fu
plasma fraction unbound
- GnRH
gonadotropin-releasing-hormone
- HBA
hydrogen-bond acceptor
- hERG
human ether-à-go-go related gene
- HPβCD
9% hydroxypropyl-β-cyclodextrin
- HPG
hypothalamic–pituitary–gonadal
- KNDy
kisspeptin-neurokinin B-dynorphin A neuron
- LH
luteinizing hormone
- LE
ligand efficiency
- LLE
ligand lipophilicity efficiency
- MC
methyl cellulose
- MED
minimum effective dose
- NKB
neurokinin B
- NK3R
neurokinin-3 receptor
- Pgp
P-glycoprotein
- PKPD
pharmacokinetic–pharmacodynamic
- POC
proof-of-concept
- T1/2
elimination half-life
- Vss
steady-state volume of distribution
Biography
Hamid Hoveyda obtained his Ph.D. from the University of British Columbia (Vancouver, Canada) followed by postdoctoral stints at Harvard University (NSERC fellow) and University of Alberta (Edmonton, Canada). He began his industrial career at the Affymax Research Institute (CA, USA) working on the applications of diversity-oriented synthesis in drug discovery. Later, at Tranzyme Pharma (Canada), he led the discovery of two ghrelin agonists that advanced into clinical development for the treatment of GI disorders. Since 2007, he has led medicinal chemistry efforts on various GPCR targets at Euroscreen (Belgium). His scientific contributions have been captured in over fifty publications and patents.
Supporting Information Available
Experimental details for the synthesis and characterization of compounds, X-ray structure of 3 (accession code: CCDC 1052911), and pharmacology and profiling assays. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00117.
This work was supported by the Ministry of Sustainable Development and Public Works, Walloon Region, Belgium.
The authors declare no competing financial interest.
Supplementary Material
References
- Hoveyda H. R.; Fraser G. L.; Roy M.-O.; Dutheuil G.; Batt F.; El Bousmaqui K. J.; Lenoir F.; Lapin A.; Noël S.; Blanc S. Discovery and optimization of novel antagonists to the human neurokinin-3 receptor for the treatment of sex-hormone disorders (Part I). J. Med. Chem. 2015, 58, 3060–3082. (Cpds 1, 2, and 8 were reported in ref (1) as 3, 31, and 39, respectively.). [DOI] [PubMed] [Google Scholar]
- Dawson L. A.; Porter R. A. Progress in the development of neurokinin 3 modulators for the treatment of schizophrenia: molecule development and clinical progress. Future Med. Chem. 2013, 5, 1525–1546. and references therein. [DOI] [PubMed] [Google Scholar]
- For a recent review, see:Skorupskaite K.; George J. T.; Anderson R. A. The kisspeptin-GnRH pathway in human reproductive health and disease. Hum. Reprod. Update 2014, 0, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall J. C.; Griffin M. L. The role of changing pulse frequency in the regulation of ovulation. Hum. Reprod. 1993, 8, 57–61. [DOI] [PubMed] [Google Scholar]
- Francou B.; Bouligand J.; Voican A.; Amazit L.; Trabado S.; Fagart J.; Meduri G.; Brailly-Tabard S.; Chanson P.; Lecomte P.; Guiochon-Mantel A.; Young J. Normosomic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS One 2011, 6, e25614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser G. L.; Hoveyda H. R.; Clarke I. J.; Ramaswamy S.; Plant T. M.; Rose C.; Millar R. P.. The NK3 receptor antagonist ESN364 interrupts pulsatile LH secretion and moderates levels of ovarian hormones throughout the menstrual cycle. Endocrinology, submitted for publication. [DOI] [PubMed] [Google Scholar]
- Riggs M. M.; Bennets M.; van der Graaf P. H.; Martin S. W. Integrated pharmacometrics and systems pharmacology model-based analysis to guide GnRH modulator development for management of endometriosis. CPT: Pharmacometrics Syst. Pharmacol. 2012, 1, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar R. P.; Newton C. L. Current and future applications of GnRH, kisspeptin and neurokinin B analogues. Nat. Rev. Endocrinol. 2013, 9, 451–466. and references therein. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Lesson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. and references therein. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Empfield J. R. Reducing the risk of drug attrition associated with physicochemical properties. Annu. Rep. Med. Chem. 2010, 45, 381–391. and references therein. [Google Scholar]
- LE = (1.37/HAC) × pKi. HAC = number of non-hydrogen atoms (ref (9)).
- Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Driscoll J.; Zhao S. X.; Walker G. S.; Shepard R. M.; Soglia J. R.; Atherton J.; Yu L.; Mutlib A. E.; Munchhof M. J.; Reiter L. A.; Jones C. S.; Doty J. L.; Trevena K. A.; Shaffer C. L.; Ripp S. L. A rational chemical intervention strategy to circumvent bioactivation liabilities associated with a nonpeptidyl thrombopoietin receptor agonist containing a 2-amino-4-arylthiazole motif. Chem. Res. Toxicol. 2007, 20, 1954–1965. [DOI] [PubMed] [Google Scholar]
- Gramec D.; Mašič L. P.; Dolenc M. S. Bioactivation potential of thiophene-containing drugs. Chem. Res. Toxicol. 2014, 27, 1344–1358. and references therein. [DOI] [PubMed] [Google Scholar]
- Trainor G. L. The importance of plasma protein binding in drug discovery. Expert Opin. Drug Discovery 2007, 2, 51–64. and references therein. [DOI] [PubMed] [Google Scholar]
- Morita S.; Miyata S. Accessibility of low-molecular-mass molecules to the median eminence and arcuate hypothalamic nucleus of adult mouse. Cell Biochem. Funct. 2013, 3, 668–677. [DOI] [PubMed] [Google Scholar]
- Kratochwil N. A.; Huber W.; Muller F.; Kansy M.; Gerber P. R. Predicting plasma protein binding of drugs: a new approach. Biochem. Pharmacol. 2002, 64, 1355–1374. [DOI] [PubMed] [Google Scholar]
- Kerns E. H.; Di L.. Drug-like Properties: Concepts, Structure Design and Methods: from ADME to Toxicity Optimization; Elsevier: Burlington, MA, 2008; pp 112–116. [Google Scholar]
- For example, see:Godbole A. M.; Ramalingam S.; Ramamurthy V. P.; Khandelwal A.; Bruno R. D.; Upreti V. V.; Gediya L. K.; Purushottamachar P.; Mbatia H. W.; Addya S.; Ambulos N.; Njar V. C. O. VN/14–1 induces ER stress and autophagy in HP-LTLC human breast cancer cells and has excellent oral pharmacokinetic profile in female Sprague-Dawley rats. Eur. J. Pharmacol. 2014, 734, 98–104. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.