

Abstract

A new subseries of ROMK inhibitors exemplified by 28 has been developed from the initial screening hit 1. The excellent selectivity for ROMK inhibition over related ion channels and pharmacokinetic properties across preclinical species support further preclinical evaluation of 28 as a new mechanism diuretic. Robust pharmacodynamic effects in both SD rats and dogs have been demonstrated.
Keywords: ROMK, hypertension, heart failure, diuresis, natriuresis
Diuretics represent an important class of therapeutics for the treatment of human diseases. Thiazide diuretics, such as hydrochlorothiazide (HCTZ), represent first line therapy for uncomplicated hypertension; either alone or in combination with other agents.1 Loop diuretics, such as furosemide, are indicated for the treatment of acute pulmonary edema and chronic heart failure.2 Approved diuretics are not without their limitations, however. The thiazide diuretics, for example, are associated with hypokalemia (serum potassium <3.5 mEq/L) and elevation in fasting glucose, side effects that can be dose limiting. Common dose-dependent adverse reactions associated with loop diuretics include hyponatremia, hypokalemia, hypomagnesaemia, and dehydration, thus requiring careful electrolyte monitoring in patients.2 Another drawback of loop diuretics is their short human half-lives (i.e., furosemide human half-life ∼2 h), which are associated with robust initial diuretic responses (impacting tolerability) followed by rebound effects; short half-lives may also be responsible for the inferior blood pressure efficacy of loop diuretics compared to longer half-life thiazide diuretics.3 Despite the established importance of these therapies and the opportunity for improvement over existing therapeutics, no new diuretics have been disclosed to be in development.
Recent efforts in our laboratory aimed at the discovery of new mechanism diuretics have focused on the development of selective inhibitors for the Renal Outer Medullary Potassium Channel (ROMK, Kir1.1, encoded by KCNJ1).4,5 This inward rectifying potassium channel is expressed at the apical membrane of epithelial cells lining two segments of the nephron: the thick ascending loop of Henle (TALH) and the cortical collecting duct (CCD).6 At the TALH, ROMK facilitates potassium recycling across the luminal membrane, which is necessary for the proper function of the furosemide-sensitive Na+/K+/2Cl– cotransporter, the rate-determining step for sodium reuptake in this part of the nephron. At the CCD, ROMK provides a route for potassium secretion that is tightly coupled to sodium uptake through the amiloride-sensitive epithelial sodium channel.7,8 Bartter’s syndrome type II patients (KCNJ1 homozygotes) lack functional ROMK expression and have a phenotype characterized by renal salt wasting, hypotension, and mild hypokalemia.9 A similar phenotype has been reported from studies with the ROMK knockout mice.9,10 In addition, KCNJ1 heterozygote carriers from the Framingham Heart Study exhibited reduced blood pressure and a reduced risk of hypertension at age 60 when compared with matched controls.11 Taken together, the evidence suggests that selective inhibitors of ROMK should represent novel diuretic/natriuretic agents with reduced kaliuresis, opportunity for improved pharmacokinetic (PK) and pharmacodynamic (PD) properties, and potential utility in the treatment of hypertension12 and heart failure.13,14
Small molecule inhibitors of ROMK were first described by Denton at Vanderbilt University15,16 and subsequently by our own group.17,18 The initial lead series in our laboratories remarkably resulted from isolation of a potent impurity 1 (Figure 1) present in an otherwise inactive high throughput screening hit. Subsequent studies detailed the development of this initial hit into a distinct subseries of potent and selective ROMK inhibitors (6). This effort resulted in the first pharmacological proof-of-biology, confirming for the first time in vivo that small molecule ROMK inhibitors represent a new class of novel mechanism diuretics with reduced kaliuresis.19 In this Letter, we will describe the parallel development of a piperazinyl diol series resulting in ROMK inhibitors with superior potency, selectivity, and preclinical PK properties suitable for further evaluation.
Figure 1.

Replacement of the two nitrophenyl groups in 1.
For all compounds listed, unless otherwise specified, ROMK potency was determined as previously described using a thallium flux functional assay20 in HEK-293 cells stably expressing ROMK and binding potency. For the outward delayed rectifier potassium channel (Kv11.1, human ether-a-go-go-related gene, hERG), potency was determined by measuring displacement of 35S MK499 from HEK-293 cells stably expressing hERG.21,22
As previously reported,17 our initial follow-up to 1 focused on replacement of the two nitro groups with an eye toward improved selectivity over hERG compared to ROMK. Differentiation of the ROMK potassium ion channel activity from the hERG potassium ion channel activity was viewed as crucial due to the association between hERG channel blockade in vitro with QTc prolongation and risk of cardiac arrhythmias seen with a broad range of drugs.23−26 To this end, phthalide and cyano groups (2 and 3) were identified as effective single nitro group replacements, able to maintain similar ROMK inhibition to 1.17 As previously described, combination of these groups led to identification of 4, which represents a >10-fold improvement in the hERG/ROMK ratio over the initial hit; further exploration of the structure–activity relationships (SARs) of substitutions on the cyano-phenyl ring resulted in 5, a potent ROMK inhibitor, which for the first time in our series provided a useful level of selectivity over hERG (18-fold).
Following the SAR successfully applied in our acyl piperazine subseries,18 incorporation of the optimal 4-methyl substitution on the phthalide ring of 5 generated compound 7 (Figure 1). This compound displayed potent ROMK inhibition (0.035 μM) with reduced binding potency for the hERG channel (2.1 μM), resulting in a 60-fold selectivity window. Unfortunately, when 7 was evaluated in a hERG electrophysiology (EP) assay the potency was found to be less than 1.0 μM. Additionally, PK evaluation in Sprague–Dawley (SD) rats revealed the compound to have high clearance, low oral exposure, and modest half-life. Nevertheless, 7 represented a potent ROMK inhibitor and an interesting starting point for further optimization.
We reasoned that structural modifications capable of attenuating the basicity of the piperazine moiety in 7 might lead to beneficial impact on both hERG selectivity and PK parameters. In addition, substitutions that could reduce lipophilicity might also provide benefit. To this end, we next explored substitution at the α and β carbons flanking the nitrogens of the central piperazine ring (Table 1). On the carbon adjacent to the phthalide phenyl ring, methyl and fluoro substitution (8 and 9) led to modest loss of ROMK potency, resulting in an overall erosion of the hERG selectivity. Hydroxy or methoxy substitution (10 and 11) also resulted in a modest loss of ROMK potency, but the corresponding loss of hERG potency resulted in an overall improvement in the selectivity margin (>150-fold in the case of 10). Further increase in the size of the alkoxy substituent to ethyl (12), however, resulted in a loss in ROMK potency and hERG margin. On the 4-CN phenyl side of the molecule, methyl substitution adjacent to the piperazine and mono- or dimethyl substitution at the benzylic position (13, 14, and 15) resulted in modest loss of ROMK potency and hERG selectivity. However, hydroxyl substitution at the benzylic position (16) largely maintained the potency and selectivity present in 7. Guided by hERG selectivity improvements observed upon heteroatom incorporation in our related acyl piperazine series,27 we also prepared the pyridyl analogue 17. In this case, the pyridyl leads to retention of ROMK potency with an increased hERG/ROMK ratio (130-fold).
Table 1. Benzylic Substitution Structure–Activity Relationshipa.
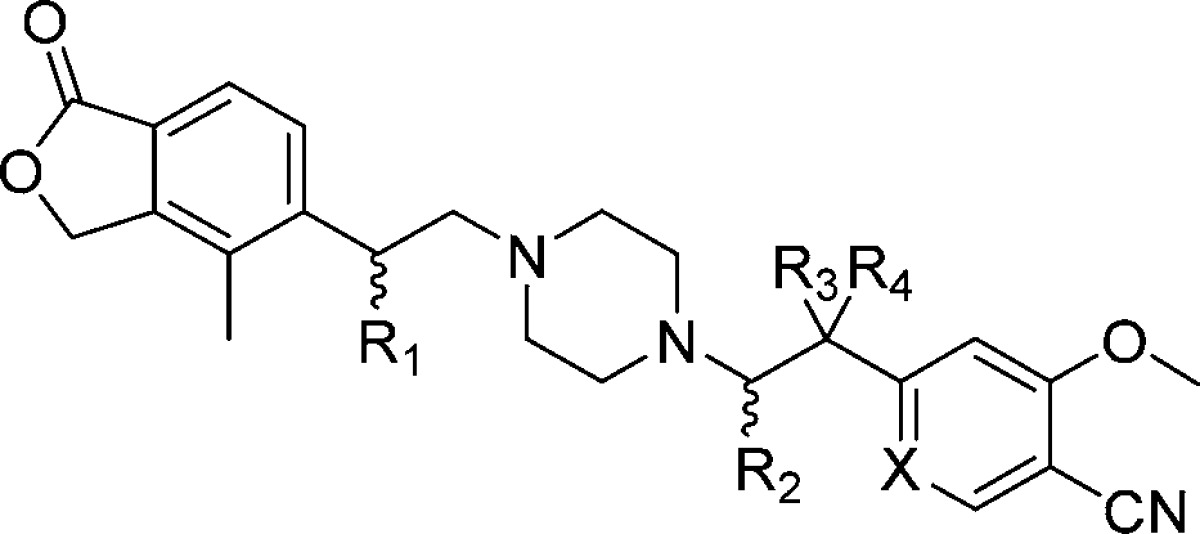
| # | R1 | R2 | R3 | R4 | X | ROMK IC50 (μM) | hERG IC50 (μM) |
|---|---|---|---|---|---|---|---|
| 8 | Me | H | H | H | C | 0.170 | 2.8 |
| 9 | F | H | H | H | C | 0.060 | 2.4 |
| 10 | OH | H | H | H | C | 0.12 | 9.0 |
| 11 | OMe | H | H | H | C | 0.11 | 9.0 |
| 12 | OEt | H | H | H | C | 0.22 | 3.5 |
| 13 | H | Me | H | H | C | 0.19 | 1.3 |
| 14 | H | H | Me | H | C | 0.15 | 1.1 |
| 15 | H | H | Me | Me | C | 0.30 | 1.2 |
| 16 | H | H | OH | H | C | 0.070 | 2.2 |
| 17 | H | H | OH | H | N | 0.049 | 6.4 |
All compounds are racemic.
Based on these results, 10, 16 and 17 were selected for PK evaluation in SD rats (0.5 mpk iv and 2 mpk po; Table 2). All three compounds showed modest improvement in clearance and oral exposure with little effect on half-life. Oral bioavailability was improved in all three cases; incorporation of the heterocyclic ring in 17 lead to the most significant effect.
Table 2. SD Rat PK Properties of 7, 10, 16, and 17a.
| 7 | 10 | 16 | 17 | |
|---|---|---|---|---|
| Cl (L/kg) | 72 | 33 | 40 | 40 |
| AUCNpo (μM h kg/mg) | 0.12 | 0.46 | 0.35 | 0.74 |
| t1/2 (h) | 1.2 | 0.83 | 1.3 | 1.5 |
| Fpo (%) | 21 | 39 | 37 | 78 |
All compounds are racemic.
Next we turned to combining the hydroxy substitution and to producing compounds as single enantiomers (Scheme 1). Suzuki cross coupling of 18 with the potassium vinyl trifluoroborate salt followed by m-chloroperbenzoic acid (mCPBA) epoxidation provided the racemic epoxide 19. Chiral supercritical fluid chromatography (SFC) separation of the enantiomers followed by reaction of the individual epoxides with Boc-piperazine led to the individual hydroxy enantiomers.28 We found that optimal selectivity for the terminal aminolysis of this and related styrene epoxides was achieved under thermal conditions in a microwave reactor in the absence of coordinating acid. Hydrolysis of the t-butyl carbamate was followed by basic workup to provide (R)- and (S)-20 as their free amine bases. Synthesis of epoxides (R)- and (S)-22 began with vinylation of the aryl bromide 21; however, epoxidation of the resulting olefins with mCPBA led to completive N-oxide formation. Alternatively, reaction with N-bromosuccinimide (NBS) and sodium hydroxide (NaOH) led to one-pot formation of the desired epoxides following in situ cyclization of the intermediate hydroxy bromides. Chiral SFC separation led to isolation of the individual epoxide enantiomers.29 Reaction of the individual epoxides (R)- and (S)-22 with (R)- and (S)-20 provided the four individual products shown in Scheme 1.
Scheme 1. Synthesis of Stereochemically Pure Diols 24–26.

Reagents and conditions: (a) potassium vinyltrifluoroborate, PdCl2(dppf)–CH2Cl2, triethylamine (TEA), ethanol (EtOH), 140 °C, MW; (b) mCPBA, CH2Cl2, 0 °C (40–70% yield, 2 steps); (c) ChiralPac AD-H SFC chromatography, 10% EtOH/CO2, flow rate 200 mL/min, 100 bar, 25 °C; (d) N-Boc-piperazine, EtOH, 150 °C, MW (50–90% yield); (e) (i) trifluoroacetic acid, CH2C2, rt, (ii) Na2CO3 (aq) (85–90% yield); (f) NBS, H2O/t-butanol (2:1), 40 °C, then addition of NaOH at 0 °C (44–85% yield); (g) 20 + 22, EtOH, 150 °C, MW (58–92% yield).
Using the chemistry outlined in Scheme 1, a series of analogues was prepared to examine the effect of substitution on the 4-CN pyridyl ring (Table 3). Because the initial analogues made with (S)-20 piperazine were substantially less potent, only (R)-20 was used for subsequent analogues. Of the initial analogues, (R,S)-23 provides the best combination of ROMK potency and hERG selectivity. In general, substitution in the para- or meta-positions relative to the pyridine nitrogen resulted in compounds with improved ROMK potency, with the exception of the p-trifluoromethyl and m-chloro groups (39, 40, 47, and 48). Of the ortho-substituents explored, only the methyl group (35 and 36) provided improved ROMK potency than (R,S)-24. Overall, the best balance of ROMK potency and hERG selectivity was achieved using the p-methoxy group ((R,S)-28). Deletion of the pyridyl nitrogen from (R,R)-27 and (R,S)-28 provided compounds with similar ROMK potency but reduced hERG selectivity (29 and 30), underscoring the importance of the heterocycle. No additivity was observed when combining the best para-substituent with the best ortho-substituent (OMe and Me, 43 and 44). Finally, anticipating the p-methoxy group might undergo metabolism in vivo, we explored the size of the alkoxy group and incorporation of the trideutero-methoxy group (45, 46, 49, and 50).
Table 3. Substitution around 5-CN pyridyl ring.
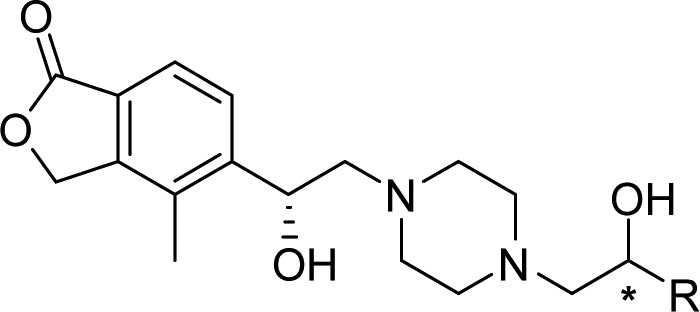
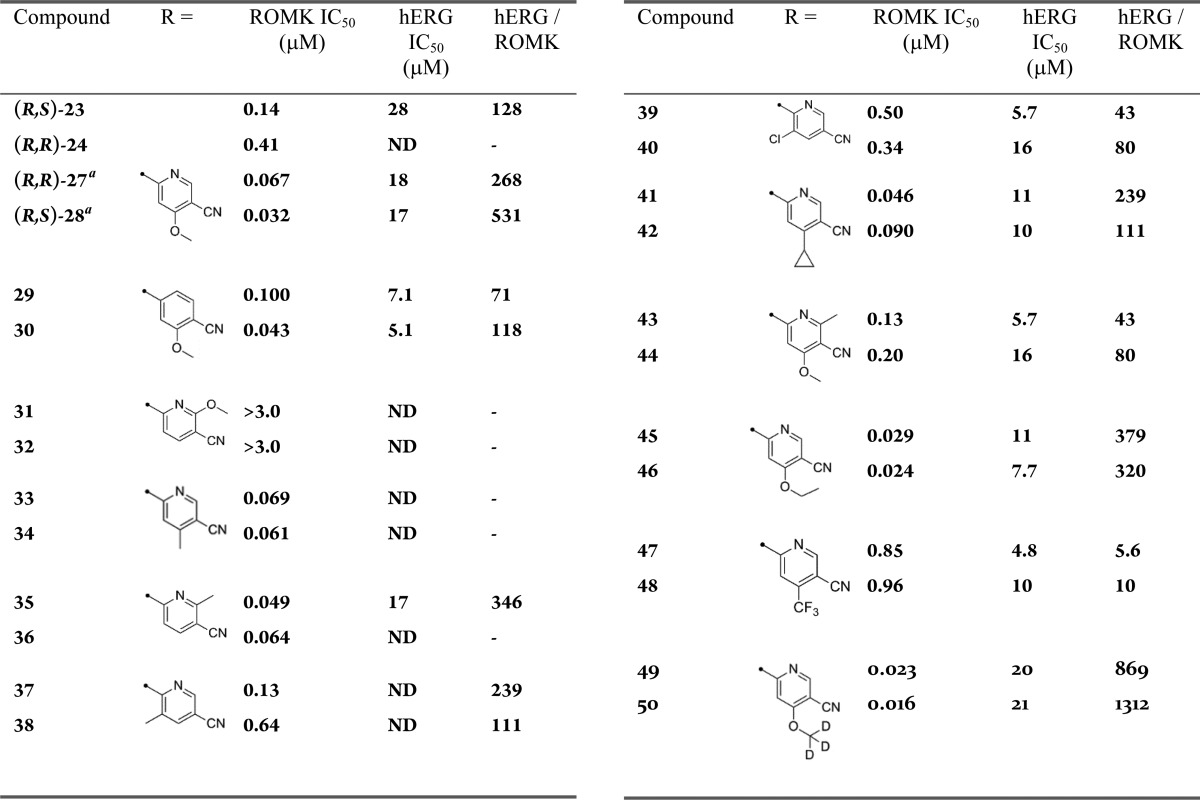
Absolute configuration assigned using vibrational circular dichroism (VCD) spectroscopy. The other compounds obtained as single isomers, but with undetermined absolute stereochemistry.
In addition to the Th flux and 35S-MK499 assays, compounds were evaluated in ROMK and hERG EP assays (Table 4). There was good correlation of potency in both the ROMK and hERG EP assays, with three ROMK inhibitors identified with EP ratios of greater than 1000-fold. To understand how this level of in vitro selectivity translated in vivo, (R,S)-28 was evaluated in anesthetized guinea pigs (n = 3). No significant change in QT interval was observed following IV infusion of (R,S)-28 at 2 mpk (average peak unbound plasma concentration = 2.2 μM).
Table 4. EP Evaluation of Selected Inhibitors.
| 6 | 23 | 28 | 35 | 41 | 46 | 50 | |
|---|---|---|---|---|---|---|---|
| ROMK EP IC50 (nM) | 64 | 18 | 27 | 12 | 54 | 40 | 13 |
| hERG EP IC50 (μM) | 4.5 | 12 | 33 | 14 | 14 | 12 | 30 |
| hERG/ROMK | 70 | 667 | 1222 | 1166 | 254 | 300 | 2307 |
With several potent and selective ROMK inhibitors identified, we next turned to evaluating the PK profiles in SD rats (1 mpk iv and 2 mpk po; Table 5). All of the diol inhibitors evaluated demonstrated improvement in Cl, t1/2, oral exposure, and oral bioavailability versus the previously reported 6.
Table 5. SD Rat PK for Selected ROMK Inhibitors.
| 6 | 23 | 28 | 35 | 50 | |
|---|---|---|---|---|---|
| Cl (L/kg) | 40 | 23 | 31 | 25 | 58 |
| AUCNpo (μM h kg/mg) | 0.34 | 1.6 | 0.84 | 1.3 | 1.2 |
| t1/2 (h) | 0.62 | 2.0 | 1.6 | 1.4 | 1.5 |
| Fpo (%) | 33 | 96 | 65 | 81 | 91 |
(R,S)-28 was further evaluated in functional assays for several related inward rectifying potassium channels (Kir 2.3. Kir2.1, IC50s >50 μM; Kir4.1, Kir7,1 IC50s >10 μM) as well as the cardiac channels Nav 1.2 and Cav 2.1 (>30 μM) and found to have excellent selectivity for ROMK. (R,S)-28 was also evaluated for inhibition of a panel of CYPs (3A4, 2D6, 2C9; IC50s >50 μM) and found to have a signal only against 2C8 (IC50 = 8.2 μM). Plasma protein binding (% free fraction) in rat and dog for (R,S)-28 was 43% and 83%, respectively, with excellent permeability (Papp = 23). Finally, rat and dog ROMK Th flux IC50s for (R,S)-28 were found to be 0.014 and 0.042 μM, respectively.
(R,S)-28 was selected for further PK profiling in two additional species (dog and rhesus monkey; Table 6). The compound was found to have PK profiles across preclinical species consistent with an oral QD therapeutic. In addition, human half-life projection of (R,S)-28 based on observed preclinical PK in rat, dog, and rhesus monkeys by applying allometric scaling is estimated to be 10 h, significantly longer than the most commonly used loop diuretics furosemide and torsemide. This potentially provides a PK–PD advantage with regard to the peak diuretic effects associated with loop diuretics, by providing a reduction in peak-to-trough exposures.
Table 6. PK Properties of (R,S)-28 in Multiple Species.
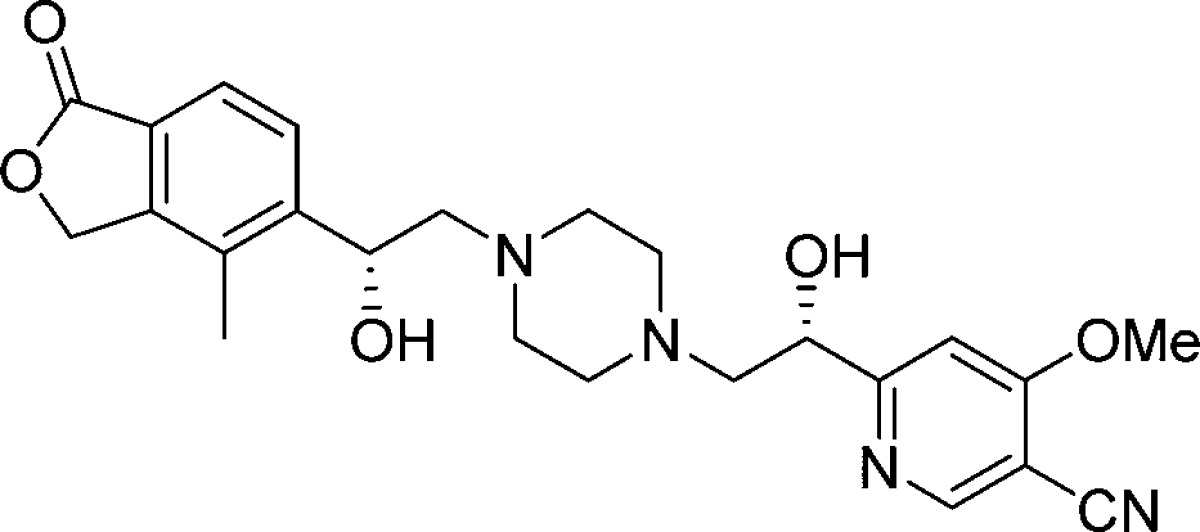
| species | dose IV/PO (mpk) | CL (L/kg) | AUCNpo (μM h kg/mg) | t1/2 (h) | Fpo (%) |
|---|---|---|---|---|---|
| dog | 0.5/1 | 5.6 | 5.6 | 5.9 | 84 |
| rhesus | 1/2 | 25 | 0.51 | 7.2 | 30 |
(R,S)-28 was dosed orally in volume loaded SD rats at 0.3 and 1 mpk to assess its pharmacodynamic effects (Table 7).19(R,S)-28 demonstrated comparable efficacy at lower dose for diuresis and natriuresis over previously reported ROMK inhibitor 6 and HCTZ. Neither ROMK inhibitor induced statistically significant kaliuresis versus vehicle control in contrast to HCTZ (1.4-fold).
Table 7. Acute 4 h Diuresis/Natriuresis of (R,S)-28 in SD Rats.
| compound dose (mpk) | 6 (3) | 28 (0.3) | 28 (1) | HCTZ (25) |
|---|---|---|---|---|
| diuresisa | 2.3 | 2.3 | 2.8 | 2.1 |
| natriuresisa | 2.5 | 2.1 | 2.5 | 2.3 |
| kaliuresisa | 1.1 | 1.1 | 1.0 | 1.4 |
Fold increase compared to vehicle; n = 5 per group.
The diuretic and electrolyte excretion effects of (R,S)-28 were also evaluated following oral administration in female mongrel dogs to assess its PD effects (Table 8).30 Robust dose-dependent increases in diuresis and natriuresis were observed over 24 h postdose, while kaliuretic response was not significantly changed versus vehicle control.
Table 8. Twenty-Four Hour Diuresis/Natriuresis of (R,S)-28 in Dogs.
| dose (mpk) | 0.1 | 0.3 | 1 | 3 | 10 |
|---|---|---|---|---|---|
| AUC (μM·h) | 0.081 | 0.51 | 1.1 | 5.0 | 15 |
| diuresisa | 1.1 | 1.7 | 2.1 | 2.8 | 4.5 |
| natriuresisa | 1.0 | 1.5 | 1.6 | 1.5 | 2.6 |
| kaliuresisa | 1.0 | 0.88 | 0.60 | 0.78 | 1.0 |
Fold increase compared to vehicle; n = 6 per group.
In summary, a new subseries of ROMK inhibitors exemplified by (R,S)-28 has been developed. Excellent selectivity for ROMK inhibition over related ion channels and PK properties of (R,S)-28 across preclinical species support continued evaluation of this compound as a new mechanism diuretic. Robust PD effects in both SD rats and dogs have been demonstrated. Translation of ROMK mediated diuresis effects into preclinical blood pressure lowering and further mechanistic studies will be the subject of future communications.
Supporting Information Available
Synthesis and characterization data for compounds 7, (R,S)-23, (R,R)-27, (R,S)-28, 35, 41, 46, and 50. Additional details regarding assay correlation. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/ml500440u.
The authors declare no competing financial interest.
Supplementary Material
References
- Tamargo J.; Segura J.; Ruilope L. M. Diuretics in the Treatment of Hypertension Part 1. Thiazide and Thiazide-like Diuretics. Expert Opin. Pharmacother. 2014, 15 (4), 527. [DOI] [PubMed] [Google Scholar]
- Felker G. M.; O’Connor C. M.; Braunwald E. Loop Diuretics in Acute Decompensated Heart Failure. Circulation: Heart Failure 2009, 2, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison D. H. Diuretic Resistance: Physiology and Therapeutics. Semin. Nephrol. 1999, 19 (6), 581–597. [PubMed] [Google Scholar]
- Ho K.; Nichols C. G.; Lederer W. J.; Lytton J.; Vassilev P. M.; Kanazirska M. V.; Hebert S. C. Cloning and Expression of an Inwardly Rectifying ATP-regulated Potassium Channel. Nature 1993, 362, 31. [DOI] [PubMed] [Google Scholar]
- Shuck M. E.; Bock J. H.; Benjamin C. W.; Tsai T. D.; Lee K. S.; Slightom J. L.; Bienkowski M. J. Cloning and Characterization of Multiple Forms of the Human Kidney ROM-K Potassium Channel. J. Biol. Chem. 1994, 269, 24261. [PubMed] [Google Scholar]
- Hebert S. C.; Desir G.; Giebisch G.; Wang W. Molecular Diversity and Regulation of Renal Potassium Channels. Physiol. Rev. 2005, 85, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinalter S. C.; Jeck N.; Peters M.; Seyberth H. W. Pharmacotyping of Hypokalemic Salt-losing Disorder. Acta Physiol. Scand. 2004, 181, 513. [DOI] [PubMed] [Google Scholar]
- Wang W. Renal Potassium Channels: Recent Development. Curr. Opin. Nephrol. Hypertens. 2004, 13, 549. [DOI] [PubMed] [Google Scholar]
- Ji W.; Foo J. N.; O’Roak B. J.; Zhao H.; Larson M. G.; Simon D. B.; Newton-Cheh C.; State M. W.; Levy D.; Lifton R. P. Rare Independent Mutations in Renal Salt Handling Genes Contribute to Blood Pressure Variation. Nat. Genet. 2008, 40, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz J. N.; Baird N. R.; Judd L. M.; Noonan W. T.; Andringa A.; Doetschman T.; Manning P. A.; Liu L. H.; Miller M. L.; Shull G. E. Impaired Renal NaCl Absorption in Mice Lacking the ROMK Potassium Channel, a Model for Type II Bartter’s Syndrome. J. Biol. Chem. 2002, 277, 37871. [DOI] [PubMed] [Google Scholar]
- Lu M.; Wang T.; Yan Q.; Yang X.; Dong K.; Knepper M. A.; Wang W.; Giebisch G.; Shull G. E.; Hebert S. C. Absence of Small Conductance K+ Channel (SK) Activity on Apical Membranes of Thick Ascending Limb and Cortical Collecting Duct in ROMK (Bartter’s) Mice. J. Biol. Chem. 2002, 277, 37881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin M. D.; Timpson N. J.; Wain L. V.; Ring S.; Jones L. R.; Emmett P. M.; Palmer T. M.; Ness A. R.; Samani N. J.; Smith G. D.; Burton P. R. Common Variation in the WNK1 Gene and Blood Pressure in Childhood. Hypertension 2008, 51, 1658. [DOI] [PubMed] [Google Scholar]
- Lifton R. P.; Gharavi A. G.; Geller D. S. Genetic Factors in the Pathogenesis of Primary (Essential) Hypertension. Cell 2001, 104, 545. [DOI] [PubMed] [Google Scholar]
- Garcia M. L.; Kaczorowski G. J. Targeting the Inward-Rectifier Potassium Channel ROMK in Cardiovascular Disease. Curr. Opin. Pharmacology 2014, 15, 1. [DOI] [PubMed] [Google Scholar]
- Lewis L. M.; Bhave G.; Chauder B. A.; Banerjee S.; Lornsen K. A.; Redha R.; Fallen K.; Lindsley C. W.; Weaver C. D.; Denton J. S. High Throughput Screening Reveals a Small-molecule inhibitor of the Renal Outer Meduallary Potassium Channel and Kir7.1. Mol. Pharmacol. 2009, 1094, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G.; Chauder B. A.; Liu W.; Dawson E. S.; Kadakia R.; Nguyen T. T.; Lewis L. M.; Meiler J.; Weaver D. D.; Satlin L. M.; Lindsley C. W.; Denton J. S. Development of a Selective Small-molecule Inhibitor of Kir1.1, the Renal Outer Meduallary Potassium Channel. Mol. Pharmacol. 2011, 79, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Walsh S. P.; Yan Y.; de Jesus R. K.; Shahripour A.; Teumelsan N.; Zhu Y.; Ha S.; Owens K. A.; Thomas-Fowlkes B. S.; Felix J. P.; Liu J.; Kohler M.; Priest B. T.; Bailey T.; Brochu R.; Alonso-Galicia M.; Kaczorowski G. J.; Roy S.; Yang L.; Mills S. G.; Garcia M. L.; Pasternak A. Discovery of Selective Small Molecule ROMK Inhibitors as Potential New Mechanism Diuretics. ACS Med. Chem. Lett. 2012, 3, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; de Jesus R. K.; Walsh S. P.; Zhu Y.; Yan Y.; Priest B. T.; Swensen A. M.; Alonso-Galicia M.; Felix J. P.; Brochu R. M.; Bailey T.; Thomas-Fowlkes B.; Zhou X.; Pai L.-Y.; Hampton C.; Hernandez M.; Owens K.; Roy S.; Kaczorowski G. J.; Yang L.; Garcia M. L.; Pasternak A. Discovery of a Novel Sub-Class of ROMK Channel Inhibitors Typified by 5-(2-(4-(2-(1H-Tetrazole-1-yl)phenyl)acetyl)piperazin-1-yl)ethyl)isobenzofuran-13H-one. Bioorg. Med. Chem. Lett. 2013, 23, 5829. [DOI] [PubMed] [Google Scholar]
- Garcia M. L.; Priest B. T.; Alonso-Galicia M.; Zhou X.; Felix J. P.; Brochu R. M.; Bailey T.; Swensen A.; Pai L.-Y.; Xiao J.; Hernandez M.; Hoagland K.; Owens K.; Tang H.; de Jesus R.; Roy S.; Kaczorowski G. J.; Pasternak A. Pharmacologic Inhibition of the Renal Outer Medullary Potassium Channel Causes Diuresis and Natriuresis in the absence of Kaliuresis. J. Pharm. Exp. Ther. 2014, 348, 153. [DOI] [PubMed] [Google Scholar]
- Felix J. P.; Priest B. T.; Solly S.; Bailey T.; Brochu R. M.; Liu C. J.; Kohler M. G.; Kiss L.; Alonso-Galicia M.; Tang H.; Pasternak A.; Kaczorowski G. J.; Garcia M. L. The Inward Rectifying Potassium Channel Kir1.1; Development of Functional Assays to Identify and Characterize Channel Inhibitors. Assay Drug Dev. Technol. 2012, 10, 417. [DOI] [PubMed] [Google Scholar]
- Wang J.; Della Penna K.; Wang H.; Karczewski J.; Connolly T. M.; Koblan K. S.; Bennett P. B.; Salata J. J.Functional and Pharmacological Properties of Canine ERG Channels. Am. J. Phys. 2003, 284, 256. [DOI] [PubMed] [Google Scholar]
- Further details regarding standard deviations and number of assay runs are available in the Supporting Information.
- Cavero I.; Mestre M.; Guillon J.-M.; Crumb W. Drugs that Prolong QT Interval as an Unwanted Effect; Assessing their Likelihood of Inducing Hazardous Cardiac Dysrhythmias. Expert Opin. Pharmacother. 2000, 1, 947. [DOI] [PubMed] [Google Scholar]
- Sanguinetti M. C.; Jiang C.; Curran M. E.; Keating M. T. A Mechanistic Link Between an Inherited and Aquired Cardiac Arrhythmia; HERG Encodes the IKr Potassium Channel. Cell 1995, 81, 299... [DOI] [PubMed] [Google Scholar]
- Fermini B.; Fossa A. A. Nat. Rev. The impact of Drug Induced QT Prolongation on Drug Discovery and Development. Nat. Rev. Drug Discovery 2003, 2, 439. [DOI] [PubMed] [Google Scholar]
- Pearlstein R.; Vaz R.; Rampe D. J. Understanding the Structure-activity Relationship of the Human Ether-a-go-go-related Gene Cardiac K+ Channel. J. Med. Chem. 2006, 49, 4801. [DOI] [PubMed] [Google Scholar]
- Full description of this related series will be the subject of another publication.
- The absolute stereochemistry of each enantiomer was inferred based on X-ray structure determination of a derivative made from (R)-20 and by Mosher ester and Trost ester 1H NMR analysis of a derivative made from the t-butyl carbamate of (R)-21.
- The absolute stereochemistry of each enantiomer was determined using vibrational chircular dichromism (VCD).
- Description of the dog diuresis model will be included separately as part of a full pharmacology paper.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.