

Abstract

Structure–activity relationship (SAR) exploration on the left-hand side (LHS) of a novel class of bacterial topoisomerase inhibitors led to a significant improvement in the selectivity against hERG cardiac channel binding with concomitant potent antimycobacterial activity. Bulky polar substituents at the C-7 position of the naphthyridone ring did not disturb its positioning between two base pairs of DNA. Further optimization of the polar substituents on the LHS of the naphthyridone ring led to potent antimycobacterial activity (Mtb MIC = 0.06 μM) against Mycobacterium tuberculosis (Mtb). Additionally, this knowledge provided a robust SAR understanding to mitigate the hERG risk. This compound class inhibits Mtb DNA gyrase and retains its antimycobacterial activity against moxifloxacin-resistant strains of Mtb. Finally, we demonstrate in vivo proof of concept in an acute mouse model of TB following oral administration of compound 19.
Keywords: Tuberculosis, type II topoisomerases, DNA gyrase, NBTIs, aminopiperidines, naphthyridones
Tuberculosis caused by Mycobacterium tuberculosis (Mtb) continues to be a global threat claiming 1.5 million lives each year.1 For treating drug susceptible TB, the World Health Organization (WHO) recommends a regimen containing four drugs administered for six months.2 The global emergence of multidrug resistant (MDR) and extremely drug resistant (XDR) strains of Mtb have greatly impeded the TB control and eradication efforts. Patients with MDR or XDR-TB require treatment with a combination of 6–8 drugs for a period of 8–24 months.3
DNA gyrase, a type II topoisomerase enzyme is involved in DNA replication and repair. This enzyme is essential in all bacteria and is absent in eukaryotes.4 DNA gyrase catalyzes the critical step of maintaining the various topological forms of DNA during DNA replication. DNA gyrase performs an ATP-dependent reaction to introduce a negative supercoiling into circular DNA.5 This enzyme exists in a heterotetrameric form, comprising a GyrA and a GyrB subunit (A2B2). The DNA breakage reunion function resides in the GyrA subunit, while the GyrB subunit catalyzes the ATP-dependent hydrolysis to generate the energy required for enzyme activity. Interestingly, the Mtb genome encodes a functional DNA gyrase, but no topoisomerase IV.6,7 Several inhibitors targeting the DNA gyrase have been reported to exhibit activity against Mtb. A class of inhibitors that target the ATP recognition site of GyrB enzyme and block ATP hydrolysis have shown antimycobacterial activity. Examples of this type include aminopyrazinamides, thiazolopyridine ureas, and pyrrolamides.6,7 A second type of inhibitor that targets the GyrA subunit has been shown to inhibit the DNA breakage–reunion function of the enzyme. This category of inhibitors is represented by fluoroquinolones (FQs), novel bacterial topoisomerase inhibitors (NBTIs), and aminopiperidines.6,8−10
The attractiveness of DNA gyrase as a high quality antibacterial target has been validated by the clinical success of several generations of FQs to treat serious bacterial infections. The widespread emergence of FQ-resistant bacterial strains is likely to reduce the clinical value of FQs in the near future.11 The clinical benefit of combining FQs in the drug regimen to treat TB have been demonstrated in preclinical animal models as well as in TB patients.12 Numerous reports have highlighted the emergence of Mtb strains resistant to FQs, thereby limiting the life of these drugs in treating TB. The existing body of evidence around the clinical safety and efficacy of FQs, provides an opportunity to identify novel GyrA inhibitors. It is envisaged that a novel GyrA inhibitor class would have a distinct binding mechanism compared to the binding mode of FQs.13 This novel class of compounds would serve as an attractive treatment option to tackle drug-resistant forms of Mtb.
The various novel bacterial topoisomerase inhibitors reported in literature are shown in Figure 1a (Supporting Information).6,8−10,14−16 The published crystal structure of NBTIs (1e, GSK299423)13 bound to Staphylococcus aureus DNA gyrase provides key insights into the key interactions involved in the gyrase active site.13 A nonoverlapping mechanism of binding distinct between FQs and NBTIs with the S. aureus DNA gyrase can be inferred from this study. The unique binding mechanism of NBTIs enables them to retain their activity against FQ-resistant strains of S. aureus. This salient observation has triggered the search for novel chemotypes and expanded the chemical repertoire of NBTIs.
Figure 1.

SAR exploration of LHS.
However, the reported cardiac ion channel (hERG) liability associated with NBTIs pose a significant challenge in the clinical advancement of several NBTIs, e.g., NXL10 “1f” was discontinued beyond Phase 1 clinical trials due to potent hERG inhibition resulting in QT prolongation.14 Therefore, there is scope to build hERG selectivity for NBTIs while retaining their antimycobacterial activity.
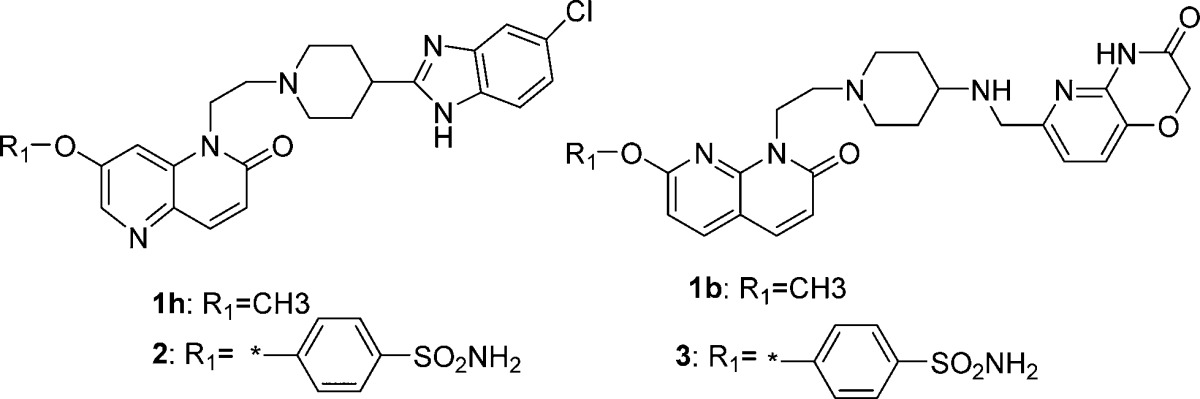
The chemical structures of NBTIs comprise a bicyclic heteroaromatic left-hand side (LHS) ring, a mono- or bicyclic hydrophobic right-hand side (RHS) ring and a middle linker part. Efforts to modulate the pKa of the linker portion by concomitantly lowering the lipophilicity (logD) of NBTIs have shown promise in reducing the hERG liability.8−10 A thorough literature analysis reveals that the chemical diversity of substituents at LHS has not been fully exploited and that most of these attempts have been confined to smaller substituents such as fluoro, cyano, and methoxy groups. Therefore, we devised a strategy to exploit bulky polar groups (R1) on LHS of naphthyridone ring system to improve the hERG selectivity while retaining antimycobacterial activity.
Herein, we report the understanding of structure–activity relationships (SARs) to improve hERG selectivity of NBTIs through bulky polar group substitutions on LHS of the naphthyridone ring (compounds 2–19 in Figure 1). A representative compound from this class (compound 7) inhibits Mtb DNA gyrase with IC50 in the nanomolar range (IC50 = 104 nM) in a DNA supercoiling assay and retains potent antimycobacterial activity against Mtb. Additionally, compound 19 kills Mtb residing in an intracellular macrophage compartment and displays efficacy in an acute mouse model of TB. Compounds from this series retain their MIC against moxifloxacin-resistant strains of Mtb and lose their MIC against NBTIs-resistant mutants of Mtb.
The synthesis of compounds with smaller substituents like fluoro or chloro at the C-7 position of naphthyridone ring were reported earlier.8−10,16
The synthesis of compounds with bulky substitutions (2–9) are illustrated in Scheme 1. Nucleophilic displacement of halo substituted naphthyridones (I) with corresponding carbinol (II) using cesium carbonate under thermal heating condition resulted in title compounds 2–19.
Scheme 1. Synthesis of Compounds 2–19.

Reagents and conditions: (a) Cs2CO3, DMSO, 130 °C.
Exploration of SARs at LHS to mitigate hERG liability: To expand the chemical diversity and build a structural handle to mitigate the hERG liability, we explored the bulky substituents as R1 instead of small fluoro or methoxy group at the C-7 position of naphthyridone LHS. This design was based on the initial SAR observations made during the lead optimization of NBTIs as novel antimycobacterial agents reported earlier.16 However, we have not explored similar substitution at C-6 position as the SAR studies suggest that a compound with 6-methoxy substitution was found to be inactive against Mtb.8 We hypothesized that by lowering the logD and modulation of polarity via bulky polar substitutions on LHS ring may provide structural diversity to mitigate hERG liability. Based on this hypothesis, several compounds with bulky polar substitutions were synthesized, and their hERG IC50s were determined. A few match pairs comparison of small versus bulky polar substituents are shown in Table 1.
Table 1. Match Pairs of Small versus Bulky Group on Napthyridones LHS.
| Compound | MIC Mtb H37Rv (μM) | Clab hERG pIC50a (μM) | hERG IC50 (μM) | ClogD7.4b | PSA (Å2) |
|---|---|---|---|---|---|
| 1h | 0.25 | 5.33 | 1 | 3.37 | 64 |
| 2 | 0.19 | 4.81 | >33 | 4 | 129 |
| 1b | 0.25 | 4.65 | 23 | 0.77 | 106 |
| 3 | 0.03 | 4.34 | >100 | 0.71 | 172 |
Predicted hERG pIC50.
Calculated logD.
From the match pair analysis, it is evident that compounds with polar bulky substitutions have shown better hERG selectivity (by both experimental determination and hERG pIC50 prediction) compared to a smaller methoxy substituted counterpart (1h versus 2, 1b versus 3 in Table1). This could be due to a higher polar surface area (PSA) combined with lower logD observed for compounds 2 and 3. Hence, the better disturbance of hERG channel binding resulted in an improved hERG selectivity for compounds 2 and 3. A thorough evaluation of lipophilicity values for compound 1b (ClogD 0.77) and 3 (ClogD 0.71) suggests that more polarity at the C-7 position of LHS confers better hERG selectivity than lipophilicity alone. Furthermore, compounds 2 and 3 retain their activity against Mtb (MIC of 0.19 and 0.03 μM, respectively).
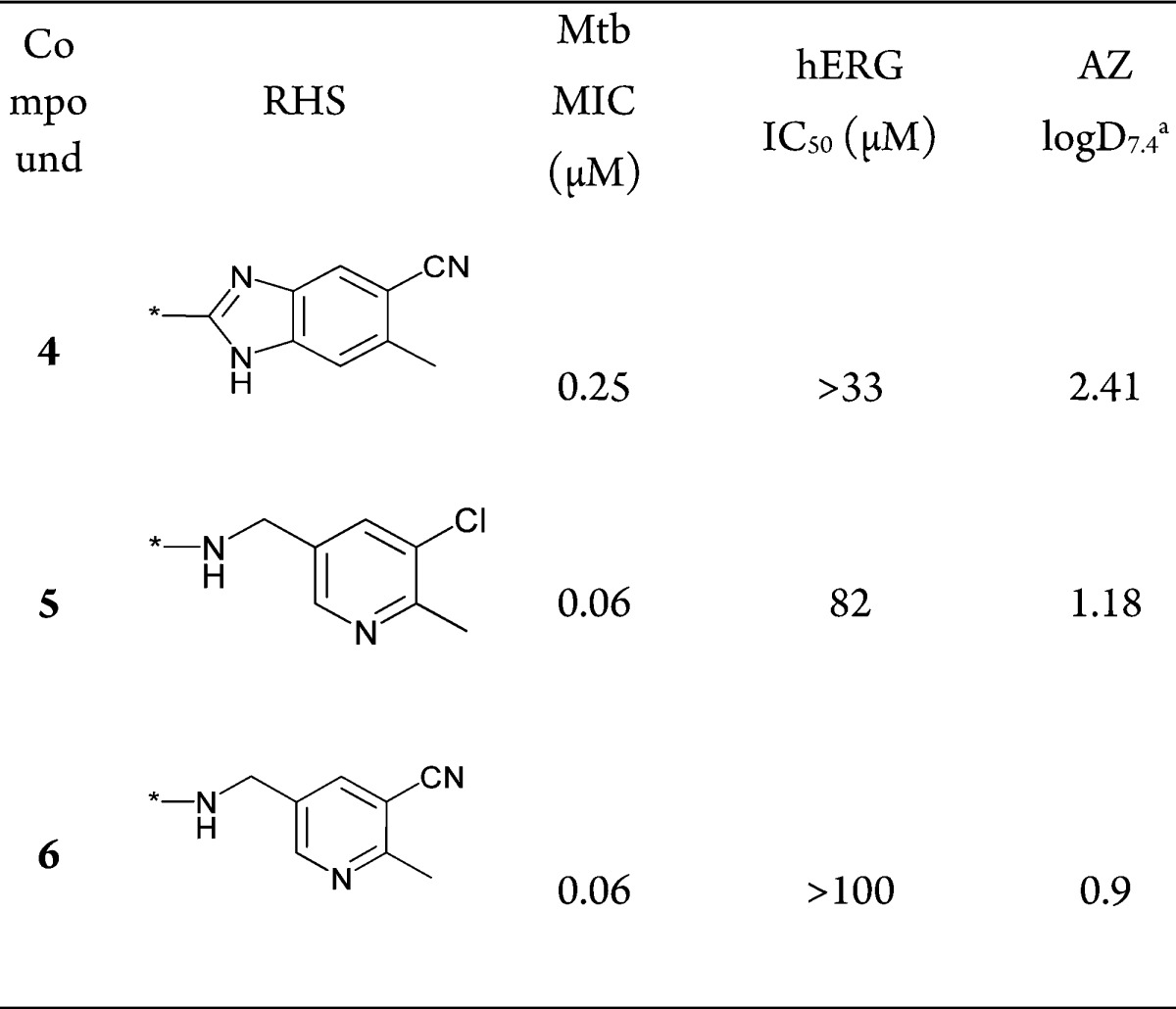
Encouraged by the improved hERG selectivity for compounds 2 and 3 with 4-sulphamido phenyl substitution, we further expanded the SAR with various RHS combinations to validate this hypothesis (Table 2).
Table 2. SAR Modification around RHS and LHS.

Calculated logD.
As shown in Table 2, moving from a benzimidazole or a pyrido oxazinone RHS ring to a monocyclic pyridine ring system (compounds 4–6, Table 2) consistently showed good hERG selectivity while retaining Mtb MIC. Thus, the large polar bulky substitution at the C-7 position does not change the potency and hERG selectivity among the compounds reported in Table 2
A homology model based on the binding mode of aminopiperidine based NBTIs to Mtb GyrA has been reported.8,16 In order to understand the effect of bulky polar substitutions on NBTIs binding to Mtb GyrA subunit, we docked compounds 2 and 3. The possible binding modes of compounds 2 and 3 in the Mtb GyrA homology model are depicted in Figure 2. The H-bonding contacts with Asp89 and occupation of hydrophobic pocket surrounded by Ala74, Val77, and Ala78 for the RHS part of compounds 2 and 3 were well maintained as reported earlier for NBTIs. The additional bulky phenoxy sulfonamide did not disturb the positioning of LHS group between the two base pairs of DNA. Additionally, the phenoxy sulfonamide group pointing toward the Asp89 residue may be involved in making additional hydrogen bonding contacts.
Figure 2.
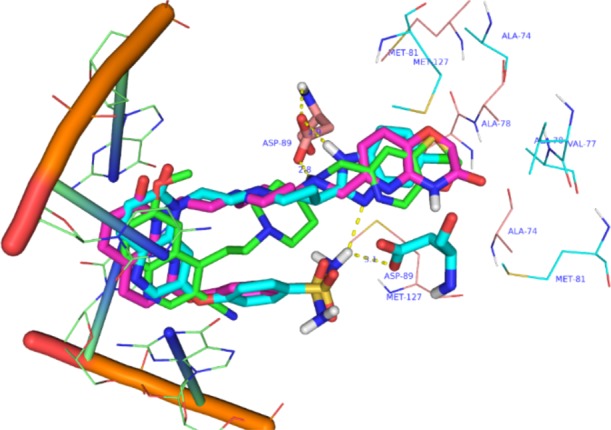
Docking pose of compounds 2 (cyan), 3 (pink), and 1e (green) on to the homology model of Mtb DNA gyrase subunit A.
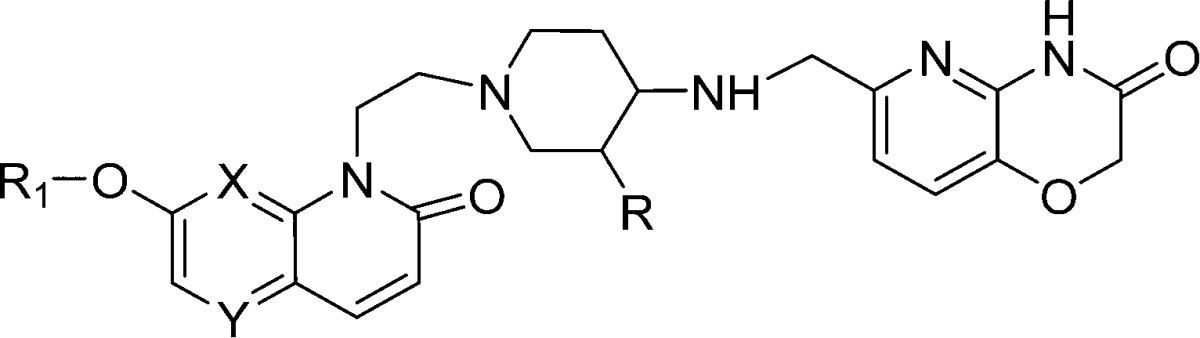
Based on the findings from the SAR study that pyridoxazinonone ring appears to be the best in terms of providing hERG selectivity and ease of synthesis for pyridoxazinone RHS, we fixed pyridoxazinone ring as the RHS and explored a variety of substituents extending from the LHS naphthyridone as well in the linker part to improve and expand the scope of SAR for hERG selectivity. The results of this modifications are presented in Table 3.
Table 3. SAR Exploration at C7-Position of LHS.

Calculated logD.
We hypothesized that the sulphonamide moiety in the R1 ring may pose permeability issues in terms of achieving good oral bioavailability due to increased polarity (high polar surface area) and increased hydrogen bonding group. Indeed, compounds 2 and 3 with sulphonamide group have shown poor Caco-2 permeability (data not shown). Hence, we started exploring the SAR without sulphonamide group in the R1 ring. Replacement of the phenoxy sulphonamide group at the C-7 position of LHS by a 3-pyridyloxy group maintained Mtb MIC and hERG selectivity (match pair: compound 3 vs compound 7). Furthermore, to expand the scope of SAR, we introduced other heterocyclic rings like pyrimidine and isoxazole as a bioisostere of pyridine at the R1 position. These modifications retained hERG selectivity with a 4-fold loss in Mtb MIC (compounds 8 and 9 versus 7, Table 3). A similar trend in Mtb MIC and hERG selectivity was observed, following replacement of a 1,8-naphthyridone LHS with a 1,5-naphthyridone LHS (compounds 10–12, Table 3). The substitution of an aromatic isoxazole ring with a saturated heterocycle like a tetrahydrofuran ring retained the Mtb MIC and hERG selectivity (compound 10 vs 13 and 14, Table 3). Further introduction of flexibility at R1 position through a methylene bridge retained the hERG selectivity, albeit with a 4-fold loss in Mtb MIC compared to direct pyridyloxy compound (compound 7 vs 15–17, Table 3). Overall, the substitutions at the R1 position with a bulky polar group showed good hERG selectivity. This data suggests that R1 substituents at the C-7 position of the LHS ring is amenable for chemical diversity
Fluoro substituted aminopiperidine based NBTIs have shown good oral bioavailability and hERG selectivity as compared to their nonfluoro counterparts.8,9 In order to identify compounds with good oral pharmacokinetic properties, we introduced a fluoro substitution in the linker portion of compounds 10 and 11 to result in compounds 18 and 19, respectively. This modification resulted in marginal loss in hERG selectivity for 19, while retaining the Mtb MIC.
The scatter plot presented in Figure 3 for hERG IC50 suggested that calculated lipophilicity (AZ logD) of <1.5 is ideal for achieving good hERG selectivity for compounds with bulky substitutions at R1 position of LHS ring (green encircled in Figure 3). This trend is not true for compounds with smaller substitutions at R1 positions (blue encircled in Figure 3). This data clearly indicates that apart from logD the polar substituents at R1 position do play a critical role in improving the hERG selectivity.
Figure 3.
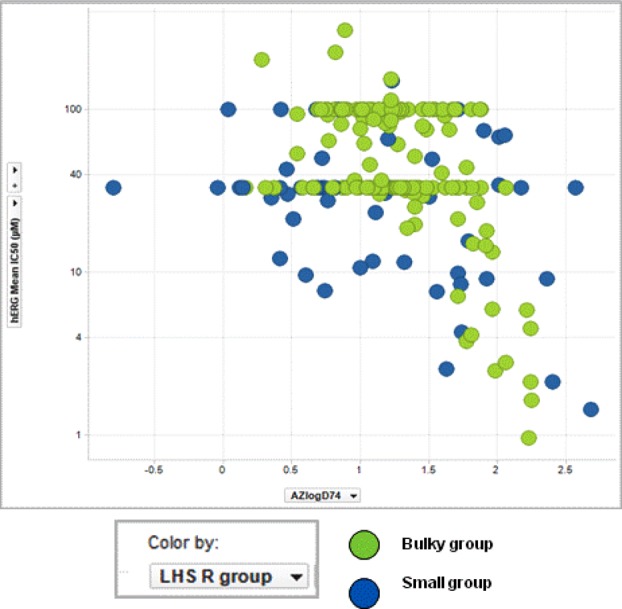
hERG IC50 (−log scale) vs AZ logD.
To link the observed Mtb MICs with gyrase inhibition, we screened representative compounds (7, 8, and 19) in a Mtb DNA gyrase supercoiling assay as described earlier8 (Table 4) as the assay is low throughput. The rationale for selecting compounds 7, 8, and 19 for Mtb DNA supercoiling was based on best hERG IC50 (compounds 7 and 8) and good mouse oral PK profile (compound 19). The data indicates that the observed Mtb MIC for these compounds can be clearly attributed to the observed Mtb gyrase inhibition.
Table 4. Activity of Naphthyridones against Resistant Mutants to Compounds 1a and 1c and Moxifloxacin.
| MIC
(μM) |
|||||
|---|---|---|---|---|---|
| entry | Mtb gyrase SC IC50 (μM) | Mtb H37Rv wild-type | Cpd 1aR mutant (A74V) | Cpd 1cR mutant (D89N) | MoxiR mutant (G88N) |
| 1a | 0.11 | 0.19 | 4 | >100 | <0.19 |
| 1c | 0.25 | 0.19 | 2 | >100 | <0.19 |
| 7 | 0.104 | 0.06 | 8 | >100 | 0.78 |
| 8 | 0.173 | 0.25 | 50 | >100 | 0.06 |
| 19 | 0.80 | 0.25 | 50 | >100 | 0.39 |
| moxifloxacin | 12.5 | 0.13 | 0.25 | 2 | 4 |
| ciprofloxacin | NDa | 0.5 | 0.5 | 16 | 8 |
| isoniazid | NDa | 0.06 | 0.06 | 0.03 | 0.06 |
| rifampicin | NDa | 0.015 | 0.008 | 0.015 | 0.008 |
Not determined.
In order to differentiate naphthyridones from FQs and NBTIs, compounds (7, 8, and 19) were screened against moxifloxacin-resistant and NBTI-resistant mutants of Mtb (1a and 1c mutants, reported earlier).8 Strikingly, these compounds retained their MIC against a laboratory generated moxifloxacin-resistant mutant, but lost their MIC against a NBTI-resistant mutant by >25-fold. This result indicates that this chemical class is likely to be active against FQ-resistant strains of Mtb and that the mechanism of inhibition is similar to that reported for other NBTIs.8−10,13,16
Compounds 10, 11, 18, and 19 exhibited solubility ranging from 665 to >1000 μM and good free plasma fractions. The compound with a fluoro substitution showed better intestinal permeability (Caco-2) as compared to their nonfluoro counterparts (compound 10 vs 18 and 11 vs 19, Table 1a, Supporting Information). Compounds 18 and 19 were selected for mouse oral pharmacokinetic profiling at 80 mg/kg by coadministration with ABT (a pan CYP inhibitor) as the compounds from the series showed high in vitro metabolic clearance in rodents. Compound 19 displayed good oral exposure at 80 mg/kg as compared to compound 11, and this is consistent with the observed intestinal permeability. Compound 19 was profiled for its activity against intracellular Mtb residing in THP-1 macrophages as well as for its activity in an acute murine model of TB following oral administration. In the macrophage model, 19 displayed a dose-dependent cidality, resulting in a 1-log kill of intracellular Mtb (Figure 2a, Supporting Information). Similarly, 19 when dosed orally at 100 mg/kg twice daily for 10 days produced >1-log kill compared to early control in an acute mouse model of TB (Figure 3a, Supporting Information).
In conclusion, exploration of polar bulky substitutions at the C-7 position of naphthyridone ring establishes a structure–activity relationship to mitigate the hERG ion channel binding activity while retaining potent antimycobacterial activity. Representative compounds from naphthyridone series showed bactericidal activity against Mtb in an intracellular macrophage model and displayed in vivo proof of concept in a murine model of TB. The naphthyridone series display good solubility with a good plasma unbound fraction. The ability to retain antimycobacterial activity against a lab-generated moxifloxacin-resistant mutant with a concomitant loss of potency against NBTIs resistant mutant suggests a mechanism of action similar to NBTIs. Further optimization of the scaffold toward improving oral bioavailability and evaluation of in vivo safety is warranted in order to progress the series toward developing a candidate drug to treat tuberculosis.
Acknowledgments
The authors would like to thank Drs. Tanjore Balganesh, Bheemrao Ugarkar, V. Balasubramanian, Pravin Iyer, and Shridhar Narayanan for their valuable suggestions and encouragement during the course of this work. We also thank the Compound Management Group, Bangalore, AZ Global DMPK, Safety, Biosciences (AZ Boston, USA), and Discovery Sciences (AZ, Alderley Park, UK) for technical support in various assays.
Glossary
Abbreviations used
- TB
tuberculosis
- FQ
fluoroquinolone
- LHS
left-hand side
- Mtb
Mycobacterium tuberculosis
- MIC
minimum inhibitory concentration
- SAR
structure–activity relationship
- hERG
ether-a-go-go related gene
- AUC
area under the curve
- cfu
colony forming units
- rt
room temperature
- DME
1,2-dimethoxyethane
- THF
tetrahydrofuran
- DMF
N,N-dimethylformamide
- ABT
aminobenzotriazole
Supporting Information Available
Figures; tables; computational methods; determining the antimycobacterial properties; determination of IC50 for the supercoiling activity of M. tuberculosis H37Rv gyrase holoenzyme; determination of resistance frequency; genetic mapping of mutations conferring resistance to N-linked aminopeperidinyl alkyl quinolones and naphthyridones; assay procedures for log D and hERG measurement; synthetic schemes and procedures. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/ml500531p.
Author Present Address
† BITS-Pilani, Hyderabad campus, Shameerpet, Hyderabad-500 078, India.
Author Contributions
S.H.P., V.K.S., A.R., and S.D. wrote the manuscript and participated in the design and execution of this study. S.H.P., P.M., V.S., J.P., G.S., M.C., and V.P. performed the chemical syntheses. S.S., P.M., N.K., R.N., and V.K.S. designed biological experiments and performed and interpreted biological data. S.R. performed the analytical experiments to assign the structure of newly synthesized compounds.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization. Global tuberculosis report. WHO 2014. http://www.who.int/tb/publications/global_report/en/.
- Zumla A.; Raviglione M.; Hafner R.; von Reyn C. F. Tuberculosis. N. Engl. J. Med. 2013, 368, 745–755. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Multidrug and extensively drug-resistant TB (M/XDR-TB); WHO 2013. http://www.who.int/tb/challenges/mdr/MDR_TB_FactSheet.pdf.
- Champoux J. J. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- Corbett K. D.; Berger J. M. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [DOI] [PubMed] [Google Scholar]
- Mayer C.; Janin Y. L. Non-quinolone inhibitors of bacterial type IIA topoisomerases: a feat of bioisosterism. Chem. Rev. 2014, 114, 2313–2342. [DOI] [PubMed] [Google Scholar]
- Hameed S. P.; Solapure S.; Mukherjee K.; Nandi V.; Waterson D.; Shandil R.; Balganesh M.; Sambandamurthy V. K.; Raichurkar A.; Deshpande A.; Ghosh A.; Awasthy D.; Shanbhag G.; Sheikh G.; McMiken H.; Puttur P.; Reddy J.; Werngren J.; Read J.; Kumar M.; Ramaiah M.; Chinnapattu1 M.; Madhavapeddi P.; Manjrekar P.; Basu R.; Gaonkar S.; Sharma S.; Hoffner S.; Humnabadkar V.; Subbulakshmi V.; Panduga V. Optimization of pyrrolamides as mycobacterial GyrB ATPase inhibitors: Structure–activity relationship and in vivo efficacy in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2013, 58, 6–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hameed P. S.; Patil V.; Solapure S. M.; Sharma U.; Madhavapeddi P.; Raichurkar A.; Murugan Chinnapattu M.; Manjrekar P.; Shanbhag G.; Puttur J.; Shinde V.; Menasinakai S.; Rudrapatana S.; Achar V.; Awasthy D.; Nandishaiah R.; Humnabadkar V.; Ghosh A.; Narayan C.; Ramya V. K.; Kaur P.; Sharma S.; Wirngren J.; Hoffner H.; Panduga V.; Naveen Kumar C. N.; Reddy J.; Mahesh Kumar K. N.; Ganguly S.; Bharath S.; Mukherjee K.; Arora U.; Gaonkar S.; Coulson M.; Waterson D.; Sambandamurthy V. K.; de Sousa S. M. Novel N-linked aminopiperidine based gyrase inhibitors with improved hERG and in vivo efficacy against Mycobacterium tuberculosis. J. Med. Chem. 2014, 57, 4889–4905. [DOI] [PubMed] [Google Scholar]
- Reck F.; Alm R.; Brassil P.; Newman J.; DeJonge B.; Eyermann C. J.; Breault G.; Breen J.; Comita-Prevoir J.; Cronin M.; Davis H.; Ehmann D.; Galullo V.; Geng B.; Grebe T.; Morningstar M.; Walker P.; Hayter B.; Fisher S. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: broad-spectrum antibacterial agents with reduced hERG activity. J. Med. Chem. 2011, 54, 7834–7847. [DOI] [PubMed] [Google Scholar]
- Reck F.; Alm R.; Brassil P.; Newman J.; Ciaccio P.; McNulty J.; Barthlow H.; Goteti K.; Breen J.; Comita-Prevoir J.; Cronin M.; Ehmann D.; Geng B.; Godfrey A.; Fisher S. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II with reduced pKa: antibacterial agents with an improved safety profile. J. Med. Chem. 2012, 55, 6916–6933. [DOI] [PubMed] [Google Scholar]
- Duong D. A.; Nguyen T. H.; Nguyen T. N.; Dai V. H.; Dang T. M.; Vo S. K.; Do D. A.; Nguyen V. V.; Nguyen H. D.; Dinh N. S.; Farrar J.; Caws M. Beijing genotype of Mycobacterium tuberculosis is significantly associated with high-level fluoroquinolone resistance in Vietnam. Antimicrob. Agents Chemother. 2009, 53, 4835–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziganshina L. E.; Titarenko A. F.; Davies G. R. Fluoroquinolones for treating tuberculosis (presumed drug-sensitive). Cochrane Database Syst. Rev. 2013, 6, 1–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax B. D.; Chan P. F.; Eggleston D. S.; Fosberry A.; Gentry D. R.; Gorrec F.; Giordano I.; Hann M. M.; Hennessy A.; Hibbs M.; Huang J.; Jones E.; Jones J.; Brown K. K.; Lewis C. J.; May E. W.; Saunders M. R.; Singh O.; Spitzfaden C. E.; Shen C.; Shillings A.; Theobald A. J.; Wohlkonig A.; Pearson N. D.; Gwynn M. N. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [DOI] [PubMed] [Google Scholar]
- Mitton-Fry M. J.; Brickner S. J.; Hamel J. C.; Brennan L.; Casavant J. M.; Chen M.; Chen T.; Ding X.; Driscoll J.; Hardink J.; Hoang T.; Hua E.; Huband M. D.; Maloney M.; Marfat A.; McCurdy S. P.; McLeod D.; Plotkin M.; Reilly U.; Robinson S.; Schafer J.; Shepard R. M.; Smith J. F.; Stone G. G.; Subramanyam C.; Yoon K.; Yuan W.; Zaniewski R. P.; Zook C. Novel quinoline derivatives as inhibitors of bacterial DNA gyrase and topoisomerase IV. Bioorg. Med. Chem. Lett. 2013, 23, 2955–2961. [DOI] [PubMed] [Google Scholar]
- Surivet J. P.; Zumbrunn C.; Rueedi G.; Hubschwerlen C.; Bur D.; Bruyère T.; Locher H.; Ritz D.; Keck W.; Seiler P.; Kohl C.; Gauvin J. C.; Mirre A.; Kaegi V.; Dos Santos M.; Gaertner M.; Delers J.; Enderlin-Paput M.; Boehme M. Design, synthesis, and characterization of novel tetrahydropyran-based bacterial topoisomerase inhibitors with potent anti-Gram-positive activity. J. Med. Chem. 2013, 56, 7396–7415. [DOI] [PubMed] [Google Scholar]
- Hameed P. S.; Raichurkar A.; Madhavapeddi P.; Menasinakai S.; Sharma S.; Kaur P.; Nandishaiah R.; Panduga V.; Reddy J.; Sambandamurthy V. K.; Sriram D. Benzimidazoles: novel mycobacterial gyrase inhibitors from scaffold morphing. ACS Med. Chem. Lett. 2014, 5, 820–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.