

Genetic and epigenetic studies of gene variants reveal a potential genomic target for treating hemoglobin disorders.
Disorders of hemoglobins are the most common monogenic diseases in the world, with substantial morbidity and mortality resulting from either defective function of the protein, such as in sickle cell anemia, or from insufficient protein production, such as the thalassemias (1). Genome-wide association studies (GWAS) have implicated two genes other than the globin genes as potential modulators of the pathology of these diseases by influencing the amounts of fetal hemoglobin (HbF). On page XXX in this issue, Bauer et al. (2) characterize common single-nucleotide polymorphisms (SNPs) in one of these genes, BCL11A. SNPs associated with mild increases in HbF amounts reside within a powerful tissue- and developmental stage-specific BCL11A enhancer (see the figure). Genome engineering reveals that this enhancer is essential for erythroid expression of BCL11A and as a consequence, for globin gene expression. This exquisite specificity points to genome editing as a plausible approach to lasting corrective cell-specific therapy for certain hemoglobinopathies.
Figure. Exploiting nature’s variants.
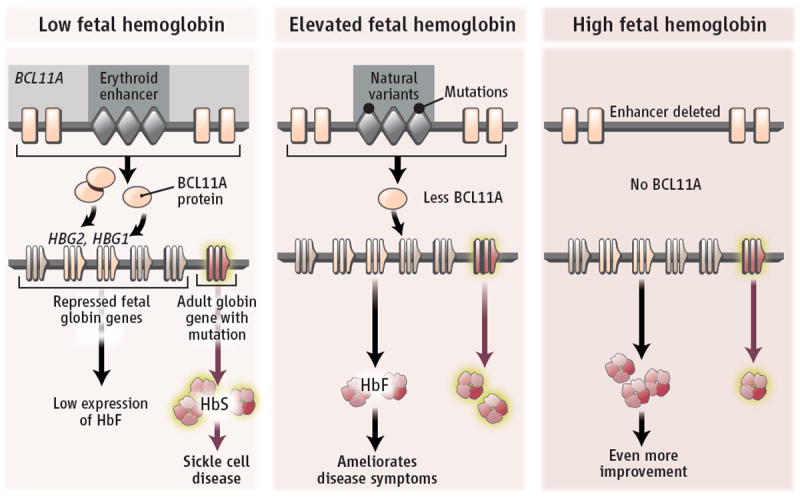
(Left) An erythroid enhancer promotes expression of BCL11A, which encodes a repressor that silences fetal globin genes HBG2 and HBG1 in adult human erythroid cells. The globin gene HBB is expressed in adult erythroid cells, but if mutated, disease can ensue. (Middle) Natural variants in the enhancer reduce BCL11A production and boost fetal globin gene expression. Production of fetal hemoglobin (HbF) can ameliorate symptoms of some hemoglobinopathies. (Right) Removal of the enhancer is expected to reduce the amount of BCL11A, thereby allowing expression of the fetal globin genes and improving the pathologies from HBB mutations.
The first disease-linked mutation defined at the molecular level was the amino acid substitution in β-globin that leads to sickle cell anemia (3). Since then, genetic alterations have been identified as underlying causes for diverse congenital hemoglobinopathies. The search for therapies based on genetic insights has driven investigation of the genetics and regulatory machinery of the globin genes. Most vertebrates produce different forms of hemoglobin during development, and humans produce a fetal stage-specific form called HbF. The concentration of HbF normally declines after birth, but the amount of HbF persisting in normal adults is a variable trait with strong heritability. Elevated HbF amounts can attenuate the symptoms in patients with sickle cell disease or thalassemias (4). GWAS initially identified two quantitative trait loci, not linked to the β-like globin genes, that determine the amount of HbF produced in normal populations (5, 6). One of these loci encodes BCL11A. Notably, ~15% of the variation in HbF in sickle cell anemia populations is accounted for by SNPs in BCL11A, which is high compared to most GWAS. BCL11A is a transcription factor that represses embryonic and fetal β-like globin transcription in human and mouse erythroid cells, and is a dominant regulator of developmental globin gene expression (7, 8). Bcl11A loss ameliorates sickle cell anemia in mouse models of the disease (9).
Bauer et al. show that a region within an intron of the BCL11A gene has epigenetic signatures indicative of a transcriptional enhancer, as reflected in hypersensitivity to deoxyribonuclease, histone modifications associated with active enhancers, and physical juxtaposition with the BCL11A promoter regions through chromatin looping. SNPs with the most pronounced effects on HbF concentration fall at or near these critical regulatory sequences, and in one case impairs binding of erythroid transcription factors. In transgenic mice this enhancer drives expression of a reporter gene predominantly in the fetal liver, the initial site of adult-type erythropoiesis in the developing mouse. Its deletion from the Bcl11a gene in a murine erythroid cell line, but not in a lymphocyte cell line, impaired BCL11A production, accompanied by an increase in embryonic globin expression, thus confirming the erythroid specificity of the enhancer.
More broadly, the results of Bauer et al. inform our understanding and expectations of the role of regulatory variants in complex traits, including disease susceptibility. Trait-associated variants discovered in GWAS are enriched in noncoding DNA segments carrying epigenetic hallmarks of regulatory regions (10, 11). However, most variants have modest effects on the phenotype, as is the case for the SNPs at the BCL11A locus. Yet, examination of the genomic context of these trait-associated variants has led to the identification of an enhancer with a powerful effect on expression of BCL11A and, indirectly, the globin genes. Thus, the modest phenotypic effects of the common variants mapped in GWAS need not be interpreted as indicative of modest effects of the regulatory region or locus in which they reside. Rather, they should be considered as components of a regulatory complex that in total could have strong effects on phenotype.
Gene replacement therapy for the hemoglobinopathies has proven to be an extraordinarily difficult prospect (12). An alternative strategy to raise HbF concentrations would be to reduce BCL11A amounts or activity. However, BCL11A is widely expressed, and its loss is lethal due to nonerythoid effects (13). These considerations and the generally undruggable nature of transcription factors led to the initial view of BCL11A as an implausible target for therapeutic intervention. The study of Bauer et al., however, raises the possibility of crippling the erythroid enhancer through genome editing in human hematopoietic stem or progenitor cells. Cells thus modified would be expected to display loss of BCL11A expression in erythroid cells while maintaining it in nonerythroid lineages. The potential benefits would be an increase in HbF production; a reciprocal decrease in expression of the defective adult globin in the case of sickle cell anemia; permanence of a one-time genetic deletion compared to gene replacement therapies that require long-term sustained expression of a transgene; and a selective growth advantage of modified erythroid cells over diseased cells.
Advances in genome editing (14, 15) are moving enhancer modifications into the arsenal of gene therapy approaches. However, additional work in animal models and primary human cells is needed to confirm and extend the characterization of the enhancer with emphasis on measuring effects on other tissues or genes. Further improvements in genome editing are essential to maximize efficiency and minimize off-target genomic alterations. Despite these challenges, the results of Bauer et al. suggest that genomic modification of an erythroid enhancer might ameliorate hemogobinopathies and the old dream of genetically guided therapies for this disease spectrum may become a reality.
Acknowledgments
We thank V. Sankaran for helpful comments. Supported by grants R37DK058044 and R01DK054937 (G.A.B.) and R01DK065806 and U54HG006998 (R.C.H.).
Contributor Information
Ross C. Hardison, Email: rch8@psu.edu.
Gerd A. Blobel, Email: blobel@email.chop.edu.
References and Notes
- 1.Williams TN, Weatherall DJ. Cold Spring Harbor Perspect Med. 2012;2:a011692. doi: 10.1101/cshperspect.a011692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauer DE, et al. Science. 2013;242 PP. [Google Scholar]
- 3.Ingram VM. Nature. 1956;178:792. doi: 10.1038/178792a0. [DOI] [PubMed] [Google Scholar]
- 4.Nagel RL. Semin Hematol. 1991;28:180. [PubMed] [Google Scholar]
- 5.Menzel S, et al. Nat Genet. 2007;39:1197. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 6.Uda M, et al. Proc Natl Acad Sci U S A. 2008;105:1620. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sankaran VG, et al. Science. 2008;322:1839. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 8.Sankaran VG, et al. Nature. 2009;460:1093. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, et al. Science. 2011;334:993. doi: 10.1126/science.1211053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The ENCODE Project Consortium. Nature. 2012;489:57. [Google Scholar]
- 11.Maurano MT, et al. Science. 2012;337:1190. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavazzana-Calvo M, et al. Nature. 2010;467:318. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu P, et al. Nat Immunol. 2003;4:525. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- 14.Boch J, et al. Science. 2009;326:1509. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 15.Mali P, et al. Science. 2013;339:823. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]