

Abstract
The kinase MEKK2 (MAP3K2) may play an important role in tumor growth and metastasis for several cancer types. Thus, targeting MEKK2 may represent a novel strategy for developing more effective therapies for cancer. In order to identify small molecules with MEKK2 inhibitory activity, we screened a collection of known kinase inhibitors using a high throughput MEKK2 intrinsic ATPase enzyme assay and confirmed activity of the most potent hits with this primary assay. We also confirmed activities of these known kinase inhibitors with an MEKK2 transphosphorylation slot blot assay using MKK6 as a substrate. We observed a good correlation in potencies between the two orthogonal MEKK2 kinase activity assay formats for this set of inhibitors. We report that ponatinib, AT9283, AZD7762, JNJ-7706621, PP121 and hesperadin had potent MEKK2 enzyme inhibitory activities ranging from 4.7 – 60 nM IC50. Ponatinib is an FDA-approved drug that potently inhibited MEKK2 enzyme activity with IC50 values of 10 – 16 nM. AT9283 is currently in clinical trials and produced MEKK2 IC50 values of 4.7 – 18 nM. This set of known kinase inhibitors represents some of the most potent in vitro MEKK2 inhibitors reported to date and may be useful as research tools. Although these compounds are not selective for MEKK2, the structures of these compounds give insight into pharmacophores that potently inhibit MEKK2 and could be used as initial leads to design highly selective inhibitors of MEKK2.
Keywords: MEKK2, MAP3K2, kinase, inhibitor, ponatinib, AT9283
Introduction
The ERK5 signaling pathway is one of the mitogen-activated protein kinases (MAPK) signaling pathways that translate extra-cellular stimuli into gene expression changes in the cell [1,2]. This pathway is activated in response to oxidative stress, hyperosmolarity, and growth factors, including epidermal growth factor (EGF) [3]. ERK5 is activated through phosphorylation by the MAPK kinase Mek5 (MAP2K5). Mek5 in turn is activated through phosphorylation by the MAPK kinase kinases MEKK2 (MAP3K2) and MEKK3 (MAP3K3). How MEKK2 is activated by external stimuli is still not fully understood, but MEKK2 autophosphorylates in response to stimuli and this autophosphorylation is required for activation of MEKK2 [4]. MEKK2 also activates the JNK MAPK pathway via phosphorylation of the MAP2K MEK7 which activates JNK [5,6]. MEKK2 knock-out mice displayed normal development and fertility [5,7]. In contrast, disruption of the MEKK3 gene in mice results in embryonic lethality due to cardiac development defects [8]. Thus, MEKK2 and MEKK3 integrate different stimuli. MEKK2 has been reported to be activated by cell attachment to fibronectin and to subsequently localize to focal adhesions. MEKK2 knockdown in breast cancer cell lines was shown to stabilize focal adhesions and inhibit cell migration in vitro [9,10].
The role of the MEKK2 in cancer has only relatively recently been explored. In one study linking MEKK2 to cancer, MEKK2 was expressed at 4.4-fold higher level in prostate cancer tissue versus benign tissue [11]. Even higher levels of MEKK2 were observed in prostate cancer cell lines. The microRNA miR-520b suppresses tumor formation in breast cancer and hepatocellular carcinoma cells by targeting MEKK2 and Cyclin D1 [12]. Knock-down of only MEKK2 expression was able to inhibit the growth of hepatocarcinoma cells in vitro and in vivo. The methylation of MEKK2 by SMYD3 was shown to increase MAP kinase signaling and promote the formation of Ras-dependent carcinomas [13]. Restoration of the expression of the tumor suppressor miR17/20a was shown to enhance tumor cell sensitivity to natural killer cell activity through suppressing MEKK2 [14]. A survey of primary colorectal cancer (CRC) lesions for MEKK2 expression level by western blotting indicated that MEKK2 was highly expressed in CRC compared to normal mucosa [15]. We used a mouse xenograft model of breast cancer to assess the role of MEKK2 in tumor growth and metastasis [16]. Using the breast cancer cell line MDA-MB-231, we observed that shRNA-mediated knockdown of MEKK2 inhibited activation of ERK5 in response to EGF. Knockdown of MEKK2 expression had no observable impact on the growth of the cells in culture, but strongly inhibited both tumor growth and metastasis in the animal model. The tumors formed by the MEKK2 knock-down cells had increased apoptosis compared to size-matched control tumors. MEKK2 shRNA knockdown in the BT474 breast cancer cell line also resulted in reduced tumor growth in vivo. Since ERK5 is activated by MEKK2 (via activation of MEK5), we determined whether shRNA knockdown of ERK5 in MDA-MB-231 cells would show similar inhibition of tumor growth and metastasis as the MEKK2 knockdown. The knockdown of ERK5 in these cells inhibited their ability to metastasize without significantly impacting tumor growth. Thus, MEKK2-mediated activation of ERK5 appeared to regulate metastasis, while another MEKK2-dependent pathway, possibly the JNK pathway, was important for tumor growth.
Overexpression or over-activation of ERK5 protein in humans coupled with in vitro and animal model data has begun to implicate a role for ERK5 in breast and prostate cancer as well as neuroblastoma, myeloma and oral squamous cell carcinoma [17,18,19,20,21]. The discovery of a potent and selective small molecule ERK5 inhibitor has been reported that inhibited tumor growth in mouse models of cancer [22]. MEK5 may also play a role in tumor development. Elevated tissue expression of MEK5 correlated with bone metastasis and poor prognosis in cases of prostate cancer and benign prostatic hypertrophy [23]. Two relatively selective MEK5 inhibitors have been reported, but no in vivo efficacy data were shown [24].
Taken together, literature data supports MEKK2 as a novel drug target for certain cancers. Targeting MEKK2 may be advantageous over inhibiting single MAPK pathways since an MEKK2 inhibitor may blunt activation of both the Erk5 and JNK pathways leading to enhanced anti-tumor efficacy. However, no specific and potent small molecule inhibitors of MEKK2 have been reported to date. Even without regard to selectivity, few potent inhibitors have been reported for MEKK2. Herein, we report the identification of 6 well-characterized kinase inhibitors that have previously un-reported potent MEKK2 inhibitory activity.
Materials and Methods
Materials
Common reagents such as HEPES, MgCl2 and dimethyl sulfoxide (DMSO) were reagent grade quality and obtained from Sigma-Aldrich (St. Louis, MO) or Thermo Fisher Scientific (Waltham, MA). The kinase inhibitor library (cat #L1200) and individual kinase inhibitors for confirmation were obtained from Selleck Chemicals (Houston, TX).
MEKK2 Enzyme Activity Assays
The MEKK2 intrinsic ATPase activity assay and the transphosphorylation assay were performed as previously described [25]. MEKK2 activity assays were performed using ATP concentrations at the apparent Km for ATP of the assay format. The Km for ATP was previously reported for the ATPase assay to be 34 uM and therefore the assay employed 30 uM ATP. The Km for ATP in the transphosphorylation assay was previously reported to be 3.3 uM and therefore we used 3 uM ATP for this assay.
IC50 value determinations
IC50 was defined as the concentration of inhibitor that generates a 50% reduction in the specific signal of the assay. Compounds were dissolved at 10 mM in 100% DMSO to make stock solutions. Serial dilutions of compounds were performed in 100% DMSO then subsequently diluted into assay buffer and used in the MEKK2 enzymatic assay. The slot blot transphosphorylation digital bands were quantified using software on the Kodak 4000R Pro imaging station. For both MEKK2 assay formats, compound concentration response curves were generated using data points that represent the average of 2 or 3 determinations per concentration and 10 compound concentrations tested. All IC50 values provided are averages of at least three (ATPase assay) or two (transphosphorylation assay) independent determinations. The IC50 values were calculated from concentration response data using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA) employing either a four- or three-parameter curve fit.
Results and Discussion
We have previously reported the development and validation of a novel intrinsic ATPase activity assay for MEKK2 and demonstrated its utility as a high throughput assay for the discovery of small molecule inhibitors of MEKK2 [25]. This assay takes advantage of intrinsic ATPase activity of MEKK2 wherein MEKK2 alone, in the absence of any protein substrates, converts ATP to ADP. After stopping the reaction, relative ADP levels were then measured, with the ADP-Glo kit (Promega, Inc.). We have used this assay to screen a commercial library of 195 known and well-characterized kinase inhibitors. We chose the 7 most potent hits for further confirmation. Bosutinib was one these top hits, but we have previously reported it to have potent activity against MEKK2, with an IC50 of 59 nM [25]. We report the identification of 6 other known kinase inhibitors with previously unknown potent MEKK2 inhibitory activity. These kinase inhibitors and their IC50 values (nM, ±SD) in the MEKK2 ATPase assay were ponatinib (16 ± 3), AT9283 (18 ± 10), AZD7762 (20 ± 7), JNJ-7706621 (24 ± 8), PP121 (30 ± 16) and hesperidin (60 ± 25) (Fig. 1 and Table 1). Average Hill slope values of the dose response curves for each compound were close to the expected value of 1.0, ranging from 0.85 to 1.18.
Figure 1.

Inhibitory activity of 6 known kinase inhibitors in the MEKK2 intrinsic ATPase assay. The compounds were tested at the indicated concentrations with the resultant raw signal normalized to maximum (DMSO only) and minimum (no MEKK2) controls, representing 100% and 0% enzyme activity, respectively. Data points represent the average of two determinations per concentration and error bars represent standard deviation (SD). This plot is representative of three independent experiments.
Table 1.
Known Kinase Inhibitors with Potent MEKK2 Inhibitory Activity
| Compound | Structure | Intrinsic ATPase Assay IC50 ± SDa |
Transphos. Assay IC50 ± SDa |
Reported Targets |
Current Status |
|---|---|---|---|---|---|
| Ponatinib | 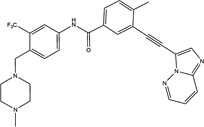 |
16 ± 3 | 10 ± 4 | Abl, Abl(T315I), FLT3, FGFR1-4, VEGFR | FDA-approved |
| AT9283 | 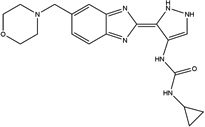 |
18 ± 10 | 4.7 ± 0.9 | Aurora A/B, JAK2/3, Abl(T315I), | Clinical Trials, Phase II |
| AZD7762 | 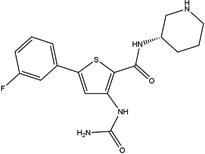 |
20 ± 7 | 14 ± 2 | Chk1/2, 9 CAMKs, 20 TKs | Terminated in Phase I Clinical Trials |
| JNJ-7706621 | 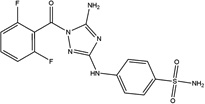 |
24 ± 8 | 17 ± 2 | CDK1,2,3, Aurora A/B | Research Tool |
| PP121 | 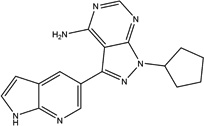 |
30 ± 16 | 7 ± 0.3 | Multi-targeted by Design, TKs, PI3Ks | Research Tool |
| Hesperadin | 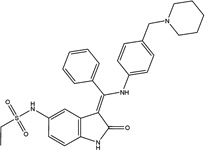 |
60 ± 25 | 34 ± 11 | Aurora B | Research Tool |
IC50 value determinations were performed with n=3 for the ATPase assay and n=2 for the transphosphorylation assay, with average IC50 values provided with standard deviations (SD).
Since the ATPase kinase activity format is still novel, a completely different transphosphorylation MEKK2 assay format was used to confirm the activity observed in the ATPase assay. In this MEKK2 activity assay, native transphosphorylation activity of MEKK2 was measured using mutant kinase-inactive MKK6 (MAP2K6) as a substrate [25]. Enzyme reactions were performed in a 96-well plate format, stopped and applied in diluted form (to dilute out detergent which prevents membrane binding) to membrane by using a slot blot apparatus. The site-specific phosphorylation of MKK6 was detected by an anti-phosphoMKK6 antibody and western blot processing. An image of a typical slot blot obtained with this method is provided as an example of the data obtained with this unique transphosphorylation assay (Fig. 2A). The intensity of the bands was quantified from software built into the image reader and used to create dose response graphs and obtain IC50 values. The IC50 values (nM, ±SD) obtained in the MEKK2 transphosphorylation assay were as follows: ponatinib (10 ± 4), AT9283 (4.7 ± 0.9), AZD7762 (14 ± 2), JNJ-7706621 (17 ± 2), PP121 (7 ± 0.3) and hesperadin (34 ± 11) (Fig. 2B and Table 1). Average Hill slope values of the dose response curves for each compound in this assay were close to the expected value of 1.0, ranging from 0.85 to 1.46. In comparing the data from the two different assay formats, there was less than a 2-fold difference between potencies obtained with the ATPase assay and those obtained with the transphosphorylation assay for 4 of the compounds and approximately a 4-fold difference for two of the compounds. Thus, all of these inhibitors confirmed activity in both MEKK2 assay formats. This comparison also strengthens the validation of the MEKK2 intrinsic ATPase assay as a homogeneous method of testing compounds for MEKK2 inhibitory activity.
Figure 2.

Inhibitory activity of 6 known kinase inhibitors in the MEKK2 transphosphorylation slot blot assay. (A) MEKK2 enzyme reactions using MKK6 as a substrate were stopped, transferred to a membrane via a slot blot apparatus and probed with phosphoMKK6-specific antibody detected by chemiluminescence. A representative slot blot image is shown with bands labeled as max (DMSO) and min (no MEKK2) controls and compound serial dilution. (B) The resultant digital image band intensities were converted to numerical data for IC50 determination. The compounds were tested at the indicated concentrations with the resultant raw signal normalized to maximum (DMSO only) and minimum (no MEKK2) controls, representing 100% and 0% enzyme activity, respectively. Data points represent the average of two determinations per concentration and error bars represent standard deviation (SD). This plot is representative of two independent experiments.
Ponatinib (AP24534, Iclusig) is a Food and Drug Administration (FDA)-approved drug with an approved indication for chronic myeloid leukemia [26]. Ponatinib was originally designed as an inhibitor of Abl (IC50 = 8.6 nM) and clinically relevant mutants of Abl, including Abl (T315I) (IC50 = 40 nM), in order to prevent drug resistance from developing [27]. It is also a multi-targeted tyrosine kinase inhibitor with low nM potency against FLT3, FGFR and VEGFR. In fact, it has been reported to be a potent pan-FGFR inhibitor with activity in animal models of FGFR-dependent cancers [28]. Remarkably, the IC50 values for inhibiting MEKK2 reported here for ponatinib are similar to the potency of ponatinib against its original target, Abl. This suggests the possibility that ponatinib could inhibit MEKK2 in patients and thus may be repurposed for cancers that are dependent on MEKK2 signaling. However, the potency of ponatinib to inhibit MEKK2 activity in cells would first need to be established.
AT9283 is a multi-targeted kinase inhibitor that has been reported to potently inhibit Aurora A and B (IC50 = 3 nM), JAK2/3 (IC50 = 1 nM), Abl (T315I) mutant (IC50 = 4 nM) and 10 other kinases (1–10 nM) [29]. This compound has been tested in multiple phase I clinical trials in humans with advanced solid tumors and non-Hodgkin’s lymphoma and it appears to be well tolerated [30,31,32]. Phase II clinical trials with AT9283 are ongoing. This is the first report of AT9283 having potent MEKK2 inhibitory activity. The potency in our in vitro assays against MEKK2 (4.7 and 18 nM) suggests the possibility that AT9283 may inhibit MEKK2 in cell-based assays and in vivo.
AZD7762 was developed to target Chk1/2 (IC50s = 5 nM), but also potently inhibits other kinases including 9 members of the CAM kinase family and 20 tyrosine kinases [33]. AZD7762 entered Phase I clinical trials, but its development was terminated due to unpredictable cardiac toxicity [34]. JNJ-7706621 is a research tool compound that was reported to be a pan-CDK inhibitor, but it most potently inhibited CDK1, CDK2, CDK3, AuroraA and AuroraB with IC50s ≤15 nM [35]. Reported profiling data on JNJ-7706621 in single point determinations indicate that this compound also inhibits many other kinases [36]. A systematic effort to discover inhibitors that potently inhibit both TKs and PI3Ks led to the discovery of the novel multitargeted tool inhibitor PP121 [37]. This broadly active compound blocked the proliferation of tumor cells [38]. Hesperadin was discovered to be a potent inhibitor of chromosome alignment and segregation [39]. This research tool compound was also shown to be an inhibitor of Aurora B (IC50 = 250 nM) and other kinases [40]. The structures of this set of kinase inhibitors are fairly diverse, except that 4 of them have in common an indole-like motif (Table 1). Also, five of these compounds have a 5-member heterocycle as part of the core of the structure.
In sum, we report the identification and confirmation of the FDA-approved drug ponatinib and 5 other known kinase inhibitors with potent in vitro MEKK2 inhibitory activity using two different MEKK2 enzymatic assay formats. Other than staurosporine-related molecules, this group of inhibitors represents some of the most potent MEKK2 enzyme assay inhibitors reported to date. These compounds may be useful tools in MEKK2 enzyme assays and possibly MEKK2-dependent cell-based assays. The ability of these compounds to inhibit MEKK2 function in cells will be assessed in future studies. Furthermore, these scaffolds may provide starting points for future structure-activity relationship (SAR) studies towards the design of highly MEKK2-selective inhibitor probes.
Supplementary Material
Highlights.
We screened a collection of known kinase inhibitors for inhibitors of MEKK2.
Six compounds confirmed in MEKK2 ATPase and transphosphorylation enzyme assays.
Good correlation observed between the two orthogonal MEKK2 activity assay formats.
Ponatinib and clinical candidate AT9283 inhibited MEKK2 with IC50 of 5–18 nM.
Compounds are among the most potent in vitro MEKK2 inhibitors reported to date.
Acknowledgements
This work was supported by a grant from NIH/NCI (U54CA156735, JES and GLJ) and in part by funds from the Golden LEAF Foundation and the State of North Carolina.
Abbreviations used
- MAPK
mitogen-activated protein kinase
- EGF
epidermal growth factor
- DMSO
dimethyl sulfoxide
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- SAR
structure-activity relationship
- SD
standard deviation
- FDA
Food and Drug Administration
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Whitmarsh AJ. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim Biophys Acta. 2007;1773:1285–1298. doi: 10.1016/j.bbamcr.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 3.Drew BA, Burow ME, Beckman BS. MEK5/ERK5 pathway: the first fifteen years. Biochim Biophys Acta. 2012;1825:37–48. doi: 10.1016/j.bbcan.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng J, Yu L, Zhang D, Huang Q, Spencer D, Su B. Dimerization through the catalytic domain is essential for MEKK2 activation. J Biol Chem. 2005;280:13477–13482. doi: 10.1074/jbc.M414258200. [DOI] [PubMed] [Google Scholar]
- 5.Garrington TP, Ishizuka T, Papst PJ, Chayama K, Webb S, Yujiri T, Sun W, Sather S, Russell DM, Gibson SB, Keller G, Gelfand EW, Johnson GL. MEKK2 gene disruption causes loss of cytokine production in response to IgE and c-Kit ligand stimulation of ES cell-derived mast cells. EMBO J. 2000;19:5387–5395. doi: 10.1093/emboj/19.20.5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kesavan K, Lobel-Rice K, Sun W, Lapadat R, Webb S, Johnson GL, Garrington TP. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. J Cell Physiol. 2004;199:140–148. doi: 10.1002/jcp.10457. [DOI] [PubMed] [Google Scholar]
- 7.Guo Z, Clydesdale G, Cheng J, Kim K, Gan L, McConkey DJ, Ullrich SE, Zhuang Y, Su B. Disruption of Mekk2 in mice reveals an unexpected role for MEKK2 in modulating T-cell receptor signal transduction. Mol Cell Biol. 2002;22:5761–5768. doi: 10.1128/MCB.22.16.5761-5768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J, Boerm M, McCarty M, Bucana C, Fidler IJ, Zhuang Y, Su B. Mekk3 is essential for early embryonic cardiovascular development. Nat Genet. 2000;24:309–313. doi: 10.1038/73550. [DOI] [PubMed] [Google Scholar]
- 9.Ameka M, Kahle MP, Perez-Neut M, Gentile S, Mirza AA, Cuevas BD. MEKK2 regulates paxillin ubiquitylation and localization in MDA-MB 231 breast cancer cells. Biochem J. 2014;464:99–108. doi: 10.1042/BJ20140420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mirza AA, Kahle MP, Ameka M, Campbell EM, Cuevas BD. MEKK2 regulates focal adhesion stability and motility in invasive breast cancer cells. Biochim Biophys Acta. 2014;1843:945–954. doi: 10.1016/j.bbamcr.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cazares LH, Troyer D, Mendrinos S, Lance RA, Nyalwidhe JO, Beydoun HA, Clements MA, Drake RR, Semmes OJ. Imaging mass spectrometry of a specific fragment of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 2 discriminates cancer from uninvolved prostate tissue. Clin Cancer Res. 2009;15:5541–5551. doi: 10.1158/1078-0432.CCR-08-2892. [DOI] [PubMed] [Google Scholar]
- 12.Zhang W, Kong G, Zhang J, Wang T, Ye L, Zhang X. MicroRNA-520b inhibits growth of hepatoma cells by targeting MEKK2 and cyclin D1. PLoS One. 2012;7:e31450. doi: 10.1371/journal.pone.0031450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazur PK, Reynoird N, Khatri P, Jansen PW, Wilkinson AW, Liu S, Barbash O, Van Aller GS, Huddleston M, Dhanak D, Tummino PJ, Kruger RG, Garcia BA, Butte AJ, Vermeulen M, Sage J, Gozani O. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014;510:283–287. doi: 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang H, Wang P, Li X, Wang Q, Deng ZB, Zhuang X, Mu J, Zhang L, Wang B, Yan J, Miller D, Zhang HG. Restoration of miR17/20a in solid tumor cells enhances the natural killer cell antitumor activity by targeting Mekk2. Cancer Immunol Res. 2014;2:789–799. doi: 10.1158/2326-6066.CIR-13-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang L, Huang M, Wang L, Fan X, Wang P, Wang D, Fu X, Wang J. Overexpression of MEKK2 is associated with colorectal carcinogenesis. Oncol Lett. 2013;6:1333–1337. doi: 10.3892/ol.2013.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cronan MR, Nakamura K, Johnson NL, Granger DA, Cuevas BD, Wang JG, Mackman N, Scott JE, Dohlman HG, Johnson GL. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene. 2011 doi: 10.1038/onc.2011.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montero JC, Ocana A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparis-Ogando A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One. 2009;4:e5565. doi: 10.1371/journal.pone.0005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carvajal-Vergara X, Tabera S, Montero JC, Esparis-Ogando A, Lopez-Perez R, Mateo G, Gutierrez N, Parmo-Cabanas M, Teixido J, San Miguel JF, Pandiella A. Multifunctional role of Erk5 in multiple myeloma. Blood. 2005;105:4492–4499. doi: 10.1182/blood-2004-08-2985. [DOI] [PubMed] [Google Scholar]
- 19.Clape C, Fritz V, Henriquet C, Apparailly F, Fernandez PL, Iborra F, Avances C, Villalba M, Culine S, Fajas L. miR-143 interferes with ERK5 signaling, and abrogates prostate cancer progression in mice. PLoS One. 2009;4:e7542. doi: 10.1371/journal.pone.0007542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCracken SR, Ramsay A, Heer R, Mathers ME, Jenkins BL, Edwards J, Robson CN, Marquez R, Cohen P, Leung HY. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene. 2008;27:2978–2988. doi: 10.1038/sj.onc.1210963. [DOI] [PubMed] [Google Scholar]
- 21.Sticht C, Freier K, Knopfle K, Flechtenmacher C, Pungs S, Hofele C, Hahn M, Joos S, Lichter P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC) Neoplasia. 2008;10:462–470. doi: 10.1593/neo.08164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR, 3rd, Gray NS, Lee JD. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010;18:258–267. doi: 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, Leung HY. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene. 2003;22:1381–1389. doi: 10.1038/sj.onc.1206154. [DOI] [PubMed] [Google Scholar]
- 24.Tatake RJ, O'Neill MM, Kennedy CA, Wayne AL, Jakes S, Wu D, Kugler SZ, Jr, Kashem MA, Kaplita P, Snow RJ. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophys Res Commun. 2008;377:120–125. doi: 10.1016/j.bbrc.2008.09.087. [DOI] [PubMed] [Google Scholar]
- 25.Ahmad S, Hughes MA, Johnson GL, Scott JE. Development and validation of a high-throughput intrinsic ATPase activity assay for the discovery of MEKK2 inhibitors. J Biomol Screen. 2013;18:388–399. doi: 10.1177/1087057112466430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prasad V, Mailankody S. The accelerated approval of oncologic drugs: lessons from ponatinib. JAMA. 2014;311:353–354. doi: 10.1001/jama.2013.284531. [DOI] [PubMed] [Google Scholar]
- 27.Huang WS, Metcalf CA, Sundaramoorthi R, Wang Y, Zou D, Thomas RM, Zhu X, Cai L, Wen D, Liu S, Romero J, Qi J, Chen I, Banda G, Lentini SP, Das S, Xu Q, Keats J, Wang F, Wardwell S, Ning Y, Snodgrass JT, Broudy MI, Russian K, Zhou T, Commodore L, Narasimhan NI, Mohemmad QK, Iuliucci J, Rivera VM, Dalgarno DC, Sawyer TK, Clackson T, Shakespeare WC. Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J Med Chem. 2010;53:4701–4719. doi: 10.1021/jm100395q. [DOI] [PubMed] [Google Scholar]
- 28.Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, Shakespeare WC, Wang F, Clackson T, Rivera VM. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther. 2012;11:690–699. doi: 10.1158/1535-7163.MCT-11-0450. [DOI] [PubMed] [Google Scholar]
- 29.Howard S, Berdini V, Boulstridge JA, Carr MG, Cross DM, Curry J, Devine LA, Early TR, Fazal L, Gill AL, Heathcote M, Maman S, Matthews JE, McMenamin RL, Navarro EF, O'Brien MA, O'Reilly M, Rees DC, Reule M, Tisi D, Williams G, Vinkovic M, Wyatt PG. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J Med Chem. 2009;52:379–388. doi: 10.1021/jm800984v. [DOI] [PubMed] [Google Scholar]
- 30.Arkenau HT, Plummer R, Molife LR, Olmos D, Yap TA, Squires M, Lewis S, Lock V, Yule M, Lyons J, Calvert H, Judson I. A phase I dose escalation study of AT9283, a small molecule inhibitor of aurora kinases, in patients with advanced solid malignancies. Ann Oncol. 2012;23:1307–1313. doi: 10.1093/annonc/mdr451. [DOI] [PubMed] [Google Scholar]
- 31.Dent SF, Gelmon KA, Chi KN, Jonker DJ, Wainman N, Capier CA, Chen EX, Lyons JF, Seymour L. NCIC CTG IND.181: phase I study of AT9283 given as a weekly 24 hour infusion in advanced malignancies. Invest New Drugs. 2013;31:1522–1529. doi: 10.1007/s10637-013-0018-9. [DOI] [PubMed] [Google Scholar]
- 32.Moreno L, Marshall LV, Pearson AD, Morland B, Elliott M, Campbell-Hewson Q, Makin G, Halford SE, Acton G, Ross P, Kazmi-Stokes S, Lock V, Rodriguez A, Lyons JF, Boddy AV, Griffin MJ, Yule M, Hargrave D. A phase I trial of AT9283 (a selective inhibitor of aurora kinases) in children and adolescents with solid tumors: a cancer research UK study. Clin Cancer Res. 2015;21:267–273. doi: 10.1158/1078-0432.CCR-14-1592. [DOI] [PubMed] [Google Scholar]
- 33.Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, Green S, Haye HR, Horn CL, Janetka JW, Liu D, Mouchet E, Ready S, Rosenthal JL, Queva C, Schwartz GK, Taylor KJ, Tse AN, Walker GE, White AM. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7:2955–2966. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- 34.Sausville E, Lorusso P, Carducci M, Carter J, Quinn MF, Malburg L, Azad N, Cosgrove D, Knight R, Barker P, Zabludoff S, Agbo F, Oakes P, Senderowicz A. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014;73:539–549. doi: 10.1007/s00280-014-2380-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emanuel S, Rugg CA, Gruninger RH, Lin R, Fuentes-Pesquera A, Connolly PJ, Wetter SK, Hollister B, Kruger WW, Napier C, Jolliffe L, Middleton SA. The in vitro and in vivo effects of JNJ-7706621: a dual inhibitor of cyclin-dependent kinases and aurora kinases. Cancer Res. 2005;65:9038–9046. doi: 10.1158/0008-5472.CAN-05-0882. [DOI] [PubMed] [Google Scholar]
- 36.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Che HY, Guo HY, Si XW, You QY, Lou WY. PP121, a dual inhibitor of tyrosine and phosphoinositide kinases, inhibits anaplastic thyroid carcinoma cell proliferation and migration. Tumour Biol. 2014;35:8659–8664. doi: 10.1007/s13277-014-2118-3. [DOI] [PubMed] [Google Scholar]
- 39.Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sessa F, Mapelli M, Ciferri C, Tarricone C, Areces LB, Schneider TR, Stukenberg PT, Musacchio A. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell. 2005;18:379–391. doi: 10.1016/j.molcel.2005.03.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.