

Significance
Pentraxins such as serum amyloid P (SAP) and C-reactive protein (CRP) have significant, and for SAP dominant, effects on the innate immune system. This report shows that contrary to the current model of how SAP and CRP are sensed by cells, Fcγ receptors are not necessary for SAP to regulate the innate immune system. This report considerably changes our understanding of the endogenous regulation of the innate immune system and connections between innate immune system signaling and epithelial cell signaling. The identification of DC-SIGN as a SAP receptor, the potential use of anti–DC-SIGN antibodies as a therapeutic, and the observation that remarkably low concentrations of a DC-SIGN ligand are therapeutic in a mouse model of fibrosis, create a new approach to treat fibrosis.
Keywords: fibrosis, pentraxin, DC-SIGN, serum amyloid P, IL-10
Abstract
Fibrosis is caused by scar tissue formation in internal organs and is associated with 45% of deaths in the United States. Two closely related human serum proteins, serum amyloid P (SAP) and C-reactive protein (CRP), strongly affect fibrosis. In multiple animal models, and in Phase 1 and Phase 2 clinical trials, SAP affects several aspects of the innate immune system to reduce fibrosis, whereas CRP appears to potentiate fibrosis. However, SAP and CRP bind the same Fcγ receptors (FcγR) with similar affinities, and why SAP and CRP have opposing effects is unknown. Here, we report that SAP but not CRP binds the receptor DC-SIGN (SIGN-R1) to affect the innate immune system, and that FcγR are not necessary for SAP function. A polycyclic aminothiazole DC-SIGN ligand and anti–DC-SIGN antibodies mimic SAP effects in vitro. In mice, the aminothiazole reduces neutrophil accumulation in a model of acute lung inflammation and, at 0.001 mg/kg, alleviates pulmonary fibrosis by increasing levels of the immunosuppressant IL-10. DC-SIGN (SIGN-R1) is present on mouse lung epithelial cells, and SAP and the aminothiazole potentiate IL-10 production from these cells. Our data suggest that SAP activates DC-SIGN to regulate the innate immune system differently from CRP, and that DC-SIGN is a target for antifibrotics.
Fibrosing diseases such scleroderma, pulmonary fibrosis, and renal fibrosis are caused by aberrant scar tissue formation in internal organs and are associated with 45% of deaths in the United States (1). At a fibrotic lesion, monocytes leave the blood, enter the tissue, and differentiate into cells such as macrophages and fibrocytes (2). Fibrocytes and macrophages then secrete extracellular matrix (ECM) proteins, ECM modifying enzymes, and/or cytokines such as IL-4 to promote scar tissue formation and fibrosis (3, 4).
Pentraxins are a family of highly conserved secreted proteins that have a profound effect on the development of fibrosis and the regulation of the innate immune system (5–7). The pentraxin serum amyloid P (SAP) reduces neutrophil activation and recruitment (8, 9), inhibits the differentiation of monocytes into fibroblast-like cells called fibrocytes (8, 10), and promotes IL-10–secreting macrophages (11–13). In animal models and two human trials (6, 14, 15), injections of SAP decrease fibrosis, indicating that SAP has a dominant effect on a disease that is mediated in part by the innate immune system. Conversely, the closely related pentraxin C-reactive protein (CRP) is proinflammatory and promotes fibrosis (5, 16). However, under some conditions, CRP decreases inflammation, indicating that much remains to be understood about this molecule (5, 17). Despite the strong effects of pentraxins on the innate immune system and fibrosis (5, 6), little is known about their mechanism of action. For instance, pentraxins such as SAP and CRP appear to bind the same Fcγ receptors (FcγR) with similar affinities (7, 8, 18), but they generally have opposite effects. What causes this functional difference is not known.
Dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN/CD209) is a C-type lectin found on dendritic cells, macrophages, and monocytes (19, 20). DC-SIGN mainly binds to mannosylated and fucosylated proteins (19, 20). Humans have DC-SIGN and L-SIGN, whereas mice have eight DC-SIGN orthologs called SIGN-R1–8 (21). SIGN-R1 most closely resembles DC-SIGN (21). DC-SIGN and SIGN-R1 also bind sialylated IgG (sIgG) (20). This interaction appears to be a protein:potein interaction and not a sialic acid:DC-SIGN interaction (22). Both sIgGs and SAP have α(2,6)-linked terminal sialic acids on the protein surface, and both sIgGs and SAP alleviate inflammation in mice (6, 20, 23).
In this report, we show that in absence of all of the FcγR, neutrophils, monocytes, and macrophages still respond to SAP, indicating that SAP uses other receptors. For SAP, we show that one of the other receptors is DC-SIGN. We also found that anti–DC-SIGN antibodies and a small-molecule DC-SIGN ligand mimic the effects of SAP. The synthetic DC-SIGN ligand shows efficacy in murine models of acute lung inflammation and pulmonary fibrosis. In contrast to SAP, we find that CRP requires the FcγR to regulate neutrophils and IL-10 secretion from macrophages, but not to increase ICAM-I+ macrophages. This finding suggests that there are additional CRP receptors that regulate macrophage polarization. Our findings suggest the presence of a previously unidentified pentraxin target that accounts for the functional difference between SAP and CRP, and which might be useful as a therapeutic target to regulate the innate immune system and fibrosis.
Results
Fcγ Receptors Are Not Necessary for SAP Effects on the Innate Immune System.
SAP and CRP both bind FcγR, but have different effects on the innate immune system (8, 18). To determine the role of FcγR in SAP and CRP function, we examined the effect of these proteins on mouse cells lacking all FcγR (FcγR quad KO). As previously observed, SAP and CRP decreased the adhesion of C57BL/6 neutrophils to fibronectin (Fig. 1A) (8, 9). However, when added to FcγR quad KO neutrophils, SAP but not CRP significantly reduced neutrophil adhesion to fibronectin (Fig. 1A).
Fig. 1.
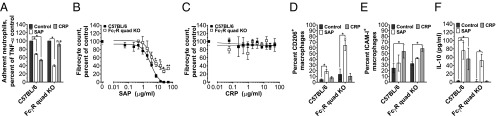
Fcγ receptors are necessary for some but not all effects of SAP and CRP. (A) Mouse neutrophils were incubated with 10 μg/mL of the indicated pentraxin and TNF-α, and neutrophil adhesion to fibronectin was assayed, n = 3. (B and C) Mouse spleen cells were incubated with the indicated concentrations of pentraxin. After 5 d, the cells were fixed, stained, and fibrocytes were counted, n = 3–6. (D and E) Mouse bone marrow-derived macrophages were polarized in 0 (control) or 10 µg/mL of the indicated pentraxin. Cells were then fixed and stained, n = 3. (F) Macrophages were polarized as in D, and IL-10 levels in the supernatants were measured, n = 3–8. n.s. (not significant relative to the control), *P < 0.05 (t test), (D) ×, P < 0.05 (t test relative to SAP in C57BL/6). All values are mean ± SEM. Data were fit to sigmoidal dose–response curves with a variable Hill coefficient (B) or a line (C).
In addition to reducing neutrophil adhesion, SAP inhibits the differentiation of monocytes into fibroblast-like cells called fibrocytes (8, 10) (Fig. 1B). In the absence of FcγR, SAP reduced but could not completely inhibit fibrocyte differentiation (Fig. 1B). CRP had no effect on fibrocyte differentiation (Fig. 1C) (10). The absence of FcγR did not alter this response (Fig. 1C).
SAP potentiates CD206+ antiinflammatory macrophages (13) (Fig. 1D). CRP, however, primarily promotes ICAM-I+ M1-like inflammatory macrophages (16) (Fig. 1E). In our assays, both SAP and CRP were able to polarize FcγR quad KO macrophages as determined by CD206 and ICAM-I expression (Fig. 1 D and E). In agreement with its antiinflammatory function, SAP also increased IL-10 secretion from C57BL/6 and FcγR quad KO macrophages (Fig. 1F). We also observed that CRP increased IL-10 secretion from C57BL/6 macrophages but not from the FcγR quad KO cells, indicating that some but not all effects of CRP on macrophages are mediated by the FcγR (Fig. 1F). The increase in IL-10 secretion in response to CRP has been observed before (24, 25) and is most likely counteracted by CRP-induced TNF-α and IL-12 under inflammatory conditions (16). The related pentraxin PTX3 was also able to decrease neutrophil adhesion and alter macrophage phenotype in absence of the FcγR, although the PTX3-induced IL-10 secretion by macrophages was absent (Fig. S1). Together these results suggest that contrary to the current model of SAP signaling, the FcγR are not necessary for SAP effects. This observation suggests the presence of additional SAP receptors. Conversely, CRP requires the FcγR to reduce neutrophil adhesion and promote IL-10 secretion by macrophages, but not to increase ICAM-I+ macrophages. This finding indicates that some aspects of the CRP effect on macrophages is mediated by an unknown receptor.
Fig. S1.

Fcγ receptors are not necessary for some PTX3 effects on neutrophils and macrophages. (A) Mouse neutrophils were incubated with 0 (control) or 1 µg/mL of PTX3, transferred to a fibronectin-coated plate, and then activated with TNF-α. After 30 min, neutrophils bound to the plate were stained and counted, n = 3. (B and C) Mouse bone marrow-derived macrophages from C57BL/6 or FcγR-deficient mice were polarized for 3 d in serum-free medium containing 0 (control) or 1 µg/mL PTX3. Cells were then fixed and stained for CD206 and ICAM-1, n = 3. (D) C57BL/6 or FcγR-deficient macrophages were polarized and then soluble IL-10 levels in supernatants were measured by ELISA, n = 3. *P < 0.05, **P < 0.01 (t test). All values are ±SEM.
SAP Glycosylation Mediates SAP Effects on the Innate Immune System.
CRP has sequence and structural similarity to SAP, and like SAP, binds FcγR (5, 6). However, SAP and CRP have different effects on monocyte and macrophage differentiation; for instance, whereas SAP inhibits fibrocyte differentiation (8) and promotes IL-10 secreting macrophages (3, 13), CRP has no effect on fibrocyte differentiation (10) and promotes proinflammatory macrophages (16). One possible reason for this functional difference is sequence divergence between SAP and CRP. However, we previously found that mutating SAP surface amino acid residues that are different between SAP and CRP has only a modest effect on SAP function (8). An alternative cause of this functional difference may be a difference in SAP and CRP glycosylation: SAP is glycosylated at N32 with α(2,6)-linked terminal sialic acids (23), which is exposed on a soluble surface, whereas CRP has no glycosylation (23). To determine whether the SAP glycosylation affects its function, we enzymatically removed the terminal sialic acids with neuraminidase. The desialylated SAP [SAP (NA)] could no longer be detected on Western blots stained with Sambucus Nigra lectin, which binds preferentially to α(2→6)-linked terminal sialic acids (Fig. 2 A and B).
Fig. 2.

Glycosylation mediates some effects of SAP. (A) Desialylated SAP [SAP (NA)], SAP, glycosylated CRP (CRP A32N), CRP, and desialylated CRP A32N [CRP A32N (NA)] were electrophoresed on SDS/PAGE gels and stained with silver stain. (B) Western blots of the samples in A were stained with Sambucus Nigra lectin to detect α(2,6)-linked terminal sialic acids. (C) Human neutrophils were incubated with 0 (control) or 10 µg/mL of the indicated pentraxin to assess neutrophil adhesion as in Fig. 1A, n = 3–5. (D and E) Human PBMC were incubated with the indicated concentrations of pentraxins. After 5 d, fibrocytes were counted, n = 3–5. (F) Mouse spleen cells were incubated in the presence or absence of 10 µg/mL of the indicated pentraxin. Fibrocyte counts were normalized to the no-pentraxin control, n = 3. (G) Mouse C57BL/6 bone marrow-derived macrophages were polarized in 0 (control) or 10 µg/mL of the indicated pentraxins. Cells were then stained for the indicated markers, n = 3. (H) Human macrophages were polarized by 3 µg/mL of the indicated protein and then stained, n = 3. *P < 0.05, **P < 0.01 (t test). (F) +, P < 0.05 (t test relative to the no-protein control). (C–H) Values are mean ± SEM. (D and E) Data were fit to sigmoidal dose–response curves with a variable Hill coefficient or a line where appropriate.
SAP and CRP as observed previously reduced human neutrophil adhesion to fibronectin (9) (Fig. 2C). However as compared with SAP, SAP (NA) had a reduced effect on human neutrophil adhesion and was unable to inhibit fibrocyte differentiation when added to human PBMCs and mouse spleen cells (Fig. 2 C–F). These findings indicate a significant functional role for SAP glycosylation and provide a possible mechanism for the immune cells to differentiate SAP from CRP. Furthermore, these results suggest that a glycosylated CRP would be able to “trick” the innate immune cells and mimic SAP effects. To examine this possibility, we mutated CRP at position 32 from an alanine to an asparagine. The mutated CRP (CRP A32N) was glycosylated and had a lower mobility on SDS/PAGE gels compared with CRP (Fig. 2 A and B). However, on average only 40% of CRP A32N monomers were sialylated (Fig. 2B). This lack of complete sialylation is likely due to problems with recombinant protein expression and/or sequence differences between SAP and CRP (26). When tested on innate immune cells, CRP A32N similar to CRP and SAP reduced human neutrophil adhesion (Fig. 2C). However, unlike CRP, CRP A32N was able to inhibit fibrocyte differentiation when added to human PBMC or mouse spleen cells (Fig. 2 E and F). Furthermore, CRP A32N similar to SAP did not require the FcγR to inhibit fibrocyte differentiation (Fig. 2F).
In addition to regulating neutrophil adhesion and fibrocyte differentiation, SAP and CRP can polarize macrophages (3, 11, 13). To examine the role of SAP glycosylation on macrophage polarization, we added pentraxins to mouse and human macrophages. SAP and CRP A32N promoted CD206+ M2 macrophages from mouse bone marrow-derived macrophages (Fig. 2G), whereas CRP and SAP (NA) increased ICAM-I+ M1 macrophages (27) (Fig. 2G). We observed a similar trend in human monocyte-derived macrophages, where SAP and CRP A32N promoted CD163+ M2 macrophages (28), whereas SAP (NA) and CRP potentiated ICAM-I+ M1 macrophages (Fig. 2H). Neuraminidase-treated CRP A32N was essentially indistinguishable from CRP in our assays (Fig. 2), indicating a role of glycosylation in the effects of CRP A32N. Together, these results indicate that SAP glycosylation allows the innate immune cells to differentiate SAP from CRP and response appropriately to different pentraxins.
SAP but Not CRP Binds to DC-SIGN To Regulate Immune Cells.
Sialylated IgG (sIgG) binds DC-SIGN to alter IgG responses (20). Because SAP shares the same type of glycosylation as sIgG (23), and SAP and the Fc domain of IgG bind to FcγR, we examined the possibility that SAP might bind to DC-SIGN. We expressed DC-SIGN on HEK293 cells and measured SAP binding to the transfected cells (Fig. 3A and Fig. S2). We used mock-transfected HEK293 cells to estimate the nonspecific binding. SAP bound to DC-SIGN with a KD of 2.3 ± 1 µg/mL (19 ± 8 nM) and a Hill coefficient of 0.7 ± 0.3 (Fig. 3A). CRP A32N also bound to DC-SIGN (KD of 3.4 ± 0.3 µg/mL, Hill coefficient = 1.9 ± 0.4) but with a lower affinity relative to SAP (Fig. 3A), suggesting that the SAP:DC-SIGN interaction may involve protein:protein interaction and, hence, not be limited to carbohydrate:lectin interaction. As expected, SAP (NA), CRP, and neuraminidase treated CRP A32N did not show detectable binding to DC-SIGN (Fig. 3A). This observation suggests that SAP and CRP A32N may bind to DC-SIGN to alter immune responses. To test this possibility, we examined the effect of SAP on spleen cells from mice lacking the mouse ortholog of human DC-SIGN, SIGN-R1. The absence of SIGN-R1 significantly decreased the inhibitory effect of SAP on fibrocyte differentiation (Fig. 3B). CRP A32N and PTX3 were also unable to alter fibrocyte differentiation from SIGN-R1–deficient spleen cells, suggesting that SIGN-R1 might mediate CRP A32N and PTX3 effects on fibrocyte differentiation (Fig. 3C and Fig. S3).
Fig. 3.

DC-SIGN activation affects neutrophils and monocyte-derived cells. (A) DC-SIGN+ HEK293 cells were incubated with fluorescently labeled SAP, CRP, CRP A32N, SAP (NA), or CRP A32N (NA), and binding was measured by flow cytometry. Mock transfected cells were used to estimate the nonspecific binding, n = 3. (B) The effect of SAP on fibrocyte differentiation in C57BL/6 and SIGN-R1–deficient cells was assessed, n = 3–5. (C) Mouse spleen cells were incubated with 0 or 10 µg/mL of the indicated pentraxin. Fibrocytes were counted as a percent of the no-pentraxin control, n = 3–5. (D) Human neutrophils were incubated with anti-DC-SIGN antibodies and then neutrophil adhesion to fibronectin was assessed, n = 3. (E) Human PBMCs were incubated with the indicated concentrations of a rabbit anti–DC-SIGN antibody or a rabbit isotype control. After 5 d, fibrocytes were counted, n = 3–5. (F) Human macrophages were polarized with 1 µg/mL of the indicated antibody. Macrophages were then fixed and stained, n = 3. (G) The effect of compound 1 on human neutrophil adhesion to fibronectin was assessed, n = 3. (H) The effect of compound 1 on human fibrocyte differentiation, n = 3. (I) Macrophage polarization by 10 pg/mL of compound 1 was examined in human macrophages, n = 3. *P < 0.05 (t test). All values are mean ± SEM. (A) Curves are fits to models of one-site binding with variable Hill coefficient where appropriate. (B, D, G, and H) Data were fit to sigmoidal dose–response curves with a variable Hill coefficient.
Fig. S2.

DC-SIGN expression on HEK293 cells. The expression of DC-SIGN (red line) on HEK293 cells (Left) and HEK293 cells expressing DC-SIGN (Right) was determined by flow cytometry. Mouse IgG1 (blue line) was used as the isotype control. Plots are representative of three independent experiments.
Fig. S3.

SIGN-R1 is necessary for the effect of PTX3 on fibrocyte differentiation. Spleen cells from SIGN-R1 null mice were incubated with the indicated concentrations of PTX3. After 5 d, cells were fixed, stained, and fibrocytes were counted, n = 3. *P < 0.05 (t test). Values are mean ± SEM.
DC-SIGN is expressed on macrophages, dendritic cells, and monocytes (19, 20) (Fig. S4). Previously, DC-SIGN mRNA has been observed in human neutrophils (29, 30). We were also able to detect cell-surface DC-SIGN on human and mouse neutrophils (Figs. S4A and S5). Because the majority of cells expressing DC-SIGN appear to respond to SAP, we examined whether DC-SIGN activation by antibodies can mimic SAP effects on neutrophils, monocytes, and macrophages. Some, but not all, anti-human DC-SIGN antibodies decreased human neutrophil adhesion to fibronectin (Fig. 3D) and inhibited human fibrocyte differentiation (IC50 = 2.4 ± 0.4 µg/mL) (Fig. 3E and Fig. S6 A and B). A different subset of anti-DC-SIGN antibodies also altered macrophage phenotype and increased CD163+ macrophages similar to SAP and CRP A32N (Fig. 3F and Fig. S6C). We observed similar effects using F(ab2) fragments of anti-DC-SIGN antibodies, suggesting that our results are Fc-independent and most likely involve DC-SIGN activation by the antibodies (Fig. S6 D–F).
Fig. S4.

DC-SIGN expression on human immune cells. (A) Human neutrophils were isolated by density centrifugation and then stained for the indicated markers (red line). DC-SIGN (9E9A8 clone, IgG1) staining was amplified by using Biotin-Avidin. (B) Human monocytes were purified by negative selection and then stained for the indicated markers (red line). DC-SIGN (9E9A8 clone, IgG1) staining was amplified by using Biotin-Avidin. (C) Human lymphocytes were gated on based on their flow characteristics and then stained for the indicated markers (red line). DC-SIGN (9E9A8 clone, IgG1) staining was amplified by using Biotin-Avidin. All plots are representative of three individual experiments. The appropriate isotype control (black line) was used for all experiments.
Fig. S5.

SIGN-R1 expression on mouse blood cells. (A) The expression of CD11b, Ly6C, Ly6G, and SIGN-R1 (red lines) on mouse peripheral blood leukocytes was determined by flow cytometry. We identified two distinct subpopulations of cells with subset 1 most likely corresponding to neutrophils and subset 2 to mononuclear cells. Appropriate isotype controls (black line) were used for each experiment. (B) Mouse peripheral leukocytes were stained for CD11b, Ly6C, Ly6G, and SIGN-R1 (or hamster IgG). We then gated on CD11b+ Ly6C+ cells and examined the expression of Ly6G on CD11b+ Ly6C+ cells. Subsequently, we gated on CD11+ Ly6C+ Ly6G+ cells and assessed SIGN-R1 expression. Similarly, we examined SIGN-R1 expression on CD11+ Ly6C+ Ly6G-negative cells. Appropriate isotype controls were used for each experiment (rat IgG2a for Anti-Ly6G, rat IgG2b for anti-CD11b, and rat IgG2c for anti-Ly6C antibodies). Plots are representative of three individual experiments.
Fig. S6.

The effect of anti-human DC-SIGN antibodies on fibrocyte differentiation and macrophage polarization. (A and B) Human PBMCs were incubated with the indicated concentrations of anti–DC-SIGN antibodies or isotype controls. After 5 d, fibrocytes were counted, n = 3. (C) Human monocyte-derived macrophages were polarized for 3 d in serum-free medium with 1 µg/mL of the indicated antibody. Macrophages were then fixed and stained for CD163, n = 3. (D) The effect of an anti–DC-SIGN F(ab2) and control rat IgG2b F(ab2) on human neutrophil adhesion was assessed, n = 3. (E) Human PBMCs were incubated with the indicated concentrations of an anti–DC-SIGN F(ab2) or F(ab2) isotype control. After 5 d, fibrocytes were counted, n = 3. (F) Human monocyte-derived macrophages were polarized for 3 d in serum-free medium with 1 µg/mL of the indicated antibody. Macrophages were then fixed and stained, n = 3. *P < 0.05, **P < 0.01 (t test, relative to no-antibody control). All values are mean ± SEM.
To further ascertain the role of DC-SIGN in SAP signaling, we examined whether DC-SIGN activation using synthetic ligands could mimic SAP effects. When added to immune cells, a polycyclic aminothiazole DC-SIGN ligand (compound 1; shown as compound 4 in figure 2 of ref. 31) decreased human neutrophil adhesion (IC50 of 1.7 ± 0.3 µg/mL and a Hill coefficient of 2.6 ± 0.2) (Fig. 3G) and inhibited human fibrocyte differentiation (IC50 = 1.2 ± 0.4 pg/mL, Hill coefficient = 0.30 ± 0.01) (Fig. 3H) without affecting cell viability below 0.1 µg/mL (Fig. S7 A and B). Other DC-SIGN ligands also inhibited human fibrocyte differentiation, although with lower potency (Fig. S8). In addition, compound 1 promoted CD163+ M2 macrophages (Fig. 3I). Compound 1 similarly reduced neutrophil adhesion, inhibited fibrocyte differentiation, and promoted M2 macrophages in FcγR quad KO cells (Fig. S9). However, in SIGN-R1–deficient cells, compound 1 did not affect fibrocyte differentiation (Fig. S7C). These data indicate that DC-SIGN activation by antibodies or a synthetic ligand is sufficient to mimic SAP effects in vitro. In addition, these results suggest that SAP:DC-SIGN interaction may contribute to the functional differences in SAP and CRP effects on the innate immune system. However, it is not clear whether SAP glycosylation directly binds DC-SIGN (carbohydrate:lectin interaction) or alters the structure of SAP so that SAP can bind DC-SIGN similar to how sIgG binds this receptor (protein:protein interaction) (32).
Fig. S7.

Compound 1 inhibits fibrocyte differentiation through SIGN-R1 without causing extensive cell death. Human PBMCs (A) or C57BL/6 mouse spleen cells (B) were incubated with the indicated concentrations of compound 1. After 5 d, Alamar blue was used to estimate cell viability, n = 3. (C) Mouse spleen cells were incubated with increasing concentrations of compound 1. After 5 d, cells were fixed, stained, and fibrocytes were counted, n = 4. Compound 1 inhibited murine fibrocyte differentiation with an IC50 of 91 ± 47 pg/mL. *P < 0.05, **P < 0.01 (t test). All values are mean ± SEM. (C) C57BL/6 data were fit to a sigmoidal dose–response curve with a variable Hill coefficient.
Fig. S8.

Compounds 2 and 3 inhibit human fibrocyte differentiation. Human PBMCs were incubated with the indicated concentrations of compound 2 (compound 5 in figure 2 of ref. 31) or compound 3 (compound 6 in figure 2 of ref. 31). After 5 d, fibrocytes were counted, n = 3. Data were fit to sigmoidal dose–response curves with variable Hill coefficients.
Fig. S9.

Compound 1 inhibits neutrophil adhesion and fibrocyte differentiation and promotes M2 macrophages in absence of FcγR. (A) Mouse neutrophils were incubated with 0 (control) or 1 µg/mL of compound 1, transferred to a fibronectin-coated plate, and then activated with TNF-α. After 30 min, neutrophils bound to the plate were stained and counted, n = 3. (B) Mouse spleen cells were incubated with 0 or 1 ng/mL of compound 1. After 5 d, cells were fixed, stained, and fibrocytes were counted. (C) Murine bone marrow-derived macrophages were polarized for 3 d in serum-free medium containing 0 or 10 pg/mL of compound 1. Cells were then stained for CD206, n = 3. *P < 0.05, **P < 0.01 (t test). All values are mean ± SEM.
Compound 1 Reduces Neutrophil Accumulation in the Lungs of Bleomycin-Treated Mice.
SAP and PTX3 regulate neutrophil recruitment in mice (9, 33). To determine whether SIGN-R1 activation similarly affects neutrophils in mice, WT mice were given bleomycin to induce acute lung inflammation. We then injected the mice with compound 1 and examined neutrophil accumulation in the lungs. We did not use anti–SIGN-R1 antibodies because they would activate both SIGN-R1 and FcγR, therefore confounding the results. As observed before (9), oropharyngeal instillation of bleomycin significantly increased the number of Ly6G+ neutrophils in the lungs (Fig. 4). When mice were injected with compound 1 at 0.1 mg/kg on days 1 and 2 after bleomycin, there was a significant decrease in Ly6G+ cells in the lungs at day 3 compared with the bleomycin control (Fig. 4). Bleomycin treatment also resulted in a significant increase in CD11b+ macrophages and CD45+ immune cells in the bronchoalveolar lavage (BAL) fluid (Fig. 4B). This increase in infiltrating cells was absent when mice were given compound 1 (Fig. 4B). Additionally in the post-BAL lungs, compound 1 reversed a bleomycin-induced decrease in CD11c+ cells (Fig. 4C). However, compound 1 did not alter the number of CD11b+ macrophages in the lungs compared with the bleomycin control (Fig. 4C). Our results indicate that DC-SIGN ligands such as compound 1 can, similar to SAP and PTX3 (9, 33), reduce neutrophil accumulation in mouse lungs following an insult.
Fig. 4.

Compound 1 decreases neutrophil accumulation in mouse lungs. (A) Mice received oropharyngeal bleomycin on day 0 to induce acute respiratory distress syndrome. The control mice received saline. Mice were then treated with i.p. injections of 0.1 mg/kg of compound 1 (C1) or an equal volume of vehicle control on days 1 and 2. On day 3, the mice were euthanized and lungs after BAL were stained for the neutrophil marker Ly6G. Images are representative of three independent experiments. (Scale bar: 100 µm.) (B) BAL cells were stained for the indicated markers, n = 4. (C) After collecting the BAL, lungs were stained for the indicated markers, n = 4. *P < 0.05 (one-way ANOVA, Holm–Bonferroni post hoc test). (B and C) Values are mean ± SEM.
Compound 1 Alleviates Pulmonary Fibrosis in Mice.
Compound 1 alters macrophage phenotype and inhibits fibrocyte differentiation similar to SAP. Because macrophages and fibrocytes are implicated in fibrosing diseases (1, 2), we determined whether DC-SIGN activation by compound 1 in a murine model of pulmonary fibrosis was sufficient to mimic SAP and alleviate fibrosis. As observed before (34), oropharyngeal instillation of bleomycin resulted in increased collagen deposition and recruitment of CD11b+ macrophages to the lungs (Fig. 5). Daily injections of compound 1 at doses as low as 0.001 mg/kg decreased collagen deposition in the lungs and improved overall health as indicated by weight change (Fig. 5 A and B and Fig. S10A). In addition, compound 1 reduced the number of CD11b+ macrophages compared with the bleomycin control (Fig. 5 C and D). These data suggest that compound 1, similar to SAP, can alleviate pulmonary fibrosis and inflammation in mice.
Fig. 5.

Compound 1 alleviates pulmonary fibrosis in mice. (A) Mice received oropharyngeal bleomycin (Bleo) on day 0 to induce pulmonary fibrosis. Control mice received saline. Mice were then injected with compound 1 (C1) or vehicle control at the indicated dose daily starting on day 1 and ending on day 20. On day 21, mice were euthanized and after collecting BAL fluid, lungs were stained with PicroSirius red to estimate collagen deposition. Images are representative of three different experiments. (Scale bar: 200 µm.) (B) Quantification of PicroSirius red staining, n = 3. (C) BAL cell were stained for CD11b, n = 3. Rat IgG1 was used as the isotype control. (D) After collecting BAL, lungs were stained for CD11b, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 (one-way ANOVA, Holm-Bonferroni post hoc test). (B–D) Values are mean ± SEM.
Fig. S10.

The effect of compound 1 on mouse weights. (A) Mice in Fig. 5 were weighed daily, n = 3. (B) IL-10–deficient mice were weighted daily, n = 3. All values are mean ± SEM.
IL-10 Is Necessary for the Antiinflammatory Effect of Compound 1.
IL-10 is an antiinflammatory cytokine that is released in response to DC-SIGN activation (21). IL-10 is also necessary for the antifibrotic effect of SAP in a mouse model of renal fibrosis (12, 13). As compound 1 activates DC-SIGN to mimic SAP, we examined the efficacy of compound 1 on pulmonary fibrosis in IL-10–deficient mice. In IL-10–deficient mice, bleomycin instillation significantly increased collagen deposition and CD11b+ and CD11c+ macrophages in lungs (Fig. 6). Oropharyngeal instillation of bleomycin also resulted in significant decrease in body weight (Fig. S10B). Daily injections of 0.1 mg/kg of compound 1 had no significant effect on collagen deposition, CD11b+ macrophage accumulation, CD11c+ cell accumulation, or body weight in IL-10–deficient mice (Fig. 6 and Fig. S10B). These results suggest that IL-10 is necessary for the antifibrotic effects of compound 1 in a mouse bleomycin model of pulmonary fibrosis.
Fig. 6.

IL-10 is necessary for the antifibrotic effect of compound 1. (A) Pulmonary fibrosis was induced by bleomycin instillation in IL-10–deficient mice. The bleomycin-treated mice were then injected with 0.1 mg/kg compound 1 (C1) or an equal volume of vehicle control daily. On day 21, mice were euthanized and lungs were stained with PicroSirius red to estimate collagen deposition, n = 3. (B) BAL cells were stained for the indicated markers, n = 3. Rat IgG1 was used as the isotype control. (C) After collecting BAL, lungs were stained for the indicated markers, n = 3. *P < 0.05 (one-way ANOVA, Holm–Bonferroni post hoc test). All values are mean ± SEM.
Lung Conducting Airway Epithelial Cells Express SIGNR-R1 and IL-10.
Because compound 1 binds SIGN-R1 to regulate monocyte functions in mice, we examined the expression of this receptor in mouse lungs. We found that SIGN-R1 was expressed on Epcam-1+ lung epithelial cells and on CD45+ immune cells (Fig. 7A). To determine the source of IL-10, we stained mouse lungs for Epcam-1, CD45, and IL-10 by immunofluorescence. In saline-treated mice, Epcam-1+ epithelial cells but not CD45+ immune cells expressed detectable levels of IL-10 (Fig. S11). However, when mice were treated with bleomycin, the number of IL-10–expressing epithelial cells (Epcam-1+) significantly decreased (Fig. 7B and Fig. S11A). This decrease in IL-10+ Epcam-1+ cells was reversed when mice were injected with compound 1 or SAP (Fig. 7B and Fig. S11A). These results suggest that compound 1 and SAP can bind to SIGN-R1 on Epcam-1+ epithelial cells to induce IL-10 expression and reduce inflammation.
Fig. 7.

Murine lung epithelial cells express SIGN-R1 and IL-10. (A) Mouse lungs following BAL were stained for the indicated markers. Images are representative of three different experiments. (Scale bar: 50 µm.) Arrows indicate CD45+ cells expressing SIGN-R1. (B) The number of Epcam-1+ cells expressing IL-10 in mouse lungs after BAL was quantified, n = 3. *P < 0.05, **P < 0.01, (one-way ANOVA, Holm–Bonferroni post hoc test). (B) Values are mean ± SEM.
Fig. S11.

Murine lung epithelial cells express SIGN-R1 and IL-10. (A) Mouse lungs were stained for Epcam-1 and IL-10. Images are representative of three different experiments. (B) The expression of CD45 and IL-10 in mouse lungs post-BAL was assessed by immunofluorescence. Images are representative of three different experiments. (Scale bars: 50 µm.)
Discussion
SAP and CRP bind FcγR and regulate the innate immune system and fibrosis (8, 12, 18). In this report, we found that in the absence of all FcγR, SAP still reduces neutrophil adhesion, inhibits fibrocyte differentiation, and alters macrophage phenotype. Conversely, CRP requires FcγR to reduce neutrophil adhesion and promote IL-10 secretion by macrophages, but not to increase ICAM-I+ M1-like macrophages. These observations suggest the presence of additional SAP and CRP receptors. We identified an additional SAP receptor as DC-SIGN, found that SAP binds to this receptor in a glycosylation-dependent manner, and observed that a DC-SIGN ligand mimics some SAP functions in vitro and in animal models of acute lung injury and pulmonary fibrosis. The DC-SIGN ligand alleviates pulmonary fibrosis in mice through an IL-10–dependent mechanism, with the IL-10 most likely originating from the epithelial cells in the lungs.
The FcγRs have been viewed as the main targets for SAP and CRP in the innate immune system (5, 18). Our data counter this view, because SAP is able to regulate the innate immune cells in absence of all FcγR. In fact, SAP is a more potent polarizer of FcγR-deficient macrophages than WT macrophages, suggesting that some of the FcγR may counteract the effect of SAP. In agreement with this observation, an anti-DC-SIGN antibody (which contains Fc regions and, thus, interacts with both FcγR and DC-SIGN) reduced, but did not abolish, neutrophil adhesion, whereas the F(ab2) fragment of the same antibody was significantly (P < 0.05 at 1 μg/mL) more potent and a DC-SIGN ligand completely abolished neutrophil adhesion. A similar trend was observed with SAP, which is a more potent inhibitor of FcγR-deficient neutrophil adhesion than WT neutrophils. This effect of FcγR appears to be cell-type dependent, because FcγR and DC-SIGN seem to act cooperatively to inhibit monocyte to fibrocyte differentiation. Both DC-SIGN and FcγR regulate the activity of Src kinases in innate immune cells (35, 36). The antagonism of DC-SIGN and FcγR signaling in some cells, and the cooperativity of DC-SIGN and FcγR signaling in other cells, may thus be due to their differential effects on Src kinases.
Although DC-SIGN/SIGN-R1 is considered to be primarily expressed by innate immune system cells, the majority of SIGN-R1 staining in mouse lungs was on Epcam-1+ epithelial cells. These SIGN-R1+ Epcam-1+ expressed high levels of IL-10. Following a bleomycin insult, at day 21, although there was no appreciable reduction in the number of Epcam-1+ cells, there was a significant decrease in the number of IL-10+ Epcam-1+ cells. IL-10 inhibits apoptosis of epithelial cells (37) and increases the clearance of cell debris (38). As such, up-regulation of IL-10 by SAP or compound 1 may have a protective effect on lungs by limiting tissue damage and inflammatory responses. A similar role has been observed for epithelial cell-derived IL-10 in mouse models of inflammatory bowel disease (39). Alternatively, it is possible that IL-10 expression in SIGN-R1+ epithelial cells is a function of their health. However, this possibility is unlikely because our studies in IL-10–deficient mice suggest a critical role for IL-10 in the antifibrotic role of compound 1.
Together, our data indicate that SAP binds DC-SIGN/SIGN-R1 to regulate innate immune cells and epithelial cells. Through its interaction with DC-SIGN, SAP distinguishes itself functionally from CRP. This observation suggests that DC-SIGN/SIGN-R1 is a key regulator of the innate immune system and is thus an interesting therapeutic target. Additionally, the functional interaction of SAP and PTX3 with DC-SIGN suggest that these pentraxins may regulate dendritic cells and, thus, the adaptive immune system.
Materials and Methods
All animals were used in accordance with National Institutes of Health guidelines and with a protocol approved by the Texas A&M University Institutional Animal Care and Use Committee. Human blood was obtained with written consent and with specific approval from the Texas A&M University human subjects Institutional Review Board. Human recombinant SAP and CRP were expressed in HEK293 cells and then purified y using affinity purification (8). Human PBMC, human neutrophils, mouse spleen cells, and mouse neutrophils were isolated and then incubated with antibodies and pentraxins, as described before (8, 9). Human monocytes were differentiated into macrophages in serum containing medium and then polarized for 3 d in serum-free medium. Pulmonary fibrosis and acute lung injury in mice were induced by bleomycin instillation (9, 40). Detailed information about mice, experimental procedures, and statistical analyses can be found in SI Materials and Methods.
SI Materials and Methods
Antibodies and Reagents.
Mouse anti-human CD209 antibody clone H200 was obtained from Santa Cruz Biotechnology. Mouse anti-human CD209, CD163, and ICAM-I antibodies; anti-mouse CD206, ICAM-I, CD11b, Ly6G, CD45, and rat IgG1 isotype control antibodies; and biotinylated IL-10 (JES5-16E3) were from Biolegend. Anti-mouse CD11c was from MBL. Anti-mouse SIGN-R1 (22D1) and Armenian hamster isotype control were from eBioscience. Anti-mouse Epcam-1 antibody was from Abcam. Anti-rat Dylight 488 and anti-Armenian hamster biotin-conjugated antibodies were from Novus. Alexa Fluor 488-anti-rabbit antibodies and IgG-free albumin were from Jackson ImmunoResearch. Streptavidin-Alexa Fluor 647, Alamar Blue cell viability reagent, and Alexa Fluor 647-NHS were from Life Technologies. Human GM-CSF, mouse M-CSF, and mouse IL-13 were from Biolegend. Sambucus Nigra lectin, streptavidin-alkaline phosphatase, and Vector Red alkaline phosphatase substrate kits were from Vector. The CRP cDNA expression plasmid was a kind gift from Jeff Crawford (Baylor College of Medicine, Houston, TX) and the DC-SIGN cDNA expression plasmid was from the NIH AIDS reagent program (Germantown, MD). SAP and bleomycin were from EMD Millipore. CRP was from Fitzgerald Industries and PTX3 from R&D Systems. Neuraminidase (catalog no. N2876) and DMSO were from Sigma-Aldrich. Compound 1 (catalog no. 5931866) was purchased from ChemBridge. Compound 2 (catalog no. 4545–1519) and compound 3 (catalog no. 4545–1530) were purchased from ChemDiv.
Mouse Strains.
C57/BL6 and IL-10-null mice were obtained from Jackson Laboratories. FcγR quad-null (32) and SIGN-R1-null (20) mice were a kind gift from Jeffery Ravetch (Rockefeller University, New York, NY). All animals were used in accordance with National Institutes of Health guidelines and with a protocol approved by the Texas A&M University Institutional Animal Care and Use Committee.
Fibrocyte Differentiation Assay.
Human blood was collected into heparin tubes (BD Bioscience) from adult volunteers who gave written consent and with specific approval from the Texas A&M University human subjects Institutional Review Board. PBMCs were then isolated and cultured to examine human fibrocyte differentiation as described (8). Mouse spleen cells were isolated and cultured to assess murine fibrocyte differentiation as described (41).
Macrophage Polarization Assay.
Cells were cultured at 37 °C in a humidified incubator with 5% (vol/vol) CO2. Human monocytes were differentiated into macrophages in 200 µL of RPMI medium 1640 (Lonza)/10% (vol/vol) FCS (Seradigm) for 6 d at a concentration of 0.5 × 106 cells per mL in a well of an eight-well slide (EMD Millipore). The medium was then replaced with fresh serum-free RPMI medium1640 (supplemented with 10 mM Hepes, 2 mM glutamine, 100 U/mL penicillin, 100 g/mL streptomycin, and ITS-3, all from Sigma-Aldrich), containing 3 µg/mL of the indicated pentraxin, 1 µg/mL of the indicated antibody, or 10 pg/mL of compound 1 for 3 d. Subsequently, the cells were dried, fixed, and stained as described (8). Mouse tibias and femurs were flushed with RPMI medium 1640, and the cells were washed three times in PBS. The cells were then resuspended in RPMI medium 1640/10% (vol/vol) FCS and 200 µL of 5 × 105 cells per mL was incubated in each well of an eight-well slide (EMD Millipore). After 1 h, the medium and nonadherent cells were removed and replaced with fresh RPMI medium 1640/10% (vol/vol) FCS/10 ng/mL GM-CSF. On day 7, the medium was replaced with serum-free RPMI medium 1640 containing 10 µg/mL of the indicated pentraxin. After 3 d, cells were dried, fixed, and stained.
Recombinant Protein Expression.
The cDNA for human CRP was mutated at residue 32 by using a QuikChange II Site-Directed Mutagenesis Kit (Agilent) and the primer 5′ AAGGCGCGCCATGGAGAAGCTGTTG 3′. CRP cDNA was inserted into a pCMV6-AC-His vector (Origene) and then expressed in HEK293 cells (Life Technologies) as described (8). CRP and glycosylated CRP were purified by using immobilized p-aminophenyl phosphoryl choline beads (Thermo Fisher Scientific) following the manufacturer’s protocol.
SAP and CRP Desialylation.
SAP (125 KDa) and glycosylated CRP (CRP A32N) were incubated with neuraminidase (Sigma Aldrich) for 2 d at 37 °C following the manufacturer’s protocol. The desialylated SAP [SAP (NA)] and CRP A32N [CRP A32N (NA)] were then buffer exchanged to 20 mM Tris, pH 7.4 by using a 100-kDa filter (EMD Millipore) to remove the 60-kDa neuraminidase.
SDS/PAGE and Western Blots.
Proteins were diluted in 20 mM sodium phosphate, pH 7.4 to a concentration of 40 µg/mL and then analyzed by SDS/PAGE gels as described (10). For Western blotting, proteins from gels were transferred to polyvinylidene difluoride membranes (Immobilon P; Millipore) in Tris/glycine/SDS buffer containing 20% (vol/vol) methanol by following the manufacturer’s protocol. Filters were blocked for 30 min at room temperature in Carbo-Free Blocking Solution (Vector). Membranes were then incubated in PBS containing 10 µg/mL of Biotinylated Sambucus Nigra Lectin (Vector) for 30 min at room temperature to detect α (2→6)-linked terminal sialic acids following the manufacturer’s protocol. Vectastain Elite ABC peroxidase kits (Vector) were used to detect the biotinylated lectin following the manufacturer’s protocol. We then used Clarity Western ECL chemiluminescence substrate (Bio-Rad) to visualize the peroxidase with a ChemiDoc XRS+ imager (Bio-Rad).
DC-SIGN Binding.
The DC-SIGN cDNA expression plasmid was transfected into HEK293 cells (Life Technologies) by using Lipofectamine 3000 (Sigma Aldrich) following the manufacturer’s protocol. After 3 d, the transfected cells were washed twice in 20 mM Tris, 140 mM NaCl, 2 mM CaCl2, pH 7.4 and then blocked for 30 min in blocking buffer (PBS/1 mM MgCl2/1 mM CaCl2/1% IgG-free albumin). Afterward, cells were washed once in binding buffer (PBS/1 mM MgCl2/1 mM CaCl2) and the cells were then incubated with Alexa Fluor 647 (Life Technologies) labeled SAP, CRP, CRP A32N, SAP (NA), or CRP A32N (NA) in the binding buffer at 4 °C. After 1 h, cells were washed three times in ice cold binding buffer and fluorescence of the cells was measured by flow cytometry as described (8). Mock transfected cells were used to estimate the nonspecific binding.
Neutrophil Adhesion Assay.
Human and murine neutrophils were isolated from blood and adhesion assays were performed as described (9).
Mouse Models of Acute Lung Inflammation and Fibrosis.
C57BL/6 mice (Jackson laboratories) were given an oropharyngeal instillation of 3 U/kg of bleomycin (EMD Millipore) to induce acute inflammation and neutrophil recruitment as described (9). Pulmonary fibrosis was induced in mice by instillation of 3 U/kg bleomycin as described (34). Briefly, on day 0, mice were given bleomycin instillation (50 µL) or saline (50 µL). Mice were then injected with vehicle control (48 µL of PBS, 2 µL of DMSO), SAP (50 µg of SAP in 50 µL of PBS), or compound 1 (48 µL of PBS, 2 µL of compound 1 in DMSO) at the indicated dose on days 1–2 for acute lung inflammation and days 1–20 for pulmonary fibrosis. On day 3 for the acute lung inflammation model and day 21 for pulmonary fibrosis model, mice were euthanized and the blood, bronchoalveolar lavage fluid (BAL), and the lungs were collected.
Immunohistochemistry and Immunofluorescence.
BAL cells and lungs were fixed and stained as described (40). Immunofluorescence images were acquired with a FluorView FV1000 Confocal Microscope (Olympus).
PicroSirius Red Staining.
PicroSirius red staining was performed as described (40).
Cell Viability Assay.
Cell viability was assessed with Alamar Blue cell viability reagent (Life Technologies) following the manufacturer’s protocol.
Statistical Analysis.
Data were analyzed by ANOVA (with Holm–Bonferroni post hoc test) or t test when appropriate by using Prism software (GraphPad). Statistical significance was defined as P < 0.05. Data were fit to the appropriate model of binding as determined by F tests.
Supplementary Material
Acknowledgments
We thank Dr. Jeffery Ravetch for his generous gift of SIGN-R1 KO spleens and FcγR quad KO mice, the staff at the Beutel Student Health Center for doing the phlebotomy work, and Michael White for his critical review of the manuscript.
Footnotes
Conflict of interest statement: Rice University has patents on the use of SAP to inhibit fibrosis. Texas A&M University has patent applications on the use of compound 1 to inhibit fibrosis. D.P. and R.H.G. are co-founders of and have equity in Promedior, a company that is developing SAP as a therapeutic. D.P. and R.H.G. receive a share of royalties paid by Promedior to Rice University.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1500956112/-/DCSupplemental.
References
- 1.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: Emerging effector cells in chronic inflammation. Nat Rev Immunol. 2011;11(6):427–435. doi: 10.1038/nri2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray LA, et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE. 2010;5(3):e9683. doi: 10.1371/journal.pone.0009683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quan TE, Cowper SE, Bucala R. The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep. 2006;8(2):145–150. doi: 10.1007/s11926-006-0055-x. [DOI] [PubMed] [Google Scholar]
- 5.Du Clos TW. Pentraxins: Structure, function, and role in inflammation. ISRN inflammation. 2013;2013:379040. doi: 10.1155/2013/379040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox N, Pilling D, Gomer RH. Serum amyloid P: A systemic regulator of the innate immune response. J Leukoc Biol. 2014;96(5):739–743. doi: 10.1189/jlb.1MR0114-068R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mantovani A, et al. The long pentraxin PTX3: A paradigm for humoral pattern recognition molecules. Ann N Y Acad Sci. 2013;1285:1–14. doi: 10.1111/nyas.12043. [DOI] [PubMed] [Google Scholar]
- 8.Cox N, Pilling D, Gomer RH. Distinct Fcγ receptors mediate the effect of serum amyloid p on neutrophil adhesion and fibrocyte differentiation. J Immunol. 2014;193(4):1701–1708. doi: 10.4049/jimmunol.1400281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maharjan AS, Roife D, Brazill D, Gomer RH. Serum amyloid P inhibits granulocyte adhesion. Fibrogenesis Tissue Repair. 2013;6(1):2. doi: 10.1186/1755-1536-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol. 2003;171(10):5537–5546. doi: 10.4049/jimmunol.171.10.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray LA, et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int J Biochem Cell Biol. 2011;43(1):154–162. doi: 10.1016/j.biocel.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 12.Castaño AP, et al. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Transl Med. 2009;1:5ra13. doi: 10.1126/scitranslmed.3000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W, Xu W, Xiong S. Macrophage differentiation and polarization via phosphatidylinositol 3-kinase/Akt-ERK signaling pathway conferred by serum amyloid P component. J Immunol. 2011;187(4):1764–1777. doi: 10.4049/jimmunol.1002315. [DOI] [PubMed] [Google Scholar]
- 14.Verstovsek S, et al. Phase 2 trial of PRM-151, an anti-fibrotic agent, in patients with myelofibrosis: Stage 1 results. Blood. 2014;124(21):713. [Google Scholar]
- 15.Dillingh MR, et al. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulm Pharmacol Ther. 2013;26(6):672–676. doi: 10.1016/j.pupt.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 16.Devaraj S, Jialal I. C-reactive protein polarizes human macrophages to an M1 phenotype and inhibits transformation to the M2 phenotype. Arterioscler Thromb Vasc Biol. 2011;31(6):1397–1402. doi: 10.1161/ATVBAHA.111.225508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu XZ, et al. Inhibition of experimental autoimmune encephalomyelitis in human C-reactive protein transgenic mice is FcγRIIB dependent. Autoimmune Dis. 2011;2011:484936. doi: 10.4061/2011/484936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu J, et al. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature. 2008;456(7224):989–992. doi: 10.1038/nature07468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Kooyk Y, Geijtenbeek TB. DC-SIGN: Escape mechanism for pathogens. Nat Rev Immunol. 2003;3(9):697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- 20.Anthony RM, Wermeling F, Karlsson MCI, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci USA. 2008;105(50):19571–19578. doi: 10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Vallejo JJ, van Kooyk Y. The physiological role of DC-SIGN: A tale of mice and men. Trends Immunol. 2013;34(10):482–486. doi: 10.1016/j.it.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Sondermann P, Pincetic A, Maamary J, Lammens K, Ravetch JV. General mechanism for modulating immunoglobulin effector function. Proc Natl Acad Sci USA. 2013;110(24):9868–9872. doi: 10.1073/pnas.1307864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pepys MB, et al. Human serum amyloid P component is an invariant constituent of amyloid deposits and has a uniquely homogeneous glycostructure. Proc Natl Acad Sci USA. 1994;91(12):5602–5606. doi: 10.1073/pnas.91.12.5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szalai AJ, Nataf S, Hu XZ, Barnum SR. Experimental allergic encephalomyelitis is inhibited in transgenic mice expressing human C-reactive protein. J Immunol. 2002;168(11):5792–5797. doi: 10.4049/jimmunol.168.11.5792. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez W, et al. C-reactive protein-mediated suppression of nephrotoxic nephritis: Role of macrophages, complement, and Fcgamma receptors. J Immunol. 2007;178(1):530–538. doi: 10.4049/jimmunol.178.1.530. [DOI] [PubMed] [Google Scholar]
- 26.Jenkins N, Curling EM. Glycosylation of recombinant proteins: Problems and prospects. Enzyme Microb Technol. 1994;16(5):354–364. doi: 10.1016/0141-0229(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 27.Martinez FO, et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: Similarities and differences. Blood. 2013;121(9):e57–e69. doi: 10.1182/blood-2012-06-436212. [DOI] [PubMed] [Google Scholar]
- 28.Mantovani A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Silva E, et al. HMGB1 and LPS induce distinct patterns of gene expression and activation in neutrophils from patients with sepsis-induced acute lung injury. Intensive Care Med. 2007;33(10):1829–1839. doi: 10.1007/s00134-007-0748-2. [DOI] [PubMed] [Google Scholar]
- 30.Radom-Aizik S, Zaldivar F, Jr, Leu SY, Galassetti P, Cooper DM. Effects of 30 min of aerobic exercise on gene expression in human neutrophils. J Appl Physiol (1985) 2008;104(1):236–243. doi: 10.1152/japplphysiol.00872.2007. [DOI] [PubMed] [Google Scholar]
- 31.Borrok MJ, Kiessling LL. Non-carbohydrate inhibitors of the lectin DC-SIGN. J Am Chem Soc. 2007;129(42):12780–12785. doi: 10.1021/ja072944v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc Natl Acad Sci USA. 2012;109(16):6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deban L, et al. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol. 2010;11(4):328–334. doi: 10.1038/ni.1854. [DOI] [PubMed] [Google Scholar]
- 34.Pilling D, Gomer RH. Persistent lung inflammation and fibrosis in serum amyloid P component (APCs-/-) knockout mice. PLoS ONE. 2014;9(4):e93730. doi: 10.1371/journal.pone.0093730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: Shaping immune responses. Nat Rev Immunol. 2009;9(7):465–479. doi: 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8(1):34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 37.Bharhani MS, et al. IL-10 protects mouse intestinal epithelial cells from Fas-induced apoptosis via modulating Fas expression and altering caspase-8 and FLIP expression. Am J Physiol Gastrointest Liver Physiol. 2006;291(5):G820–G829. doi: 10.1152/ajpgi.00438.2005. [DOI] [PubMed] [Google Scholar]
- 38.Xu W, et al. IL-10-producing macrophages preferentially clear early apoptotic cells. Blood. 2006;107(12):4930–4937. doi: 10.1182/blood-2005-10-4144. [DOI] [PubMed] [Google Scholar]
- 39.Olszak T, et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature. 2014;509(7501):497–502. doi: 10.1038/nature13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pilling D, et al. Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J Immunol. 2007;179(6):4035–4044. doi: 10.4049/jimmunol.179.6.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crawford JR, Pilling D, Gomer RH. Improved serum-free culture conditions for spleen-derived murine fibrocytes. J Immunol Methods. 2010;363(1):9–20. doi: 10.1016/j.jim.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]