

Significance
The programmed cell death (PCD) of mammalian cells plays important roles in fighting bacterial infections. Relatively little is known about the adaptive role of PCD in bacteria. Here we report the discovery of a potential PCD pathway in Pseudomonas aeruginosa. We show that activation of the system can occur in a subset of cells in response to DNA damage through cleavage of an essential transcription regulator that controls a cell lysis program. Although this is lethal to the individual cell in which it occurs, we find that PCD enhances the ability of the bacterium to cause disease. Our findings suggest that PCD is a strategy used by both host and pathogen to promote survival during an infection.
Keywords: Pseudomonas aeruginosa, essential regulator, cell lysis, lung colonization, programmed cell death
Abstract
In mammalian cells, programmed cell death (PCD) plays important roles in development, in the removal of damaged cells, and in fighting bacterial infections. Although widespread among multicellular organisms, there are relatively few documented instances of PCD in bacteria. Here we describe a potential PCD pathway in Pseudomonas aeruginosa that enhances the ability of the bacterium to cause disease in a lung infection model. Activation of the system can occur in a subset of cells in response to DNA damage through cleavage of an essential transcription regulator we call AlpR. Cleavage of AlpR triggers a cell lysis program through de-repression of the alpA gene, which encodes a positive regulator that activates expression of the alpBCDE lysis cassette. Although this is lethal to the individual cell in which it occurs, we find it benefits the population as a whole during infection of a mammalian host. Thus, host and pathogen each may use PCD as a survival-promoting strategy. We suggest that activation of the Alp cell lysis pathway is a disease-enhancing response to bacterial DNA damage inflicted by the host immune system.
Programmed cell death (PCD) refers to cell suicide that results from a genetically encoded program (1). It is well recognized that PCD occurs in mammalian cells, where it plays important roles in development, in the removal of damaged cells, and in fighting bacterial infections (1–4). In contrast to the situation in multicellular organisms, the proposal that PCD occurs in bacteria has been controversial, largely because the evolutionary advantage of PCD to a single-celled organism is unclear (5, 6). In bacteria, PCD appears to have roles in limiting the spread of bacteriophage (7), in the formation of biofilms (8, 9), and has been suggested to prevent the proliferation of compromised cells (10, 11). Whether PCD of pathogenic bacteria influences the ability of these organisms to cause disease has remained unclear.
Pseudomonas aeruginosa is a Gram-negative bacterium and an important opportunistic pathogen of humans (12). The organism is one of the most common causes of ventilator-associated and hospital-acquired pneumonia (13) and it is notorious as the principal cause of morbidity and mortality in cystic fibrosis (CF) patients (12). Chronic colonization of the CF lung by P. aeruginosa typically leads to progressive lung damage and, eventually, respiratory failure and death (12). A distinctive feature of P. aeruginosa is that it encodes a large number of putative transcription regulators (14). These regulators are speculated to facilitate adaptation of the organism to varied environments, including those within the human host.
Here we identify an essential transcription regulator in P. aeruginosa that we call AlpR. We show that AlpR is essential because it represses a previously undocumented PCD pathway. We present evidence that the AlpR-regulated PCD pathway can be activated in a subset of cells in response to DNA damage and promotes colonization of the murine lung. Our findings suggest that bacterial PCD can enhance the virulence of P. aeruginosa and that PCD functions altruistically during the course of an infection. In addition, these findings have implications for the role of PCD pathways in other pathogens. PCD may represent a survival strategy common to both host and pathogen that each mounts during the course of an infection.
Results
Loss of AlpR Results in Cell Lysis.
Analysis of the P. aeruginosa strain PAO1 genome revealed a putative transcription regulator encoded by the PA0906 gene that is highly conserved among different strains of P. aeruginosa and exhibits homology to the CI repressor protein from bacteriophage λ (λCI), and the LexA protein from Escherichia coli (Fig. S1A) (15). Although no transcription regulator has been definitively shown to be essential for growth of P. aeruginosa under standard laboratory conditions, PA0906 is among the set of candidate essential genes in P. aeruginosa strains PAO1 and PA14 (16, 17). We therefore reasoned PA0906, which we refer to here as AlpR, might be essential in P. aeruginosa because it represses the expression of genes whose products can be lethal. We first used a ClpXP-based protein depletion system to explicitly test whether or not AlpR is essential in P. aeruginosa strain PAO1 (18). Depletion of AlpR resulted in at least a 105-fold decrease in CFUs (Fig. 1A). Thus, loss of AlpR results in a loss of viability or inhibition of cell growth in PAO1. AlpR therefore appears to be essential in this strain of P. aeruginosa.
Fig. S1.

AlpR is related to the CI protein from bacteriophage λ (λCI) and to the LexA protein from E. coli, and is highly conserved among P. aeruginosa isolates. (A) Alignment indicating the relatedness of AlpR to LexA and to λCI. AlpR and LexA share 19.9% identity and 34.5% similarity, whereas AlpR and CI share 21.9% identity and 34.2% similarity. Conserved residues are indicated by a bar (|) symbol, whereas a period (.) and a colon (:) denote similar and highly similar residues, respectively. Gaps are shown with a hyphen (-). Pairwise alignments were produced using EMBOSS Needle using default parameters (43). (B) Alignment indicating the degree of conservation of AlpR (PA0906) among P. aeruginosa isolates. A conserved residue is denoted by an asterisk (*), and a period (.) and a colon (:) denote similar and highly similar residues, respectively. Alignment was generated by T-Coffee using default parameters (44).
Fig. 1.
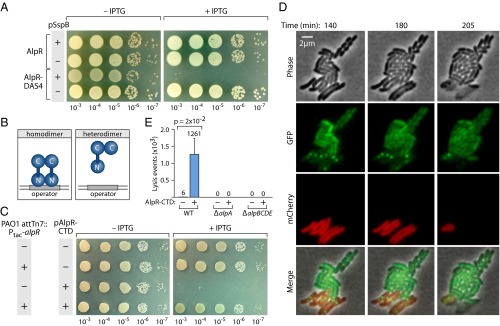
Loss of AlpR function results in cell lysis. (A) Depletion of AlpR with a ClpXP-based system. AlpR with a C-terminal DAS4 epitope tag is degraded by the ClpXP protease complex when the ClpXP adaptor protein SspB is supplied (18). IPTG-induced synthesis of SspB (supplied by pSspB) resulted in inhibition of growth or in cell death only in cells that synthesized AlpR-DAS4. (B) Genetic strategy for inhibition of AlpR function with the AlpR-CTD; the AlpR-CTD sequesters full-length AlpR into inactive heterodimers. (C) Ectopic synthesis of the AlpR-CTD results in cell death or the inhibition of cell growth in wild-type PAO1 cells but not in cells that contain an additional copy of alpR (PAO1 attTn7::Ptac-alpR). Plasmid pAlpR-CTD directs the synthesis of the AlpR-CTD in an IPTG-inducible manner. Wild-type PAO1 cells and PAO1 attTn7::Ptac-alpR cells containing pAlpR-CTD (pAlpR-CTD+) or the empty control vector (pAlpR-CTD–) were serially diluted and incubated on media lacking or containing IPTG. (D) Ectopic synthesis of the AlpR-CTD results in cell lysis. TLFM image sequence of mCherry-labeled cells (PAO1 attTn7::mCherry) synthesizing the AlpR-CTD and GFP-labeled control cells (PAO1 attTn7::gfp) that do not synthesize the AlpR-CTD. Images were acquired at 5-min intervals. (E) Quantification of the effect of the AlpR-CTD on the lysis of wild-type, ΔalpA mutant, and ΔalpBCDE mutant cells. Counting of lysis events in TLFM image sequences of mCherry-labeled cells that do or do not synthesize the AlpR-CTD.
Proteins related to AlpR, such as λCI and LexA, must be dimeric to bind the DNA and contain dimerization determinants in their C-terminal domains (CTDs) (15, 19). We found that the predicted CTD of AlpR could (i) interact with itself in a bacterial two-hybrid assay (Fig. S2A), and (ii) functionally substitute for the CTD of λCI (Fig. S2B), suggesting that the AlpR-CTD contains a dimerization determinant. We thought we might be able to interfere with the activity of AlpR in wild-type cells of P. aeruginosa by synthesizing the AlpR-CTD; our expectation was that the AlpR-CTD would sequester an AlpR monomer into an inactive heterodimer and function as a dominant-negative mutant (Fig. 1B). Indeed, synthesis of the AlpR-CTD in E. coli interfered with the binding of a hybrid repressor, in which the CTD of λCI had been replaced with the CTD of AlpR, to a λ operator (Fig. S2C). Consistent with the idea that AlpR is essential, ectopic synthesis of the AlpR-CTD appeared to be lethal to wild-type cells of PAO1 (Fig. 1C); similar results were observed when the AlpR-CTD was ectopically synthesized in P. aeruginosa strains CF18, PA14, and in the CF epidemic strain LESB58 (Fig. S3 A and B). Furthermore, ectopic synthesis of the AlpR-CTD did not result in lethality or impair growth in cells that contained a second copy of alpR (Fig. 1C), suggesting that the deleterious effect of the AlpR-CTD observed in wild-type cells is mediated by sequestration of AlpR.
Fig. S2.
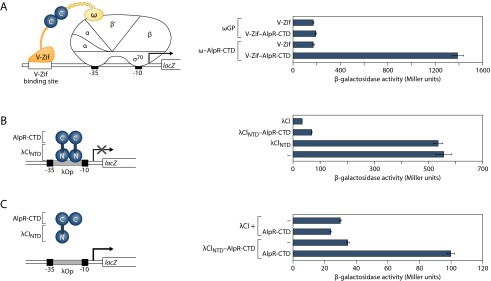
The predicted CTD of AlpR contains a dimerization determinant. (A) Bacterial two-hybrid analysis demonstrates interaction between AlpR-CTDs. Contact between AlpR-CTDs (denoted as “C”) fused to both the ω subunit of E. coli RNAP and to V-Zif activates transcription from the test promoter driving expression of lacZ. The diagram depicts test promoter placZif1, which bears a V-Zif-binding site positioned upstream of the lac core promoter. Effect of the ω–Gal11P (indicated ωGP), or ω–AlpR-CTD fusion protein on expression of the lacZ reporter in the presence of either the unfused V-Zif or the V-Zif–AlpR-CTD fusion protein. KDZif1ΔZ cells harboring compatible plasmids directing the IPTG-inducible synthesis of the indicated proteins were grown in the presence of 50 µM IPTG and assayed for β-galactosidase activity. This finding indicated that only the AlpR-CTD fusion proteins are capable of interacting. (B) The AlpR-CTD can functionally substitute for the dimerization domain of the CI protein from bacteriophage λ (λCI). FW123 cells containing a plasmid directing the IPTG-dependent synthesis of either the N-terminal domain and linker of λCI (λCINTD), λCI, or the λCINTD–AlpR-CTD fusion protein were grown in the presence of 5 µM IPTG and assayed for β-galactosidase activity. In this system the AlpR-CTD functionally replaces the dimerization domain of λCI. Occupancy of the λ operator (located between the promoter −35 and −10 elements) by λCI prevents RNAP from binding the promoter and represses transcription. (C) Ectopic synthesis of the AlpR-CTD specifically interferes with the ability of the λCINTD–AlpR-CTD fusion protein to bind the λ operator in E. coli, resulting in de-repression of the test promoter.
Fig. S3.

Ectopic synthesis of the AlpR-CTD is toxic in cells of clinical P. aeruginosa isolates, and causes a decrease in culture density and an increase in DNA in the culture supernatant of strain PAO1. (A) Ectopic synthesis of the AlpR-CTD results in cell death or the inhibition of cell growth in P. aeruginosa clinical isolates CF18 and PA14. CF18 and PA14 cells containing pAlpR-CTD were serially diluted and incubated on media lacking or containing IPTG. (B) Ectopic synthesis of the AlpR-CTD results in cell death or the inhibition of cell growth in the Liverpool Epidemic Strain, LESB58. LESB58 cells containing pL-AlpR-CTD (in which synthesis of the AlpR-CTD is under the control of a weaker promoter than in pAlpR-CTD) or the empty control vector (pEV) were streaked and incubated on media lacking or containing IPTG. (C) In wild-type PAO1 cells grown in liquid culture, induction of the AlpR-CTD resulted in a decrease in the OD600 of the culture over time that corresponded with an increase in the abundance of DNA in the culture supernatant (AlpR-CTD+), in comparison with wild-type cells that contained the empty vector control (pPSPK; AlpR-CTD−). Experiments were repeated three times and a representative dataset is shown.
We next sought to determine whether ectopic synthesis of the AlpR-CTD results in lethality or prevents cell growth. In wild-type PAO1 cells grown in liquid culture, induction of the AlpR-CTD resulted in a decrease in the OD600 of the culture and an increase in the abundance of DNA in the culture supernatant (Fig. S3C). To directly observe the phenotypic consequences of AlpR sequestration, we used time-lapse fluorescence microscopy (TLFM) (20). The growth of cells that synthesized mCherry and the AlpR-CTD was visualized alongside control cells that synthesized GFP alone. Following induction of the AlpR-CTD, most red fluorescent cells lysed, with some noticeably losing structural integrity or forming spheroplasts before lysis (Fig. 1D and Movie S1). Before cell lysis, green fluorescent foci appeared despite the absence of GFP in these cells (Fig. 1D and Movie S1). These fluorescent foci that appeared as a prelude to cell lysis might represent a marker for cell death, or may form in a manner that is mechanistically unrelated to the onset of cell death. Cell lysis did not occur in control cells that synthesized GFP (Fig. 1 D and E and Movie S1). Time-lapse microscopy revealed that the ClpXP-based depletion of AlpR also resulted in cell lysis (Movies S2 and S3). These findings indicate that the loss of AlpR function, either through ectopic synthesis of the AlpR-CTD or through depletion of AlpR, results in cell death through cell lysis. The name AlpR stands for “P. aeruginosa lysis phenotype repressor.”
AlpR Undergoes Autocleavage in Response to DNA Damage and Functions as a Repressor.
Like the related λCI and LexA proteins, AlpR contains a so-called Ser-Lys dyad that in LexA and in λCI mediates autocleavage (19). The autocleavage of LexA and λCI occurs in response to DNA damage, leading to inactivation of these regulators and the de-repression of target genes. We therefore asked whether AlpR undergoes autocleavage in response to DNA damage. Exposure of P. aeruginosa cells to the antibiotic ciprofloxacin, which leads to DNA damage, resulted in cleavage of wild-type AlpR but not the AlpR mutant AlpR(S153A) that contained amino acid substitution S153A that is predicted to prevent autocleavage (19) (Fig. 2B). Exposure of cells to hydrogen peroxide, which also damages DNA, resulted in cleavage of AlpR but did not result in cleavage of AlpR(S153A) (Fig. S4). These findings suggest that AlpR undergoes cleavage in response to DNA damage and establish that substitution S153A results in a mutant version of AlpR that cannot undergo autocleavage.
Fig. 2.

AlpR is a repressor that undergoes cleavage in response to DNA damage. (A) Schematic of alp gene cluster. (B) AlpR is cleaved in cells exposed to ciprofloxacin. Western blot analysis of AlpR with a vesicular stomatitis virus-glycoprotein (VSV-G) epitope tag fused to its N terminus (V-AlpR) and V-AlpR(S153A) following exposure of cells to ciprofloxacin for the indicated amounts of time. Arrow indicates AlpR cleavage product. (C) AlpR represses expression of the alp gene cluster. Quantification of transcript abundance by qRT-PCR in cells of the indicated strains following exposure to ciprofloxacin. Relative transcript abundance is shown in cells treated with ciprofloxacin compared with untreated cells. Expression of the PA0642 gene was induced in both the wild-type and AlpR(S153A) mutant cells, indicating that AlpR does not control the expression of all genes that are induced upon exposure to ciprofloxacin. (D) ChIP-Seq reveals that AlpR regulates the alp genes directly. ChIP-Seq with V-AlpR shows AlpR associates with the alpR-alpA and alpA-alpB intergenic regions.
Fig. S4.

AlpR is cleaved in cells exposed to hydrogen peroxide. Western blot analysis of AlpR with a vesicular stomatitis virus-glycoprotein (VSV-G) epitope tag fused to its N terminus (V-AlpR) and V-AlpR(S153A) following exposure of cells to hydrogen peroxide for the indicated amounts of time. Arrow indicates AlpR cleavage product.
Exposure of PAO1 cells to ciprofloxacin was shown previously to result in an increase in expression of alpR and the PA0907–PA0911 genes (named here alpABCDE) (21), which are in a putative operon that is located adjacent to and transcribed divergently from alpR (Fig. 2A). Exposure to ciprofloxacin induced expression of the alpR, alpA, alpB, alpC, and alpE genes in wild-type cells but not in cells that synthesized AlpR(S153A) (Fig. 2C). These findings suggest that AlpR, either directly or indirectly, represses expression of the alpR and alpABCDE genes.
ChIP-Seq Reveals That AlpR Regulates the alp Genes Directly.
To determine whether the effect of AlpR on expression of the alp genes might be direct or indirect we used chromatin immunoprecipitation coupled with high-throughput DNA-sequencing (ChIP-Seq) to identify those regions of the PAO1 chromosome that AlpR associates with. ChIP-Seq with an epitope-tagged version of AlpR revealed that AlpR associates with only two regions of the PAO1 chromosome: the alpR-alpA and alpA-alpB intergenic regions (Fig. 2D). Global transcription start-site mapping experiments in PAO1 (22) reveal the existence of promoters in each of these regions, and the expression of alpR promoter–, alpA promoter–, and alpB promoter–lacZ fusions was repressed by ectopic synthesis of AlpR (Fig. S5 A and B). AlpR therefore exerts its effect on expression of these target genes directly.
Fig. S5.

Repression of alp promoter-lacZ fusions by AlpR and activation of the alpB promoter-lacZ fusion by AlpA. (A) Ectopic synthesis of AlpR in E. coli resulted in repression of alpR promoter– and alpA promoter–lacZ reporter fusions, whereas ectopic synthesis of λCI did not. (B) Ectopic synthesis of AlpR in P. aeruginosa resulted in repression of an alpB promoter–lacZ reporter fusion, but did not repress a control orn promoter–lacZ reporter fusion. Plasmid pPSV38 served as an empty vector control. (C) Ectopic synthesis of AlpA in E. coli resulted in activation of an alpB promoter–lacZ reporter fusion, whereas activity did not increase in cells that contained the empty vector. Error bars denote SD from the mean.
The Essential Function of AlpR Is to Repress Expression of alpA.
Having established that AlpR represses expression of the alpABCDE genes, we wondered whether this might be the essential function of AlpR. That is, we wondered whether de-repression of alpABCDE results in cell lysis. Although the alp genes have not previously been shown to influence cell lysis, AlpB is annotated as a holin (14), which are small pore-forming proteins typically produced by bacteriophage that facilitate cell wall degradation and cell lysis at the end of a phage lytic cycle (23). Consistent with this idea, we found that AlpB could functionally substitute for the holin from bacteriophage λ in E. coli (Fig. S6) (24).
Fig. S6.

AlpB can functionally substitute for the holin from bacteriophage λ. Plasmid pS105 contains the λSRRzRz1 lysis cassette placed downstream from the pR′ promoter. The λS105 gene (except the portion that overlaps with the R gene) in the plasmids pS105 and pS105R− was replaced with the alpB gene to create pAlpB and pAlpBR−. Cultures of MC4100 λΔ(SR) carrying the pS105-derived plasmids were grown at 30 °C to A550 of 0.3, thermally induced for 15 min at 42 °C, and aerated at 37 °C thereafter. Lysis curves were obtained by measuring A550 after thermal induction. Time in minutes is shown. Cells containing pS105 (dark blue line) undergo rapid lysis after thermal induction, and pAlpB-containing cells also show a drop in turbidity after thermal induction (red line). The drop in turbidity is endolysin dependent, as cells that contain the plasmids pS105R− (purple line) and pAlpBR– (light blue line), that lack a functional R gene, do not show the same phenotype. Additionally, the lysis is holin dependent, as the cells containing the pSam7 plasmid (green line) that produces a truncated holin also does not undergo lysis.
AlpC may be an antiholin, a protein that counters the activity of a holin. In keeping with this notion, genes that encode antiholins can be found immediately downstream of those encoding the cognate holin, and AlpC is predicted to contain transmembrane domains, which are a feature common to antiholins (23). AlpD and AlpE are hypothetical proteins of unknown function, and AlpB, AlpC, and AlpD homologs are encoded by P. aeruginosa phage DE3 (25). De-repression of alpABCDE might therefore influence cell lysis through a holin-dependent mechanism and the alp genes, which are highly conserved in strains of P. aeruginosa, may have originated from a phage.
If the essential function of AlpR is to repress expression of the alpABCDE genes then a strain that lacked these genes would be expected to tolerate ectopic synthesis of the AlpR-CTD. Synthesis of the AlpR-CTD did not result in lethality in cells that lacked the alpABCDE cluster (Fig. 3A), suggesting that AlpR is essential because it represses expression of one or more of the alpABCDE genes. To assess the individual contribution of each of the genes in this operon to the lethality that results upon ectopic synthesis of the AlpR-CTD we constructed strains in which each of these genes (except alpB alone) was deleted. The results depicted in Fig. 3A suggest that alpA is essential for the cell lysis that occurs upon ectopic synthesis of the AlpR-CTD and that genes alpB, alpD, and alpE also contribute. Consistent with the possibility that AlpC might serve to limit the holin-like activity of AlpB, cells that lack alpC were found to be more sensitive to the lethal effects of the AlpR-CTD than wild-type cells (Fig. 3A).
Fig. 3.

The essential function of AlpR is to repress expression of alpA, which encodes a positive regulator of the alpBCDE lysis genes. (A) Effect of ectopic synthesis of the AlpR-CTD. Wild-type and mutant PAO1 cells containing plasmid pAlpR-CTD were serially diluted and incubated on media lacking or containing IPTG. (B) AlpA positively regulates expression of an alpB promoter–lacZ fusion. Cells harboring the indicated promoter–lacZ fusions and containing plasmid pAlpA or the empty control plasmid pPSV38 were assayed for β-galactosidase activity. (C) AlpA positively regulates expression of the alpBC genes. Quantification of transcript abundance by qRT-PCR in cells following exposure to ciprofloxacin. [Cells of the PAO1 alpA(S) mutant strain contain a premature stop codon early in the alpA ORF.] Relative transcript abundance is shown in cells treated with ciprofloxacin compared with cells of the PAO1 AlpR(S153A) mutant strain treated with ciprofloxacin.
AlpA Is a Positive Regulator of the alpBCDE Genes.
Structural prediction algorithms suggest that AlpA may contain a winged-helix DNA-binding domain. We therefore asked whether AlpA might be a positive regulator that in turn is required for expression of the alpBCDE genes. Ectopic expression of alpA strongly activated expression of an alpB promoter-lacZ fusion in PAO1 cells, demonstrating that AlpA is a positive regulator of the alpB promoter (Fig. 3B), and in cells of E. coli, suggesting that AlpA regulates the alpB promoter directly (Fig. S5C). Furthermore, comparing the effects of ciprofloxacin on the abundance of transcripts in cells of the PAO1 AlpR(S153A) mutant strain to those in wild-type cells and to those in cells that contain a premature stop codon early on in the alpA ORF, indicates that genes in the putative alpBCDE operon are positively regulated by AlpA (Fig. 3C). Consistent with the hypothesis that the principal role of AlpA in promoting cell lysis is to positively regulate expression of the alpBCDE genes, we found that cells lacking either alpBCDE or alpA did not lyse following ectopic synthesis of the AlpR-CTD (Fig. 1E and Fig. S7A). In addition, we found that ectopic expression of the alpBCDE cassette resulted in lethality (Fig. S7B). Inactivation of AlpR therefore initiates a regulatory cascade that de-represses alpA expression resulting in the production of a positive regulator that then goes on to drive expression of the alpBCDE genes.
Fig. S7.

Cells lacking alpBCDE tolerate ectopic synthesis of the AlpR-CTD and ectopic synthesis of alpBCDE results in lethality. (A) Cells lacking alpBCDE tolerate ectopic synthesis of the AlpR-CTD almost as well as cells that lack alpA. Wild-type PAO1 cells, cells of the PAO1 ΔalpA mutant strain, and cells of the PAO1 ΔalpBCDE mutant strain containing pAlpR-CTD were serially diluted and incubated on media lacking or containing IPTG. (B) Ectopic synthesis of alpBCDE results in lethality in strain PAO1. Wild-type PAO1 cells containing the empty vector pPSV38 or the alpBCDE expression plasmid pAlpBCDE were serially diluted and incubated on media lacking or containing IPTG.
DNA Damage Induces the Alp System in a Subset of Cells and Results in Cell Lysis.
We next asked whether treatment of PAO1 cells with ciprofloxacin resulted in a homogeneous or heterogeneous response. The analysis of cells containing an alpB promoter-yfp fusion by TLFM revealed that the addition of ciprofloxacin to cells for 1 h resulted in induction of alpB promoter activity in a subset of cells (Fig. 4 and Movies S4–S11). P. aeruginosa possesses a pyocin-associated cell death mechanism that can respond to DNA damage (26). Because we sought to define the causal link between alpBCDE induction and cell death upon DNA damage, we inactivated this potentially confounding pathway by deletion of prtN, a gene required for its activity (27). Importantly, essentially all of the ΔprtN mutant cells in which the alpB promoter-yfp reporter was induced lysed (Fig. 4 and Movie S8), whereas few of the ΔprtN ΔalpBCDE mutant cells in which the yfp reporter was induced lysed (Fig. 4 and Movie S10). These findings indicate that DNA damage-dependent induction of the Alp system results in cell lysis. Moreover, the strong association of alp induction and cell death indicates that triggering the Alp system commits a cell to death. Indeed, even in the presence of the pyocin system, we see that following DNA damage a significantly greater fraction of Alp-induced cells survive when alpBCDE are absent, implying that the Alp system contributes to the lysis of wild-type cells (Fig. S8 and Movies S4 and S6). In total, our findings suggest that the Alp system represents a PCD pathway that can be activated in a subset of cells in response to DNA damage.
Fig. 4.

Induction of the Alp system occurs in a subset of cells treated with a DNA-damaging agent and results in alpBCDE-dependent cell lysis. Representative cropped regions from TLFM image sequences of ΔprtN mutant cells and ΔprtN ΔalpBCDE mutant cells containing an alpB promoter–yfp fusion following exposure to ciprofloxacin. The addition of ciprofloxacin resulted in induction of alpB promoter activity in a subset of cells. Expression of the alpB promoter–yfp fusion was not induced in cells that were not exposed to ciprofloxacin (Movies S9 and S11). Yellow arrowheads point to alp-induced cells; red arrowheads point to cells that undergo lysis in the subsequent frame. Cells of the ΔprtN mutant strain in which the yfp reporter was induced lysed (Upper). Cells of the ΔprtN ΔalpBCDE mutant strain in which the yfp reporter was induced did not lyse (Lower). Images were normalized for background fluorescence. (Scale bar, 2 µm.)
Fig. S8.

The alpBCDE genes influence the survival of wild-type cells in which the alp genes are induced in response to DNA damage. Quantification of remaining alp-induced regions (a measure of surviving alp-induced cells) from time-lapse microscopy sequences taken after a 1-h ciprofloxacin pulse, or taken after no ciprofloxacin treatment. Wild-type and ΔalpBCDE mutant strains harbored an alpB promoter–yfp fusion. Final YFP-positive cells were normalized to the starting cell number. Error bars, SD; n = 3. See also Movies S4–S7.
The AlpR-Regulated PCD Pathway Enhances Colonization of the Host Lung.
P. aeruginosa is a notorious lung pathogen and is the principal cause of chronic and intractable pulmonary infections in individuals with CF, as well as a major cause of acute pneumonias in compromised individuals, such as those on mechanical ventilation (12, 13). P. aeruginosa elicits a strong neutrophil response, which includes the generation of reactive oxygen species including H2O2 as an antimicrobial defense (28, 29). We posited that P. aeruginosa present in the lung would experience DNA damage resulting in cleavage of AlpR and induction of the Alp PCD pathway in a subset of cells. We therefore asked whether the alp system influences the ability of P. aeruginosa to colonize the murine lung in a model of acute infection. Fig. 5A indicates that cells of the ΔalpA mutant strain, cells of the ΔalpBCDE mutant strain, and cells of the AlpR(S153A) mutant strain were unable to colonize the murine lung as well as wild-type cells. Furthermore, cells of a derivative of the ΔalpA mutant strain in which the alpA gene was restored (PAO1 alpA-IN), colonized the murine lung similarly to wild-type cells, demonstrating that the effect of the ΔalpA deletion on lung colonization was not the result of a secondary mutation introduced during construction of the ΔalpA mutant strain (Fig. 5A). Note that cells containing deletions of alpA or alpBCDE, or that synthesized AlpR(S153A), grew similarly to wild-type cells when grown in vitro (Fig. S9), ruling out the possibility that the colonization defects observed in vivo could be attributed to any differences in growth rate observed in vitro. These findings suggest that alpA, the alpBCDE genes, and the ability of AlpR to undergo autocleavage are all important for lung colonization.
Fig. 5.

The AlpR-regulated PCD pathway promotes P. aeruginosa colonization of the murine lung. (A) Ability of the indicated strains to colonize the murine lung. Male C57BL/6 mice were inoculated via the oropharyngeal route with equivalent numbers of wild-type PAO1 cells and the indicated mutant derivatives. Graphs indicate the mean and error bars indicate SD. After 24 h mice were killed and CFUs in the lungs were enumerated. (B) Ability of the indicated marked strains to colonize the murine lung when mixed with an excess of wild-type cells. PAO1 cells marked with a gentamicin-resistance cassette (GmR PAO1) and PAO1 ΔalpA cells marked with a gentamicin resistance cassette (GmR ΔalpA) were each mixed with an excess of unmarked wild-type PAO1 cells and inoculated into male C57BL/6 mice via the oropharyngeal route. After 24 h mice were killed and the percent gentamicin-resistant (GmR) cells compared with the total number of cells in the lungs was determined. Experiments were performed twice with similar results. A representative dataset is shown.
Fig. S9.

Cells of the ΔalpA mutant strain, the ΔalpBCDE mutant strain, and the AlpR(S153A) mutant strain grow identically to wild-type cells in vitro. Growth curves of cultures of cells of the ΔalpA mutant strain, the ΔalpBCDE mutant strain, the AlpR(S153A) mutant strain, and the wild-type PAO1 strain. Growth curves were obtained by measurement of the culture OD600 over a 24 h period. Error bars denote SD from the mean.
We hypothesized that the virulence defect in the ∆alpA strain was because of the fact that PCD-mediated lysis of a subset of the P. aeruginosa cells in the lung benefitted the remaining cell population rather than because of intrinsic defects in the virulence of individual cells upon the loss of this regulator. To test this, we compared lung colonization by tagged wild-type and tagged ΔalpA mutant cells in an excess of wild-type cells. When wild-type PAO1 with an intact PCD pathway dominated the population, colonization by cells of the tagged wild-type and tagged ΔalpA strains was indistinguishable (Fig. 5B). Taken together, these findings strongly suggest that in the acute lung infection model used here, PCD that occurs in a subset of cells through the de-repression of AlpR-controlled genes promotes colonization by P. aeruginosa. Although both the Alp and pyocin systems can contribute to the lysis of cells that occurs in response to DNA damage in vitro, we do not know their relative contributions to the lysis of P. aeruginosa that appears to occur in the host lung. Nonetheless, our findings demonstrate that the Alp system is functionally relevant in this context.
Discussion
The findings we have described reveal a possible PCD pathway in P. aeruginosa that can be activated in response to DNA damage in a subset of cells. DNA damage results in de-repression of alpA expression. In turn, AlpA positively regulates expression of the alpBCDE lysis genes. The cell lysis that ensues could provide nutrients or liberate toxins or other bacterial factors that might facilitate lung colonization by the remaining P. aeruginosa cells in the population (30, 31). PCD pathways in other pathogenic bacteria could enhance virulence in the same fashion.
The PCD of mammalian cells is thought to control bacterial infections through a variety of mechanisms. In particular, neutrophil extracellular traps that promote the killing of pathogenic bacteria are generated through apoptosis (3). In addition, eradication of infected cells through death-receptor–induced apoptosis is thought to limit disease caused by attaching and effacing pathogens, such as enteropathogenic E. coli (4). Our findings with the Alp system suggest that the PCD of a subpopulation of P. aeruginosa cells can enhance colonization of the host by the remainder of the cell population. PCD may therefore represent a survival-promoting strategy common to both pathogen and host during infection.
The Alp pathway has distinct similarities to—as well as distinct differences from—other PCD pathways that have been defined in both eukaryotes and prokaryotes. In Staphylococcus aureus, a holin encoded by cidA is thought to be responsible for mediating cell lysis in a PCD pathway that contributes to biofilm formation and might play a role in virulence (8, 32). We have found that AlpB can functionally substitute for the λ holin, suggesting that in P. aeruginosa AlpB might contribute to PCD in a manner related to CidA or holin-like proapoptotic factors of the BCL-2 family in eukaryotes (24). In E. coli, LexA has been shown to not only mediate the classic SOS response, but also to mediate a PCD pathway referred to as apoptosis-like death (ALD) in response to extensive DNA damage (10, 11). The Alp system is analogous to the ALD system in E. coli in that both are under the control of a repressor that becomes inactivated in response to DNA damage through autocleavage. However, in P. aeruginosa, LexA does not appear to control expression of the alp genes (21). Furthermore, in E. coli, ALD is not mediated by genes analogous to alpABCDE, and LexA does not exclusively control the expression of a dedicated death cassette like AlpR does in P. aeruginosa. The Alp system is therefore distinct from the ALD response with respect to both the genes that directly mediate PCD and the specific DNA damage-responsive regulator used to control PCD.
We cannot exclude the possibility that in the host lung, expression of the alpABCDE genes becomes induced, but to a degree that is insufficient to promote cell lysis. In this case, induction of the alp genes in a subset of cells would facilitate colonization of the lung by a lysis-independent mechanism. In relation to this, it is noteworthy that in Serratia marcescens, a protein with features similar to that of a holin has been implicated in promoting the secretion of chitinases (33). It is therefore conceivable that expression of the alp genes might promote colonization of the host lung through an effect on protein secretion. However, because we find that the vast majority of cells in which the alp system is induced in response to DNA damage undergo lysis, we favor the hypothesis that the lysis of Alp-induced cells plays a key role in facilitating infection of the host lung by the remainder of the cells in the population.
Pathogens present in the host can experience DNA damage as a result of the host immune response (28). Although host processes that generate DNA-damaging agents are principally thought to limit infections, our findings suggest that they can also act to promote the virulence of P. aeruginosa by activating the alp genes. Conceivably, treatment of P. aeruginosa infections with antibiotics, such as ciprofloxacin, that damage the DNA may also inadvertently promote the virulence of any surviving cells through the activation of the Alp system. Control of the alp genes by a DNA damage-responsive transcription regulator may provide a facile way for P. aeruginosa to respond to these insults. In addition, the Alp system may facilitate the survival of P. aeruginosa in environments other than those of the host. Damage of P. aeruginosa DNA resulting from phage infection or from the actions of competing microbes could result in Alp-mediated lysis in a subset of cells, thereby limiting the spread of phage or promoting biofilm formation, and in turn enhancing the survival of the population as a whole (5–9).
Materials and Methods
Plasmids and Strains.
All bacterial strains and plasmids used in this study are described in SI Materials and Methods.
AlpR Sequestration Assays on LB Agar Plates.
The pAlpR-CTD plasmid, synthesizing the AlpR-CTD, was introduced into the indicated strains by electroporation. Colonies of plasmid-containing cells were selected on LB agar containing gentamicin and resuspended in PBS to an OD600 of 0.01 (considered the 10−2 dilution). Tenfold serial dilutions of cells (10 µL) were spotted onto LB agar plates containing gentamicin with or without 10 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Plates were incubated overnight at 37 °C before being photographed.
Time-Lapse Microscopy.
Time-lapse microscopy sequences were acquired on a Nikon Ti-E inverted microscope with a 60× oil objective, automated focusing (Perfect Focus System, Nikon), a Xenon light source (Sutter Instruments), a CCD camera (Clara series, Andor), and image acquisition software (NIS Elements, Nikon), essentially as described previously (20). Additional experimental details are provided in SI Materials and Methods.
Western Blot Analyses.
Equal numbers of cells were lysed by sonication and separated by SDS/PAGE on 12% Bis-Tris NuPAGE gels (Invitrogen). Western blotting was performed essentially as described previously (18).
Quantitative PCR.
Triplicate cultures were grown from separate single colonies. Ciprofloxacin was added to cultures at OD600 0.5, and samples were harvested 120 min later. TriReagent was used for RNA isolation (Molecular Research Center) and cDNA synthesis was conducted as described previously for P. aeruginosa (22). Transcript abundances were determined relative to the 23S rRNA transcript by quantitative RT-PCR (qRT-PCR) with the iTaq SYBR Green Supermix (Bio-Rad) and an Applied Biosystems StepOnePlus detection system. The specificity of the PCR primers was verified by melting curve analyses. Relative transcript abundances are the average of three biological replicates. Error bars represent SD.
ChIP-Seq.
Strains PAO1 V-AlpR and PAO1 (the mock control) were grown in LB to an OD600 of 0.5. ChIP, library preparation, DNA sequencing, read mapping and peak calling is detailed in SI Materials and Methods.
β-Galactosidase Assays.
Cells were permeabilized with SDS and CHCl3 and assayed for β-galactosidase activity, as described previously (34). Assays were performed in triplicate. Values are averages based on three independent measurements from one experiment. Error bars represent the SD from the mean.
P. aeruginosa in Vivo Infection.
All animal experiments were performed as approved by the Dartmouth College Institutional Animal Care and Use Committee. Male C57BL/6 mice (Jackson Labs) 8–10 wk of age were inoculated with 2 × 107 CFU of P. aeruginosa suspended in sterile PBS via oropharyngeal aspiration essentially as described previously (35). Additional details are provided in SI Materials and Methods.
SI Materials and Methods
Bacterial Strains and Growth Conditions.
Pseudomonas aeruginosa strains PAO1 and PA14 were provided by Arne Rietsch (Case Western Reserve University, Cleveland, OH). PAO1 ΔsspB, LESB58, and CF18 have been described previously (14, 18, 36). Escherichia coli DH5 F′IQ (Invitrogen) was used for plasmid construction and maintenance, CSH100 and FW102 were used for lacZ reporter analyses (37), SM10 (λpir) was used for conjugal transfer of plasmids into P. aeruginosa, FW123 was used for the λCI-CTD functional substitution assays (37), KDZif1ΔZ was used for the bacterial two-hybrid assays (34), and MC4100 λΔ(SR) was used for the λ holin functional replacement assay (24). For introducing in-frame deletions by conjugal transfer, P. aeruginosa was selected for on Pseudomonas isolation agar (PIA), and grown on low-salt LB agar supplemented with 5% (wt/vol) sucrose at 37 °C for sacB counter selection. E. coli strains were supplemented with 15 µg/mL gentamicin, 10 µg/mL tetracycline, 100 µg/mL carbenicillin, 30 µg/mL kanamycin, and 25 µg/mL chloramphenicol, as needed. E. coli strains that contained plasmids with IPTG-inducible promoters were supplemented with the indicated IPTG concentration. For P. aeruginosa, gentamicin (LB: 30 µg/mL; PIA: 60 µg/mL) and tetracycline (LB: 35 µg/mL; PIA: 200 µg/mL) were added as needed. Liquid P. aeruginosa cultures were inoculated at a starting OD600 of 0.01 and grown with aeration at 37 °C in LB broth. For induction of IPTG-inducible promoters in liquid-grown P. aeruginosa cultures, IPTG was added at a concentration of 5 mM. Growth was monitored by measurement of optical density at 600 nm.
DNA Manipulations.
Standard molecular cloning procedures were followed. Primers used in this study were obtained from MWG and Sigma Life Sciences. DNA amplification was carried out using KOD polymerase (Novagen). DNA sequencing was performed by Genewiz Inc. Restriction enzymes were obtained from New England Biolabs.
Mutant Strain Construction.
The suicide vector pEXG2 (38) was used to make the in-frame deletion constructs pEXG2-∆alpR, pEXG2-∆alpA, pEXG2-∆alpBC, pEXG2-∆alpC, pEXG2-∆alpD, pEXG2-∆alpE, pEXG2-∆alpABCDE, pEXG2-∆alpBCDE, and pEXG2-∆prtN. In-frame deletion constructs were synthesized by amplification of regions of ∼400 bp flanking the deletion, ligation, and subsequent cloning into pEXG2. Allelic replacement in PAO1 with pEXG2-∆alpR, pEXG2-∆alpA, pEXG2-∆alpC, pEXG2-∆alpD, pEXG2-∆alpE, and pEXG2-∆prtN resulted in replacement of the ORF in question with ATGGCGGCCGCN-stop codon (where N is any base). As the genes of alpB and alpC overlap, the alpC ORF was replaced in PAO1 with ATGGTTGATCCGGCGGCCGCTTGA by allelic exchange using plasmid pEXG2-∆alpC, thereby leaving intact the alpB ORF (stop codon of alpB underlined). Allelic replacement in PAO1 using both pEXG2-∆alpABCDE resulted in replacement of alpABCDE, from the start codon of alpA to the stop codon of alpE, whereas pEXG2-∆alpBCDE resulted in replacement of the alpBCDE, from the start codon of alpB to the stop codon of alpE, with ATGGCGGCCGCCTAA. Deletions were confirmed by PCR.
Plasmid pEXG2 was also used to make mutant genes by allelic replacement (38). Plasmid pEXG2-alpA-STOP was generated by amplification of regions that flank and overlap base pairs 181–186 of alpA by the PCR, using oligonucleotides that integrate an in-frame BspHI restriction site (TCATGA; coding for Ser followed by a stop codon) in place of base pairs 181–186. The resulting products were ligated together and cloned into pEXG2. Allelic replacement in PAO1 with pEXG2-alpA-STOP resulted in replacement of the alpA gene with one that contains a premature stop codon early in the alpA ORF. Mutations were confirmed by the PCR, followed by digestion of the PCR product with BspHI.
Plasmid pEXG2-V-AlpR was generated by amplification of regions flanking alpR by the PCR, using oligos that also contained an in-frame VSV-G tag that was codon optimized for use in P. aeruginosa (ATGTATACCGATATCGAAATGAATCGCCTGGGCAAA). The resulting products were spliced together by overlap extension PCR and cloned into pEXG2. Allelic replacement in PAO1 with pEXG2-V-AlpR resulted in replacement of the alpR gene with one that contains an in-frame N-terminal VSV-G tag. Mutations were confirmed by the PCR, followed by digestion of the PCR product with EcoRV (restriction site underlined above).
Plasmid pEXG2-AlpR(S153A) was generated by amplification of regions that flank and overlap base pairs 457–458 of alpR by the PCR, using oligonucleotides that introduce a mutation that results in both the Ser153Ala substitution and the introduction of an NcoI restriction site. The resulting products were spliced together by overlap extension PCR and cloned into pEXG2. Allelic replacement in PAO1 with pEXG2-AlpR(S153A) resulted in replacement of the alpR gene with one that encodes the substitution Ser153Ala. Mutations were confirmed by the PCR, followed by digestion of the PCR product with NcoI.
Plasmid pEXG2-alpA-IN was generated by amplification of a 1.2-kb region centered on the alpA gene, and subsequent ligation into pEXG2. Allelic replacement in PAO1 ∆alpA with pEXG2-alpA-IN resulted in restoration of the ∆alpA lesion with the wild-type alpA lesion. Mutations were confirmed by PCR.
Plasmid Construction.
The AlpR-CTD expression plasmids were made as follows: oligonucleotides that contained a restriction site, a Shine–Dalgarno sequence, spacer region, and an in-frame start codon fused to the final 327 bp of alpR were used to generate a DNA fragment, which was ligated into pPSV38 (22) to generate plasmid pL-AlpR-CTD. DNA was subcloned from pL-AlpR-CTD into pPSPK (18) to generate pAlpR-CTD. Expression of the 110-aa CTD peptide was under the control of an IPTG-inducible promoter.
The expression plasmid pAlpA, used in Fig. 3B, was constructed by amplification by the PCR of the alpA gene using an oligonucleotide that added a Shine–Dalgarno motif and spacer to the alpA ORF, followed by ligation into pPSV38. The expression plasmid pAlpR, used in Fig. S5, was constructed by amplification by the PCR of the alpR gene using an oligonucleotide that added a Shine–Dalgarno motif and spacer to the alpR ORF, followed by ligation into pPSV38. The expression plasmid pAlpBCDE, used in Fig. S7B, was constructed by amplification by the PCR of the alpBCDE genes using an oligonucleotide that added a Shine–Dalgarno motif and spacer to the alpB ORF, followed by ligation into pPSV38.
Plasmid pUC18-mini-Tn7T-Plpp-alpR was generated by amplification of the lpp promoter from pBRα1 and ligation with pUC18-mini-Tn7T-LAC (39) to replace the lac promoter. A Shine–Dalgarno sequence, spacer region, and the alpR gene were placed downstream of the lpp promoter. The alpB fluorescent reporter plasmid pUC18-mini-Tn7T-alpB-yfp was derived as follows: the lac promoter and lacI gene of pUC18-mini-Tn7T-LAC was replaced with the yfp gene, which was amplified from pEYFP creating the resulting plasmid, pKHT190. The alpB regulatory region was amplified by PCR and ligated into pKHT190, upstream of the yfp gene and controlling its expression. The plasmids pUC18T-mini-Tn7T-Gm-GFP and pUC18T-mini-Tn7T-Gm-mCherry, which were used to generate P. aeruginosa attTn7::GFP and attTn7::mCherry strains, were described previously (20, 39).
The DAS4-tag integration vector pDIVT was generated by cloning a DNA fragment containing a sequence that specifies the peptide AANDENYSENYADAS followed by a stop codon, into the plasmid pP30ΔFRT (34). The peptide consists of the DAS4 SspB-dependent degradation tag (18). The aac1 gene that confers gentamicin resistance was replaced with the tet gene from plasmid mini-CTX-lacZ (40), which confers resistance to tetracycline. Plasmid pDIVT carries the mobilization region from RP4 (mob), the ColE1 origin of replication, the tet gene, FRT sites for efficient Flp recombinase-mediated excision, and DNA specifying the DAS4 tag. Plasmid pDIVT-alpR was generated by ligation of ∼300 bp of the 3′ portion of alpR into pDIVT in-frame with the DNA specifying the DAS4 tag.
Plasmids PalpA-lacZ, PalpB-lacZ, and PalpR-lacZ, the F′ episome lacZ reporter plasmids, were produced by amplification and ligation of the alpA, alpB, and alpR promoter regions into pFW11 (37), respectively. Plasmid pLX-AlpR was generated by amplification of the alpR gene followed by ligation into pLX10 (37), and it directs the synthesis of AlpR under the control of the lac promoter. Plasmid pCI was derived from plasmid pLX10 and directs the synthesis of the CI protein from bacteriophage λ under the control of the lac promoter (37).
The plasmid mini-CTX-lacZ (40) was used to make lacZ reporter fusions to the promoter regions of alpB and the control promoter, orn, by amplification of promoter regions by the PCR and ligation into mini-CTX-lacZ.
Induction of DNA Damage.
Cultures were grown to an OD600 of 0.5 and ciprofloxacin (Sigma-Aldrich) was added at a final concentration of 1 µg/mL for the specified times before harvesting. Hydrogen peroxide (Sigma-Aldrich) was added at a final concentration of 1 mM for the indicated times. For ciprofloxacin pulse experiments, growth was continued for 1 h after addition of ciprofloxacin. Cells were then washed and resuspended in LB. Slides were prepared and imaged in the phase and YFP channels as described below. To quantify the surviving, alp-induced cells in the wild-type PAO1 background, YFP pixels with intensity exceeding an empirically determined threshold in the final frame (485 min) were divided by the cell-associated pixels from the phase image in the initial frame using ImageJ. The mean values for three fields of view are displayed (Fig. S8).
Depletion Strain Construction and Depletion Assay.
The alpR depletion strain used in Fig. 1A was generated as follows: plasmid pUC18-mini-Tn7T-Plpp-alpR was used to integrate an lpp promoter-controlled alpR into the Tn7 attachment site in PAO1 ΔsspB, as described previously (39). Plasmid pEXG2-ΔalpR was used to generate a deletion of the native alpR gene in the resulting strain by allelic exchange. Deletions were confirmed by the PCR. Plasmid pDIVT-alpR, together with recipient strain PAO1 ΔalpR ΔsspB Plpp-alpR, was used to create strain PAO1 ΔalpR ΔsspB Plpp-alpR-DAS4 by integration of the plasmid into the chromosome. Plasmid pSspB directs the IPTG-inducible synthesis of SspB and confers resistance to gentamicin (18). The pSspB plasmid, synthesizing SspB, or the empty vector were introduced into the strains described above by electroporation. Colonies of plasmid-containing cells were selected on LB agar containing gentamicin and resuspended in PBS to an OD600 of 0.01 (considered the 10−2 dilution). Tenfold serial dilutions of cells (10 µL) were spotted onto LB agar plates containing gentamicin with or without 10 mM IPTG. Plates were incubated overnight at 37 °C before being photographed. For microscopy, cells were prepared in an identical manner, but were spotted on agarose pads containing 10 mM IPTG and imaged in the phase channel as described below.
Construction of Gentamicin-Resistance Marked Strains.
Gentamicin-resistant strains used in the mixing experiment (Fig. 5B) were generated by integrating the plasmid pUC18-mini-Tn7T-LAC (39) into the chromosomes of wild-type PAO1 and the ΔalpA deletion mutant, as previously described (39).
ChIP-Seq.
ChIP was performed with 80 mL of culture by using anti–VSV-G agarose beads (Bethyl Laboratories) for VSV-G–tagged proteins as previously described (18), except that cells were lysed and chromatin fragmented using a Bioruptor (Diagenode). Library preparation for ChIP-Seq was performed according to the TruSeq DNA Sample Preparation Guide (Illumina) with the following modification: adapters were diluted 10-fold before ligation and adapter-ligated DNA fragments were amplified by 15-cycle PCR using KOD polymerase (Novagen). ChIP libraries were sequenced by Elim Biopharmaceuticals, using an Illumina Genome Analyzer Ilx generating 36-bp reads. Read mapping and peak calling was essentially as described previously (41), except that reads were mapped to the P. aeruginosa PAO1 genome allowing up to one mismatch per seed. All of the mock IP data were merged and used as background for each biological replicate. Regions in each biological replicate were considered peaks if they were twofold enriched for reads over background, had a positive peak shift and strand correlation, and had a q-value of less than 0.01. Peaks were defined as the minimal region identified in all biological replicates.
λCI Dimerization and Bacterial Two-Hybrid Assays.
For the bacterial two-hybrid assays shown in Fig. S2A, the AlpR-CTD was amplified by the PCR and ligated in frame into the vectors pACTR-ω-GP and pBR-V-Zif-AP (42), resulting in pACTR-ω-AlpR-CTD and pBR-V-Zif-AlpR-CTD, respectively. Plasmids were introduced into the E. coli strain KDZif1ΔZ (34), and the assays were performed as described previously (34).
For the λCI-CTD dimerization-determinant assays in Fig. S2 B and C, the AlpR-CTD was subcloned from pAC-ω-AlpR-CTD into pACλCILN (42), to make pAC-λCINTD-AlpR-CTD. Plasmid pACλCILN encodes the N-terminal domain and linker (residues 1–132) of λCI followed by three alanine residues (λCINTD) under the control of the lacUV5 promoter (42). Plasmid pACλCI encodes full-length λCI, and pACΔCI is a version of pACλCI where a portion of the cI gene has been deleted. The indicated plasmids were introduced into the E. coli strain FW123 (37) and the assays were performed as described previously (34).
λ Holin Functional Replacement Assay.
Plasmids pS105, pS105R– and pSam7 were provided by Ry Young (Texas A&M University). Plasmid pS105 contains the λSRRzRz1 lysis cassette placed downstream from the pR′ promoter. Expression of genes in the lysis cassette can be induced by thermal induction. The plasmid pS105R– contains two stop codons in the R gene resulting in a truncated, nonfunctional endolysin, and the pSam7 plasmid contains a stop codon in the S105 gene, resulting in the production of a truncated, nonfunctional holin (24). The λ S105 gene (except the portion that overlaps with the R gene) in the plasmids pS105 and pS105R– was replaced with the alpB gene using overlap extension PCR, to create pAlpB and pAlpBR–. Cultures of MC4100 λΔ(SR) carrying the pS105-derived plasmids were grown at 30 °C to A550 of 0.3, then thermally induced for 15 min at 42 °C, and aerated at 37 °C thereafter. Lysis curves were obtained by measuring A550 after thermal induction (24).
Time-Lapse Microscopy.
For all microscopy experiments, midlog (OD600 = 0.5–0.8) P. aeruginosa cultures were concentrated 5× in LB. One microliter of each bacterial suspension was spotted onto a 1.5% (wt/vol) agarose growth pad [prepared using Vogel Bonner minimal media containing 0.2% (wt/vol) sodium nitrate, and 0.01% (wt/vol) casamino acids]. Automated image acquisition was performed for 8 h at 5-min intervals using image acquisition software (NIS Elements, Nikon) on a Nikon Ti-E inverted microscope with a 60× oil objective, automated focusing (Perfect Focus System, Nikon), a Xenon light source (Sutter Instruments), and a CCD camera (Clara series, Andor) in the indicated channels. For the lysis analysis in Fig. 1 E, P. aeruginosa attTn7::mCherry bearing pAlpR-CTD was compared with the same strain carrying pPSPK for the indicated genotypes (parental, ∆alpBCDE, and ∆alpA). Experiments were performed on agarose pads containing 5 mM IPTG and 30 μg/mL gentamicin, and data were acquired for phase and mCherry channels. For Movie S1, P. aeruginosa attTn7::mCherry (pAlpR-CTD) and P. aeruginosa attTn7::GFP (pPSPK) were mixed at a 1:1 ratio, spotted onto agarose pads containing 5 mM IPTG and 30 μg/mL gentamicin, and images were acquired in the phase, GFP, and mCherry channels. Data analysis was performed using custom Matlab based software as previously described (20). Briefly, cells were identified using a watershed algorithm and linked between frames. A rapid, sustained drop in cellular mCherry intensity was defined as a lysis event. Mean lysis events from four frames were determined; experiments were repeated independently at least three times.
P. aeruginosa in Vivo Infection.
Male C57BL/6 mice (Jackson Labs) 8–10 wk of age were inoculated with 2 × 107 CFU of P. aeruginosa suspended in sterile PBS via oropharyngeal aspiration following brief anesthesia with isofluorane (35). Animals were killed and lung tissue harvested 24 h postinfection. Whole lungs were homogenized in 1 mL of cold sterile PBS using a Tissue-Tearor (BioSpec Products, MODEL 398) at medium speed for 15 s. Lung homogenate was then diluted and plated on PIA. In the strain mixing experiment, the input comprised of unmarked wild type and either wild-type or alpA mutant carrying a gentamicin-resistance marker (50 unmarked, 1 marked). After 24 h, the lungs were harvested, homogenized, and the ratios of gentamicin-resistant bacteria to total bacteria were determined. The experiment was performed twice with no apparent or statistically significant differences in relative abundance detected between wild-type or alpA mutant in ability to colonize in the presence of a larger population of wild-type cells. Means were compared using Student’s t test with GraphPad Prism.
Supplementary Material
Acknowledgments
We thank Thomas Hampton (Geisel School of Medicine) for statistical support; Ry Young and Irina Chen (Texas A&M University) for plasmids and strains; Stephen Lory (Harvard Medical School) for strains; Ann Hochschild (Harvard Medical School) for comments on the manuscript; and Renate Hellmiss for artwork. This work was supported by National Institutes of Health Grants AI105463 (to S.L.D.), AI091702 (to D.A.H), and AI105268 (to J.D.M.); an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund (to J.D.M.); and a Graduate Research Fellowship from the National Science Foundation (to T.K.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1506299112/-/DCSupplemental.
References
- 1.Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24(23):2592–2602. doi: 10.1101/gad.1984410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raff M. Cell suicide for beginners. Nature. 1998;396(6707):119–122. doi: 10.1038/24055. [DOI] [PubMed] [Google Scholar]
- 3.Brinkmann V, Zychlinsky A. Beneficial suicide: Why neutrophils die to make NETs. Nat Rev Microbiol. 2007;5(8):577–582. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- 4.Pearson JS, et al. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature. 2013;501(7466):247–251. doi: 10.1038/nature12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engelberg-Kulka H, Amitai S, Kolodkin-Gal I, Hazan R. Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet. 2006;2(10):e135. doi: 10.1371/journal.pgen.0020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayles KW. Bacterial programmed cell death: Making sense of a paradox. Nat Rev Microbiol. 2014;12(1):63–69. doi: 10.1038/nrmicro3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hazan R, Engelberg-Kulka H. Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol Genet Genomics. 2004;272(2):227–234. doi: 10.1007/s00438-004-1048-y. [DOI] [PubMed] [Google Scholar]
- 8.Rice KC, et al. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc Natl Acad Sci USA. 2007;104(19):8113–8118. doi: 10.1073/pnas.0610226104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma L, et al. Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog. 2009;5(3):e1000354. doi: 10.1371/journal.ppat.1000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erental A, Sharon I, Engelberg-Kulka H. Two programmed cell death systems in Escherichia coli: An apoptotic-like death is inhibited by the mazEF-mediated death pathway. PLoS Biol. 2012;10(3):e1001281. doi: 10.1371/journal.pbio.1001281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erental A, Kalderon Z, Saada A, Smith Y, Engelberg-Kulka H. Apoptosis-like death, an extreme SOS response in Escherichia coli. MBio. 2014;5(4):e01426-14. doi: 10.1128/mBio.01426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15(2):194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chastre J, Fagon JY. Ventilator-associated pneumonia. Am J Respir Crit Care Med. 2002;165(7):867–903. doi: 10.1164/ajrccm.165.7.2105078. [DOI] [PubMed] [Google Scholar]
- 14.Winsor GL, et al. Pseudomonas Genome Database: Improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011;39(Database issue):D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sauer RT, Jordan SR, Pabo CO. Lambda repressor: A model system for understanding protein-DNA interactions and protein stability. Adv Protein Chem. 1990;40:1–61. doi: 10.1016/s0065-3233(08)60286-7. [DOI] [PubMed] [Google Scholar]
- 16.Jacobs MA, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2003;100(24):14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liberati NT, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci USA. 2006;103(8):2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castang S, McManus HR, Turner KH, Dove SL. H-NS family members function coordinately in an opportunistic pathogen. Proc Natl Acad Sci USA. 2008;105(48):18947–18952. doi: 10.1073/pnas.0808215105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo Y, et al. Crystal structure of LexA: A conformational switch for regulation of self-cleavage. Cell. 2001;106(5):585–594. doi: 10.1016/s0092-8674(01)00479-2. [DOI] [PubMed] [Google Scholar]
- 20.LeRoux M, et al. Quantitative single-cell characterization of bacterial interactions reveals type VI secretion is a double-edged sword. Proc Natl Acad Sci USA. 2012;109(48):19804–19809. doi: 10.1073/pnas.1213963109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cirz RT, O’Neill BM, Hammond JA, Head SR, Romesberg FE. Defining the Pseudomonas aeruginosa SOS response and its role in the global response to the antibiotic ciprofloxacin. J Bacteriol. 2006;188(20):7101–7110. doi: 10.1128/JB.00807-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldman SR, et al. NanoRNAs prime transcription initiation in vivo. Mol Cell. 2011;42(6):817–825. doi: 10.1016/j.molcel.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang IN, Smith DL, Young R. Holins: The protein clocks of bacteriophage infections. Annu Rev Microbiol. 2000;54:799–825. doi: 10.1146/annurev.micro.54.1.799. [DOI] [PubMed] [Google Scholar]
- 24.Pang X, et al. Active Bax and Bak are functional holins. Genes Dev. 2011;25(21):2278–2290. doi: 10.1101/gad.171645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kropinski AM. Sequence of the genome of the temperate, serotype-converting, Pseudomonas aeruginosa bacteriophage D3. J Bacteriol. 2000;182(21):6066–6074. doi: 10.1128/jb.182.21.6066-6074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakayama K, et al. The R-type pyocin of Pseudomonas aeruginosa is related to P2 phage, and the F-type is related to lambda phage. Mol Microbiol. 2000;38(2):213–231. doi: 10.1046/j.1365-2958.2000.02135.x. [DOI] [PubMed] [Google Scholar]
- 27.Matsui H, Sano Y, Ishihara H, Shinomiya T. Regulation of pyocin genes in Pseudomonas aeruginosa by positive (prtN) and negative (prtR) regulatory genes. J Bacteriol. 1993;175(5):1257–1263. doi: 10.1128/jb.175.5.1257-1263.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nathan C, Cunningham-Bussel A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13(5):349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang FC. Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nat Rev Microbiol. 2004;2(10):820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 30.Wagner PL, et al. Bacteriophage control of Shiga toxin 1 production and release by Escherichia coli. Mol Microbiol. 2002;44(4):957–970. doi: 10.1046/j.1365-2958.2002.02950.x. [DOI] [PubMed] [Google Scholar]
- 31.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. Extracellular DNA required for bacterial biofilm formation. Science. 2002;295(5559):1487. doi: 10.1126/science.295.5559.1487. [DOI] [PubMed] [Google Scholar]
- 32.Thomas VC, et al. A central role for carbon-overflow pathways in the modulation of bacterial cell death. PLoS Pathog. 2014;10(6):e1004205. doi: 10.1371/journal.ppat.1004205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamilton JJ, et al. A holin and an endopeptidase are essential for chitinolytic protein secretion in Serratia marcescens. J Cell Biol. 2014;207(5):615–626. doi: 10.1083/jcb.201404127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vallet-Gely I, Donovan KE, Fang R, Joung JK, Dove SL. Repression of phase-variable cup gene expression by H-NS-like proteins in Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2005;102(31):11082–11087. doi: 10.1073/pnas.0502663102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson AA, et al. Anr and its activation by PlcH activity in Pseudomonas aeruginosa host colonization and virulence. J Bacteriol. 2013;195(13):3093–3104. doi: 10.1128/JB.02169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolfgang MC, et al. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2003;100(14):8484–8489. doi: 10.1073/pnas.0832438100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whipple FW. Genetic analysis of prokaryotic and eukaryotic DNA-binding proteins in Escherichia coli. Nucleic Acids Res. 1998;26(16):3700–3706. doi: 10.1093/nar/26.16.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2005;102(22):8006–8011. doi: 10.1073/pnas.0503005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi K-H, Schweizer HP. Mini-Tn7 insertion in bacteria with single attTn7 sites: Example Pseudomonas aeruginosa. Nat Protoc. 2006;1(1):153–161. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 40.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. Integration-proficient plasmids for Pseudomonas aeruginosa: Site-specific integration and use for engineering of reporter and expression strains. Plasmid. 2000;43(1):59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 41.Ramsey KM, et al. Ubiquitous promoter-localization of essential virulence regulators in Francisella tularensis. PLoS Pathog. 2015;11(4):e1004793. doi: 10.1371/journal.ppat.1004793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castang S, Dove SL. High-order oligomerization is required for the function of the H-NS family member MvaT in Pseudomonas aeruginosa. Mol Microbiol. 2010;78(4):916–931. doi: 10.1111/j.1365-2958.2010.07378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 44.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302(1):205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.