

Significance
Prostate cancer has an unpredictable natural history: While most tumors are clinically indolent, some patients display lethal phenotypes. Serum prostate-specific antigen is the most often used test in prostate cancer but screening is controversial. Treatment options are limited for metastatic disease, hence the need for early diagnosis. Prostate cancer antigen 3 (PCA3), a long noncoding RNA, is the most specific biomarker identified and approved as a diagnostic test. However, its inherent biological function (if any) has remained elusive. We uncovered a negative transdominant oncogenic role for PCA3 that down-regulates an unrecognized tumor suppressor gene, PRUNE2 (a human homolog of the Drosophila prune gene) thereby promoting malignant cell growth. This work defines a unique biological function for PCA3 in prostate cancer.
Keywords: PRUNE2, PCA3, long noncoding RNA, ADAR, prostate cancer
Abstract
Prostate cancer antigen 3 (PCA3) is the most specific prostate cancer biomarker but its function remains unknown. Here we identify PRUNE2, a target protein-coding gene variant, which harbors the PCA3 locus, thereby classifying PCA3 as an antisense intronic long noncoding (lnc)RNA. We show that PCA3 controls PRUNE2 levels via a unique regulatory mechanism involving formation of a PRUNE2/PCA3 double-stranded RNA that undergoes adenosine deaminase acting on RNA (ADAR)-dependent adenosine-to-inosine RNA editing. PRUNE2 expression or silencing in prostate cancer cells decreased and increased cell proliferation, respectively. Moreover, PRUNE2 and PCA3 elicited opposite effects on tumor growth in immunodeficient tumor-bearing mice. Coregulation and RNA editing of PRUNE2 and PCA3 were confirmed in human prostate cancer specimens, supporting the medical relevance of our findings. These results establish PCA3 as a dominant-negative oncogene and PRUNE2 as an unrecognized tumor suppressor gene in human prostate cancer, and their regulatory axis represents a unique molecular target for diagnostic and therapeutic intervention.
Several lines of evidence demonstrate that long noncoding RNAs (lncRNAs) are functional in carcinogenesis through regulatory mechanisms such as promoter looping, alternative splicing, antisense gene silencing, transcriptional regulation, and DNA repair, thus potentially serving as tumor markers. A few lncRNA species have emerged as potential prostate cancer biomarkers such as prostate cancer gene expression marker-1 (PCGEM1) and prostate cancer noncoding RNA1 (PRNCR1), which enhance androgen receptor (AR)-dependent gene activation, and prostate cancer-associated ncRNA transcript-1 (PCAT1), which silences BRCA2 via posttranscriptional homologous recombination (1). Notably, the most specific biomarker in human prostate cancer identified to date is an lncRNA, prostate cancer antigen 3 (PCA3, also known as PCA3DD3 or DD3PCA3), which is up-regulated in human prostate cancer (2). Since its discovery more than 15 y ago, PCA3 has been extensively investigated (3) and has been approved for clinical applications to aid the diagnosis of prostate cancer in both the European Union and the United States. Paradoxically—despite its striking clinical specificity—the inherent cellular role of the lncRNA PCA3 in human prostate cancer, if any, remains completely unknown (1). Here we report a unique biological function for PCA3. Within a single functional genetic unit, we show that PCA3 is an antisense intronic lncRNA that down-regulates an as yet unrecognized tumor suppressor gene, a human homolog of the Drosophila prune gene, PRUNE2, through a process that involves RNA editing mediated by a supramolecular complex containing adenosine deaminase acting on RNA (ADAR) family members. We propose a working model in which PCA3 acts as a dominant-negative oncogene in prostate cancer and show consistent results in therapeutic preclinical models and in patient-derived human samples. Therefore, the molecular interaction of PRUNE2 and PCA3 is a candidate target for translational applications.
Results
PCA3 Is an Antisense Intronic lncRNA Within a Single PRUNE2 Transcriptional Unit.
Certain mammalian lncRNAs are embedded in the intronic-antisense regions of protein-coding genes (4–6). PCA3 is a spliced intronic antisense lncRNA embedded within intron 6 of the corresponding sense gene PRUNE2 (2, 7–10) (Fig. 1A). We hypothesized the existence of a functional role between PCA3 and PRUNE2, and their involvement in prostate cancer progression. To study this possibility, we investigated PRUNE2 as well as the PCA3 intronic antisense transcripts, which we cloned from MDA-PCa-133, a patient-derived xenograft (PDX) of bone metastasis from prostate cancer (11) (Fig. 1 A and B). We next analyzed the expression of PRUNE2 in representative panels of human tumors and nonmalignant cell lines by quantitative gene expression profiling with primers located in the PRUNE2 exons that flank PCA3 (Tables S1 and S2 and Fig. S1 A and B). PRUNE2 was detectable in prostate cancer cell lines, with the highest levels in androgen-dependent (LNCaP) cells, as well as in several brain and breast lines. We also analyzed PRUNE2 levels alongside PCA3 lncRNA in prostate cancer cells and observed differential expression of the two genes: LNCaP cells displayed the highest levels of both PRUNE2 and PCA3 relative to androgen-independent (DU145 and PC3) cells (Fig. S1C). We confirmed the expression of native or recombinant V5-tagged PRUNE2 by immunoblot analysis, and the predicted endogenous protein (∼337 kDa) was observed in LNCaP but not in PC3 cells (Fig. S1 D and E).
Fig. 1.
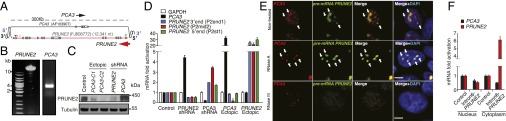
PRUNE2/PCA3 cloning, genomic structure, and colocalization. (A) Genomic context of intron/exon boundaries of PCA3 and PRUNE2 (GenBank accession no. FJ808772). Stars indicate missing or new exons; arrowheads indicate initiation (green) or stop (red) codons. Arrows indicate transcript orientation (black, PCA3; red, PRUNE2). (B) RT-PCR with RNA from the PDX MDA-PCa-133 used to clone/sequence PRUNE2. (C) Analysis of PRUNE2 in LNCaP cells stably expressing ectopic PCA3, PCA3-silenced, PRUNE2-silenced, or control. (D) qRT-PCR assays with primers (Table S1) amplifying PCA3 or different regions of PRUNE2 in LNCaP cells with silenced or ectopic PRUNE2 and PCA3. (E) Combined RNase resistance and RNA-FISH analysis. Before hybridization, LNCaP cells were treated with RNase A or RNase III. Hybridization was performed with specific probes against PCA3 and PRUNE2 transcripts. Nuclei are stained with DAPI. Arrows indicate foci. Confocal images are shown (bar, 10 µm). Fig. 1E represents 100× magnifications (from Fig. S3A). (F) Expression effects of intron6-PRUNE2 on nuclear and cytoplasmic PCA3 and PRUNE2 levels in LNCaP cells. Shown data are mean ± SD.
Table S1.
Oligonucleotide and probe sequences
| Oligonucleotide ID | Sequence |
| PRUNE2 amplification | |
| P2.rv | 5′ CCAAAACGAAGTCTAACAGACA 3′ |
| P2.fw | 5′ ACCCCGCTCGTCTTCCTT 3′ |
| PRUNE2- C1/C2 coding sequences subcloning | |
| P2-1enter.fw | 5′ CACCATGGAAGAATTTTTGCAACG 3′ |
| P2-1enter.rv | 5′ AGGCTTTTCTTTCAGCTTCAAGTC 3′ |
| P2-2enter.fw | 5′ CACCATGGAATCAGAGAAGATCTCAG 3′ |
| P2-2enter.rv | 5′ GAGAAGGTTACCTGAATCTCCTCC 3′ |
| Primers for RNA CHIP | |
| PRUNE2 118540.fw | 5′ TTTACGTTCTGGGATACATGTGC 3′ |
| PRUNE2 121974.rv | 5′ AGATCCCTGGGAGAAATGCCCGGCC 3′ |
| PCA3 ex3.fw | 5′ AAGAAAGGCTGCTGACTTTACCATCTGAGGC 3′ |
| PCA3 ex4.rv | 5′ GATCTTGAGATGCTTCCCAGCCTGTTCACAG 3′ |
| Primers for oligo hybridization (5′ biotinylated) | |
| PCA3 ex4.rv | 5′ GATCTTGAGATGCTTCCCAGCCTGTTCACAG 3′ |
| PRUNE2.rv | 5′ CTGTGAACAGGCTGGGAAGCATCTCAAGATC 3′ |
| Tubulin.rv | 5′ ATGCGCGAGATCGTGCACATCCAGGCGGGCCA 3′ |
| 5′-PRUNE2 | |
| P2st1.fw | 5′ CCACGACATGGAAGAATTTTTG 3′ |
| P2st1.rv | 5′ GCGTTTGCTTCGATTCAGTTT 3′ |
| P2st2.fw | 5′ TCTCCATCTGACAGCTCCTTTA 3′ |
| P2st2.rv | 5′ CTCATCACCGAGCAACTGGCT 3′ |
| PRUNE2 | |
| P2mid.fw | 5′ GGAGACCCAGTTCAGTGCTC 3′ |
| P2mid.rv | 5′ TGTAAATGCTTTCAAGTCACTGGT 3′ |
| 2mid2.fw | 5′ TCAGACGGTGAAATAAAAGTG 3′ |
| P2mid2.rv | 5′ AGGATCTCATCACAGCCAC 3′ |
| P2mid3.fw | 5′ AATCGAAGCAAACGCTTGGAG 3′ |
| P2mid3.rv | 5′ TGTAAATGCTTTCAAGTCACTG 3′ |
| PRUNE2_BMCC1 | |
| P2end1.fw | 5′ CGTTTATTTGCCGGTAGGAG 3′ |
| P2end1.rv | 5′ GCTCAGGCTCTTTGGTAGGA 3′ |
| P2end2.fw | 5′ GGGAAATGCTTTCACCACAG 3′ |
| P2end2.rv | 5′ CTCTTCAAAGGGGATGTCCA 3′ |
| P2end3.fw | 5′ TCAATAGCTTATCAGAACTCAGTGG 3′ |
| P2end3.rv | 5′ TCAACAGAACCATGAACCAGA 3′ |
| PCA3 | |
| AS1.fw | 5′ AGAAGCTGGCATCAGAAA 3′ |
| AS1.rv | 5′ CTGGAAATGTGCAAAAACAT 3′ |
| AS2.fw | 5′ TGGGAAGGACCTGATGATACA 3′ |
| AS2.rv | 5′ CCCAGGGATCTCTGTGCTT 3′ |
| PSA AS.fw | 5′ AGCATTGAACCAGAGGAGTTCT 3′ |
| PSA AS.rv | 5′ CCCGAGCAGGTGCTTTTG 3′ |
| GAPDH AS.fw | 5′ GAAGGTGAAGGTCGGAGTC 3′ |
| GAPDH AS.rv | 5′ TCAGAAGATGGTGATGGGATTTC 3′ |
| B2M.fw | 5′ AGCAGAGAATGGAAAGTCAAA 3′ |
| B2M.rv | 5′ TGCTGCTTACATGTCTCG 3′ |
| HPRT.fw | 5′ CTCAACTTTAACTGGAAAGAATGTC 3′ |
| HPRT.rv | 5′ TCCTTTTCACCAGCAAGCT 3′ |
| Probes for FISH RNA | |
| CY3-PCA3 Probe-A | 5′ AGTTTAGGCAGCAGGGCCAGAATCCTGACCCTCTGCCCCGTGGTTATCTCC 3′ |
| CY3-PCA3 Probe-B | 5′ CCCTGGAAAGATCTTGAGATGCTTCCCAGCCTGTTCACAGATCCCCTGG 3′ |
| CY3-PCA3 Probe-C | 5′ TCTAAACTCTTATAATCAAATTACACTTTTAGTATTTGCTGTCTC 3′ |
| CY3-PCA3 pre-mRNA | 5′ GAATAAAATTCCAAACCCATTCCCATGGCCTACAGACCTCTG 3′ |
| CY3-GFP Neg Ctl | 5′ ATGGAGAGCGACGAGAGCGGCCTGCCCGCCATGGAGATCGAGTGCCGCATCACCGGCAC 3′ |
| CY5-PRUNE2 | 5′ CCAGGGGATCTGTGAACAGGCTGGGAAGCATCTCAAGATCTTTCCAGGGTTATACTTACTAGC 3′ |
| CY5- GFP Neg Ctl | 5′ TTACGTCGCCGTCCAGCTCGACCAGGATGGGCACCACCCCGGTGAACAGCTCCTCGCCCT 3′ |
| Primers used for the synthesis of RNA probes | |
| T3BMCC1.fw | 5′ ATCGAAATTAACCCTCACTAAAGGGAGCTGTCAGATGGAGAAATAAAAGTG 3′ |
| T7BMCC1.rev | 5′ ATCGATAATACGACTCACTATAGGGCACCAGGATCTCATCACAGCCA 3′ |
| T3HESPCA3.fw | 5′ ATCGAAATTAACCCTCACTAAAGGGTGGGAAGGACCTGATGATACA 3′ |
| T7HESPCA3.rev | 5′ ATCGATAATACGACTCACTATAGGGGAGGGTCCCTAGAGAGACACGAA 3′ |
| T7GFP Neg ctl | 5′ ATCGATAATACGACTCACTATAGGGGCTACTTGTACATTATTCTT 3′ |
| Probes for Northern blot | |
| LNA-DIG PRUNE2 (NB-probe 2) | /5DigN/AGCAATCTGTCGTCTCGGCT/3Dig_N/ |
| LNA-DIG BMCC1 (3′PRUNE2) | /5DigN/TATCCTGAATCTCCTCCGTGA/3Dig_N/ |
| LNA-DIG PRUNE2 (5′ PRUNE2 NB-probe 1) | /5DigN/TGGTATGTTCAGCACTGGTAAA/3Dig_N/ |
ACTIN-DIG Probe Cytoplasmic RNA (Purchased from ROCHE).
Table S2.
Primers for RT-PCR, PCR, cloning, and editing analysis of dsRNA PCA3/Intron6-PRUNE2
| Primer name | Forward sequence (5′→3′) | Primer name | Reverse sequence (5′→3′) |
| Intron6-PRUNE2-1 | AGAAGAAATAGCAAGTGC | PCA3-1 | TCGGCTGCAGCCACACAA |
| Intron6-PRUNE2-2 | GAAAACGATGCCATAGAA | PCA3-2 | CATTCAAAACAATTGAC |
| Intron6-PRUNE2-3 | GGTTGCAGTGAGCCAAGA | PCA3-3 | TTTTTGAGGCAGAGTCT |
| Intron6-PRUNE2-4 | TCTACCTCTCCCACCTCG | PCA3-4 | GGCAAGGTTAAGATTTC |
| Intron6-PRUNE2-5 | ATGGCTATTCTCATCTTC | PCA3-5 | TCCTTGCTCTTTTACACC |
| Intron6-PRUNE2-6 | ATTTAATCACAAATCACT | PCA3-6 | TATTTTAATGTAGCATAG |
| Intron6-PRUNE2-7 | GTGCTGAGATTACAGGCG | PCA3-7 | CCTACAGAAATACAAGCA |
| Intron6-PRUNE2-8 | GAAACTAACATTTCAACC | PCA3-8 | ATTGTTGCAATAAACCTA |
| Intron6-PRUNE2-9 | ACAATGAGGAAGACTACG | PCA3-9 | AAAGTAGCCATTATTACC |
| Intron6-PRUNE2-10 | TTGCTACGTAGTCTTGAA | PCA3-10 | TGGTTTCCTTTAAGAGTT |
| Intron6-PRUNE2-11 | AGAATGGGAAGAAAGGTT | PCA3-11 | ATTACAGGAAGCCAGAT |
| Intron6-PRUNE2-12 | CGATCCTCCCACCTGACCC | PCA3-12 | AAAAATTAACTGGGCATG |
| Intron6-PRUNE2-13 | AAGAAAGGACTTTTTCACT | PCA3-13 | TCCCAAATTCTAAGACTC |
| Intron6-PRUNE2-14 | AGACAATCTCACGTGCACA | PCA3-14 | GCTTACTGTCTTTACTGA |
| Intron6-PRUNE2-15 | ACTCAGCGGAGATTCTCGGC | PCA3-15 | GGTGCAGTGGCAGATGTG |
| Intron6-PRUNE2-16 | TGTTTTCTAGTCAGAGAGG | PCA3-16 | CTAGAACACAGCAAGCAT |
| Intron6-PRUNE2-17 | AACTGAGAAATGGCATAACA | PCA3-17 | TATGAGATTGAATGATAA |
| Intron6-PRUNE2-18 | TCCTGACTGTGGGCTCAGAT | PCA3-18 | TGAAACATTCATTATCC |
| Intron6-PRUNE2-19 | GTGAACCCAGGAGGCAGAGC | PCA3-19 | GCTCTGTCGCCCAGGCTG |
| Intron6-PRUNE2-20 | TAAACTTAAAAATTACTTTC | PCA3-20 | TGTATATAACCTATTTTA |
| Intron6-PRUNE2-21 | TTTTTACAGCCTTGGGAAAG | PCA3-21 | GTAGTTGTCTTCATGTTTC |
| Intron6-PRUNE2-22 | TATTGGCCAGGCTGGTCTTG | PCA3-22 | CCCAGCACTTTGGGAGGCT |
| Intron6-PRUNE2-23 | TCCTGTTGTGGATATTTATT | PCA3-23 | ATTGGTAATGCTCACTTT |
| Intron6-PRUNE2-24 | TAACTTGTAATTCCTTGAA | PCA3-24 | AGAGACAAGGAAGAGCTT |
| Intron6-PRUNE2-25 | AGCTGGGGCTGTGCATCAGG | PCA3-25 | AGGGAATTCAAATTCTGG |
| Intron6-PRUNE2-26 | CTTTACGTTCTGGGATAC | PCA3-26 | CTTTACGTTCTGGGATAC |
| Intron6-PRUNE2-27 | AGAAGCTGGCATCAGAAAAA | PCA3-27 | CATTCAGTTAACAGTTTGGAAGG |
| Intron6-PRUNE2-28 | TATGTGGGTTGGCATTCTTG | PCA3-28 | TTTACGTTCTGGGATACATGTGC |
| Intron6-PRUNE2-29 | GGGAAGGACCTGATGATACA | PCA3-29 | CTGGAAATGTGCAAAAACAT |
| Intron6-PRUNE2-30 | AGTCCGCTGTGAGTCT | PCA3-30 | GCCTCAGATGGTAAAGTCAGC |
| Intron6-PRUNE2-31 | ATCGACGGCACTTTCTGAGT | PCA3-31 | CCCAGGGATCTCTGTGCTT |
| Intron6-PRUNE2-32 | AAGAAAGGCTGCTGACTTTACCATCTGAGGC | PCA3-32 | CCATTTCAGCAGATGTGTGG |
| Intron6-PRUNE2-33 | CTGTGAACAGGCTGGGAAGCATCTCAAGATC | PCA3-33 | AGATCCCTGGGAGAAATGCCCGGCC |
| Intron6-PRUNE2-34 | CCGAGAAGCTGGCATCA | PCA3-34 | GATCTTGAGATGCTTCCCAGCCTGTTCACA |
| Intron6-PRUNE2-35 | GCTGCAGCCGAGGGAGAC | PCA3-35 | TTTCAAATCTGTAATCCCGTTCAA |
| Intron6-PRUNE2-36 | TGGTGGGAAGGACCTGATGA | PCA3-36 | TTAAAGGGGCTGGAAATGTGC |
| Intron6-PRUNE2-37 | CCTTCTGGGCCCAACATTCT | PCA3-37 | GATCTTGAGATGCTTCCCAGCCTGTTCACAG |
| Intron6-PRUNE2-38 | CCCATCCCTCCAGCCTTATC | PCA3-38 | AGATCCCTGGGAGAAATGCCCGGCC |
| Intron6-PRUNE2-39 | TCATGCAGTGCAAATCCCCA | PCA3-39 | GCCCAGAAGGAACCGTAGAG |
| Intron6-PRUNE2-40 | CCTCGCATTTGTGGGTTCTC | PCA3-40 | TTTGCTTCACATCCCAGGCT |
| PCA3-41 | GAACCCACAAATGCGAGGTG | ||
| PCA3-42 | GGCCAGAAGCTAGCATCCAT | ||
| PCA3-43 | ACAAGGAGCCACTGGGTTTC |
Fig. S1.
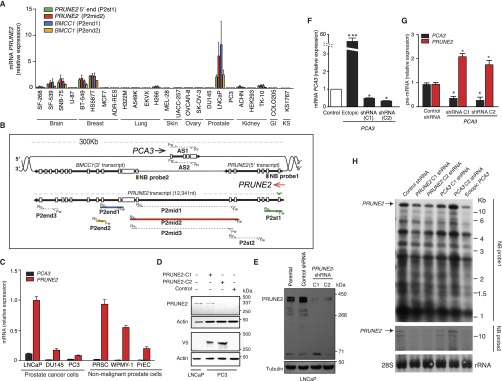
Expression of PRUNE2 and PCA3 transcripts and related constructs in human cells. (A) PRUNE2-related mRNA levels in a representative panel of human tumor cell lines. Transcripts were amplified through qRT-PCR with a set of specific primers indicated in parentheses (Table S1). (B) Genomic scheme depicting intron and exon boundaries of the locus. Arrows indicate orientation of transcripts (red, PRUNE2; black, PCA3). Representation of PRUNE2 pre-mRNA with the location of primer sets (Table S1) targeted to different regions encompassing the PRUNE2 and PCA3 sequences. Boxes (brown) represent Northern blot (NB) probes (NB probe 1, 5′-PRUNE2; NB probe 2, PRUNE2; Table S1) shown in H. (C) Relative mRNA expression of PRUNE2 and PCA3 in a panel of nonmalignant prostate-derived cells and prostate cancer-derived cells. Relative expression levels were compared against a panel of standard endogenous controls (Materials and Methods). Mean ± SD is shown. (D) Immunoblots probed with anti-PRUNE2 or anti-V5 epitope tag antibodies showing endogenous PRUNE2 expressed in prostate cancer LNCaP cells and ectopic expression of PRUNE2 in prostate cancer PC3 cells (PRUNE2-deficient). A GFP-expressing vector served as a negative control. (E) Immunoblotting analysis of whole cell extracts from LNCaP cells stably transduced with lentiviral shRNA constructs: two independent shRNA clones against PRUNE2 along with a negative control (nontargeting) shRNA were used. (F) Changes in PCA3 mRNA levels in prostate cancer LNCaP cells, from baseline (control shRNA), ectopic PCA3 expression, or endogenous PCA3-silencing by two independent shRNA constructs (termed PCA3-shRNA-C1 and PCA3-shRNA-C2. (G) Relative expression of PRUNE2 and PCA3 pre-mRNA levels in LNCaP cells stably expressing PCA3-shRNA or nontargeting shRNA control. (H) Northern blot analysis of RNA extracted from LNCaP cells stably expressing constructs as indicated. Corresponding primers depicted in A and B are color-coded.
PCA3 lncRNA Binds PRUNE2 Pre-mRNA and Regulates Its Levels.
Given that PCA3 is embedded within intron 6 of PRUNE2, and is transcribed in the antisense direction, we hypothesized that a double-stranded (ds)RNA forms between PCA3 lncRNA and PRUNE2 pre-mRNA to regulate PRUNE2 levels in prostate cancer. To evaluate this possibility, we first generated prostate cancer cell lines (LNCaP and PC3) stably transduced with ectopic PCA3, PCA3-shRNA, ectopic PRUNE2, PRUNE2-shRNA, or the corresponding controls. Levels of endogenous PRUNE2 protein, pre-mRNA and mRNA increased with PCA3 silencing and decreased with ectopic PCA3 expression (Fig. 1 C and D and Fig. S1 F–H). We confirmed these findings in prostate- and prostate cancer-derived cells, where ectopic PCA3 expression induced down-regulation of endogenous PRUNE2 expression (Fig. S2A). To determine whether PRUNE2 and PCA3 form a dsRNA, we used co-RNA-FISH assays. PCA3 and PRUNE2 hybridized in the same nuclear foci (Fig. 1E and Fig. S2A). These foci were completely depleted on treatment with RNase III, which degrades only dsRNA, but not with RNase A, which degrades only single-stranded (ss)RNA (Fig. 1E and Fig. S2B), indicating the formation of dsRNA from the physical association of PCA3 and PRUNE2 pre-mRNA. Next, to evaluate whether binding of PRUNE2 mRNA to PCA3 was required for the regulation of PRUNE2 levels, we assessed the effect of PCA3 on exogenous mature PRUNE2 cDNA, which has no sequence complementarity to PCA3 and therefore would be unable to form a dsRNA. Indeed, ectopic PCA3 did not affect the exogenous expression of PRUNE2 mRNA and protein (Fig. S3A). To complement this finding, we also designed and expressed a PRUNE2 construct that contains no protein-coding sequence but is still fully complementary to PCA3 (termed intron6-PRUNE2) and should therefore be able to bind PCA3 and possibly sequester it from PRUNE2. Consistent with this, overexpression of intron6-PRUNE2 caused an increase in endogenous PRUNE2 mRNA in the cytoplasm and a concomitant reduction in the nucleus (Fig. 1F). We confirmed a direct interaction between PCA3 and its corresponding antisense sequence (intron6-PRUNE2) by using RNase-resistant assays and co-RNA-FISH in tumor cells expressing both sequences (Fig. S3 B–E). These data suggest that PCA3 binding to PRUNE2 pre-mRNA controls PRUNE2 levels.
Fig. S2.

PRUNE2 protein expression in human prostate- and prostate cancer-derived cells and nuclear colocalization of PRUNE2/PCA3 RNA duplex. (A) Evaluation of PRUNE2 protein expression by immunoblot in a representative panel (n = 9) of human prostate- and prostate cancer-derived cells (Materials and Methods) stably transduced with either PRUNE2-shRNA, ectopic PCA3, or control constructs. (B–D) Prostate cancer LNCaP cells were subjected to either RNase A or RNase III treatment followed by labeled oligonucleotide (Table S1) hybridization as described (Materials and Methods). PCA3-cy3 (located either in PCA3 exon 4 for mature mRNA or in PCA3 intron 1 for pre-mRNA; as indicated) and PRUNE2-cy5 (located in PRUNE2 intron 6) oligomers were used. Pre-mRNA of PRUNE2 (green) and mRNA of PCA3 (red) are shown. Yellow color (merge panels) indicates PCA3 and PRUNE2 colocalization within the nucleus (DAPI, shown in blue). Labeled oligonucleotide-cy3 and -cy5 GFP probes served as negative controls (B). Ribonuclease digestion controls of the pre-mRNA (red) or mRNA (green) for PCA3 and PRUNE2 are shown (C and D).
Fig. S3.

Effects of PCA3 expression on PRUNE2 coding sequence and nuclear colocalization of PRUNE2 pre-mRNA and PCA3. (A) Analysis of combinatorial transfections in human prostate cancer PC3 cells (PRUNE2-deficient). Empty vector, V5-PRUNE2, intron6-PRUNE2, and ectopic PCA3 were used as indicated. Immunoblots with either a V5-tag antibody or an anti-PRUNE2 antibody demonstrate that PCA3 levels have no detectable effect on exogenous PRUNE2 expression compared to controls (outlined in red). (B and C) LNCaP cells stably overexpressing PCA3. RNase-resistance assay on nuclear RNA (B) followed by RT-PCR by using specific oligonucleotides for PCA3 and PRUNE2 pre-mRNA (Table S1). RNA from PC3 cells was used as negative control (C). (D) Cells were subjected to co-RNA-FISH using labeled oligonucleotides for PCA3 (located in exon 4; red) and PRUNE2, in red (located in intron 6; green). (E) HeLa cells stably coexpressing ectopic PCA3 and intron6-PRUNE2 RNA were subjected to co-RNA-FISH using labeled oligonucleotides for PCA3 (located in exon 4, red) and PRUNE2 (located in intron 6, green). (D and E) Yellow color (merge panels) indicates PCA3 and PRUNE2 colocalization within the nucleolus (DAPI, shown in blue). Representative images are shown. (Scale bars, 10 µm.)
ADARs Bind PRUNE2/PCA3 dsRNA and Regulate PRUNE2 Levels.
ADAR proteins are key regulatory enzymes for RNA editing and sequestering of noncoding RNA sequences, such as introns and untranslated mRNAs (5, 11–13), derived from the hybridization of retroinverted Alu elements (5, 13), with conversion of adenosine-to-inosine (A-to-I) RNA editing after nuclear dsRNA formation. Thus, we hypothesized that PCA3-PRUNE2 dsRNA may be regulated by ADAR-mediated RNA editing. To test this possibility, we used quantitative RT-PCR (qRT-PCR), co-RNA-FISH, and RNA-ChIP. We found that endogenous PCA3 and PRUNE2 pre-mRNAs colocalize to nuclear foci associated with ADAR proteins, which were sensitive to RNase III treatment (Fig. 2 A–C and Fig. S4). PRUNE2/PCA3 dsRNA and ADAR1 formed a complex only when both RNA species were coexpressed; the corresponding signals for PRUNE2/PCA3 dsRNA decreased after PCA3 or PRUNE2 silencing and increased with ectopic expression of PCA3 in a UV-induced RNA-protein cross-linking assay (Fig. 2D). To determine whether ADAR proteins regulate PRUNE2 and PCA3 levels, we silenced ADAR1 in human tumor cells and found increased PRUNE2 mRNA and protein levels (Fig. 2 E–G). We also found that ADAR-depleted prostate cancer cells have increased cytosolic PRUNE2 and PCA3 levels (Fig. 2 F and G and Fig. S5 A and B), revealing the importance of ADAR members in the regulation of both genes, consistent with functions of A-to-I editing in the regulation of noncoding RNA species (14). To gain functional insight into the regulation of PRUNE2 and PCA3, we established sensor/reporter assays in which either PCA3 or the PCA3 antisense sequence (i.e., intron6-PRUNE2) was fused to reporters to generate PCA3-luciferase or intron6-PRUNE2-GFP. Reporter expression (by FACS and luminescence assays) showed that the coexpression of intron6-PRUNE2-GFP plus PCA3 or intron6-PRUNE2 plus PCA3-luciferase results in reduction of the corresponding reporter signals compared to controls (Fig. S5 A–F). Thus, in addition to PCA3 regulating PRUNE2 levels, and consistent with our earlier results, intron6-PRUNE2 could also down-regulate PCA3 (Fig. 1F). Silencing of ADAR1 or ADAR2 increased the reporter signals, confirming that these enzymes are required for a coregulatory effect on both RNAs (Fig. S5 E–H).
Fig. 2.

PRUNE2/PCA3 colocalization to ADAR1. (A) RNA-ChIP and PCA3 and PRUNE2 binding by qRT-PCR in LNCaP cells. (B) Combined RNase resistance and RNA-FISH analysis. Before hybridization, LNCaP cells were treated with RNase A. Hybridization and immunostaining were performed with specific probes and an anti-ADAR1 antibody. (C) PCA3 and PRUNE2 binding to ADAR1 by RNA-ChIP. (D) Hybridization with biotin-labeled oligomers (Table S1) against PCA3 and PRUNE2 in LNCaP cells after UV-induced RNA-protein cross-linking. Immunoblot against ADAR1 is shown. (E and F) Evaluation of PCA3 and PRUNE2 expression in LNCaP cells stably expressing two independent lentiviral ADAR1-shRNAs: immunoblots against PRUNE2, ADAR1, and control protein (YY1) (E) and qRT-PCR (F) are shown. (G) Cytosolic (C) and nuclear (N) RNA fractionation followed by qRT-PCR; specific oligonucleotides served for amplification of nuclear pre-mRNA and cytosolic mRNA of PCA3 and PRUNE2. Shown data are mean ± SD. *P < 0.05; **P < 0.01.
Fig. S4.

Nuclear colocalization of PRUNE2/PCA3 RNA duplex with ADAR1/2. (A–D) LNCaP cells were subjected to either DNase or RNase treatment as indicated, followed by oligonucleotide hybridization. Combined RNA-FISH and immunofluorescence with an anti-ADAR1 (A–C) or anti-ADAR2 (D) antibodies. Labeled oligonucleotides PCA3-cy3 (located in exon 4) and PRUNE2-cy5 (located in intron 6) were used. PCA3 and PRUNE2 (red) and ADAR1or ADAR2 (green) are shown. Yellow color (merge panels) indicates either PCA3 and ADAR1 or ADAR2 colocalization or PRUNE2 and ADAR1 or ADAR2 colocalization within the nucleolus (DAPI, shown in blue). Labeled oligonucleotide-cy3 and -cy5 GFP probes served as negative controls (C). (E) HeLa cells stably coexpressing PCA3 and intron6-PRUNE2 RNA were subjected to RNase A treatment followed by oligonucleotide hybridization. Combined RNA-FISH and immunofluorescence with an anti-ADAR1 antibody. Labeled oligonucleotides PCA3 (located in exon 4) and PRUNE2 (located in PRUNE2 intron 6) were used. PCA3 and PRUNE2 (red) and ADAR1 (green) are shown. Yellow color (merge panels) indicates either PCA3 and ADAR1 colocalization or PRUNE2 and ADAR1 colocalization within the nucleolus (DAPI, shown in blue). Representative images are shown. (Scale bars, 10 µm.)
Fig. S5.
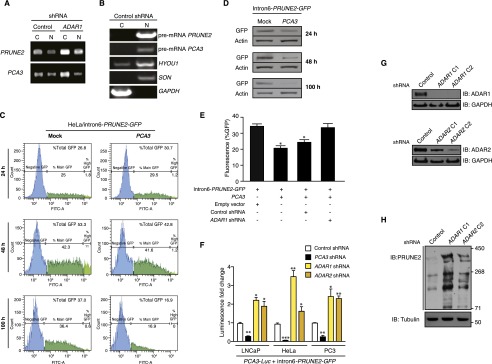
Analysis PRUNE2 and PCA3 regulation through ADAR-mediated mechanisms. (A) RT-qPCR on fractionated RNA from cytosol (C) or nucleus (N) from LNCaP cells stably expressing ADAR1-shRNA or negative control constructs. (B) Quality control of cytosolic and nuclear RNA-fractionation by agarose gel electrophoresis after RT-PCR amplification. PRUNE2 and PCA3 pre-mRNA species are detected mostly in the nucleus as well as the controls HYOU1 and SON (control nuclear mRNAs); in contrast, GAPDH mRNA (control cytosolic mRNA) is detected mostly in the cytosol. (C–E) HeLa cells stably expressing intron6-PRUNE2-GFP were transduced with either PCA3 or control constructs. Cells were analyzed for reporter GFP expression by FACS (C) and by immunoblotting with anti-GFP antibodies (D) after 24, 48, and 100 h. HeLa cells stably expressing intron6-PRUNE2-GFP, PCA3, ADAR1-shRNA, nontargeting shRNA control, or negative control (empty) lentivirus were analyzed after 48 h for the reporter GFP expression (E). (F) Human tumor cell lines stably coexpressing PCA3-luciferase (Luc) were transduced with intron6-PRUNE2-GFP or negative control expression vector. Tumor cells were lysed and luciferase activity was measured at 36 h after transduction. *P < 0.05, **P < 0.01, ***P < 0.001. (G and H) Evaluation of PRUNE2 levels in LNCaP cells stably expressing ADAR1- or ADAR2-shRNA, and control lentivirus. Quality control of two individual ADAR1- and ADAR2-shRNA constructs (G). Whole cell extracts from LNCaP cells expressing ADAR1- and ADAR2-shRNA constructs were probed with antibodies against PRUNE2 and tubulin (H).
RNA Editing of PRUNE2 and PCA3 RNA Species.
Our results thus far have indicated that ADAR proteins associate with PRUNE2/PCA3 dsRNA and regulate PRUNE2 and PCA3 levels via A-to-I RNA editing. To test this possibility directly, we evaluated the presence of A-to-I editing throughout the genomic coordinates of PCA3 and its corresponding antisense pre-mRNA intron6-PRUNE2 by RNA capture followed by next-generation sequencing. Although RNA editing is found largely within Alu elements, we carefully filtered out repetitive elements (such as Alu sequences) to avoid erroneous alignments. We showed that A > G/T > C changes, which reflect A-to-I editing, were the most frequent substitutions. Editing sites were distributed in intronic and exonic regions (Fig. 3 A and B), suggesting that a dsRNA hybrid is formed between pre-mRNA species of both genes, as observed in the RNA colocalization experiments. Given that the Drosophila behavior human splicing (DBHS) protein P54NRB preferentially binds to inosine-containing RNA (RNA-I) and regulates gene expression, we investigated a potential role for P54NRB and other DBHS proteins in regulating PRUNE2/PCA3. Both, PCA3 and PRUNE2 pre-mRNA species associated with P54NRB and the other two known mammalian family members (PSF and PSPC-1) compared with negative control RNA by RNA-ChIP (Fig. 2A) or combined co-RNA-FISH and immunofluorescence assays (Fig. S6). In addition, P54NRB-silenced prostate cancer cells had increased levels of PCA3 and PRUNE2 mature RNA (Fig. 3 C and D) and a concomitant increase of PRUNE2 protein levels relative to controls (Fig. 3E). These data confirm that PRUNE2 and PCA3 RNAs undergo A-to-I editing and reveal a functional role for DBHS proteins in their regulation.
Fig. 3.

Functional role of RNA editing and androgen receptor (AR) activation in PRUNE2/PCA3 regulation. (A and B) Identification, quantification, and distribution of A > G/T > C changes (features pathognomonic of A-to-I editing in both strands of the PRUNE2/PCA3 dsRNA) analyzed after RNA capture followed by high-throughput sequencing. Reads were aligned against hg19 of the region. Only nondbSNP variations indicated by at least three reads, and out of repetitive elements were considered. (A) Distribution and percentage of all possible alterations for the PCA3 genomic coordinates in LNCaP cells are shown. (B) RNA editing map for LNCaP cells showing the precise location of each A > G (green) or T > C (red) sites over PCA3 and intron6-PRUNE2 pre-mRNA species. Each square represents one individual base from the PCA3 locus (23,112 nt). Black borders delimit the bases of the four annotated exons (3,923 nt). Repeats (RepeatMasker) are shown in gray (B). (C–E) Evaluation of PCA3 and PRUNE2 levels in LNCaP cells stably expressing two independent P54NRB-shRNA clones (C1 and C2) or controls (NT). Detection of PCA3 and PRUNE2 mRNA cytosolic levels by RNA-FISH (C) and by qRT-PCR (D) are shown. Analysis of PRUNE2 expression in LNCaP P54NRB-silenced cells or negative control is shown (E). (F) Analysis of PRUNE2, AR, and phosphorylated AR (P-AR) expression in after concentration-dependent androgen stimulation with R1881. Representative PAGE 3–8% shown. (G) Relative mRNA expression levels of PCA3 and PRUNE2 transcript under R1881 stimulation. (H) Relative mRNA expression of PRUNE2, PCA3, and PSA (positive control) measured by qRT-PCR in LNCaP cells after dose-dependent R1881 stimulation. (I) RNA-FISH analysis for PCA3 and pre-mRNA of PRUNE2 in LNCaP cells under steroid-depleted conditions or after androgen stimulation.
Fig. S6.

Colocalization of PRUNE2/PCA3 with P54NRB. LNCaP cells were subjected to RNase pretreatment as indicated followed by oligonucleotide hybridization and immunofluorescence as described (Materials and Methods). (A–C) Detection of PCA3-cy3 (located in exon 4, red) and PRUNE2-cy5 (located in intron 6, green) by RNA-FISH and immunofluorescence with an anti-P54NRB antibody. Yellow color (merge panels) indicates PCA3 and pre-mRNA PRUNE2 colocalization with P54NRB; labeled oligonucleotide-cy3 against Actin mRNA (red) served as a negative control. (D and E) Combined RNA-FISH and immunofluorescence with an anti-PSF antibody. Labeled oligonucleotides PCA3-cy3 and PRUNE2-cy5 (located in intron 6) were used. PCA3 and pre-mRNA of PRUNE2 (red) and PSF (green) are shown. Either PCA3 and PSF or PRUNE2 and PSF colocalize (yellow) within the nucleus (DAPI, blue). (F) RNA-FISH and combined immunofluorescence with an anti–PSPC-1 antibody. A labeled PCA3 oligonucleotide (located in exon 4) and a PRUNE2 oligonucleotide (located in intron 6), both shown in red, and an anti–PSPC-1 antibody (shown in green) were used. Yellow color (merge panels) indicates either PCA3 and PSPC-1 colocalization or PRUNE2 and PSPC-1 colocalization within the nucleus (DAPI, shown in blue). (Scale bars, 10 µm.) Representative images are shown.
Function of the PRUNE2/PCA3 Regulatory Axis in Prostate Cancer.
Androgen dependence and resistance to androgen deprivation therapy are central to the biological and clinical features of prostate cancer. Thus, we investigated whether AR activation regulates PCA3 and PRUNE2 expression in androgen-dependent LNCaP cells, which had lower PCA3 and higher PRUNE2 levels than androgen-independent PC3 cells, when grown in steroid-depleted serum (Fig. 3 F and G and Fig. S1C). Androgen stimulation of LNCaP cells with a synthetic testosterone homolog (R1881) induced a concomitant increase of PCA3 and decrease of PRUNE2 levels (Fig. 3 F–H), consistent with a report that PCA3 modulates prostate cancer through AR signaling (15). We also observed an increase in nuclear localization of PRUNE2 and PCA3 along with androgen-induced responses (Fig. 3I). Thus, PRUNE2/PCA3 regulation appears to be sensitive to AR activation, a molecular hallmark of prostate cancer. To further assess the functional role(s) of the PRUNE2/PCA3 regulatory axis in prostate cancer, we generated LNCaP cells (PRUNE2-expressing) or PC3 cells (PRUNE2-deficient) stably expressing lentiviral constructs to silence or ectopically express PRUNE2 and PCA3 (Figs. S1 D–F and S3A). PCA3 silencing or ectopic PRUNE2 expression decreased cell proliferation and transformation in vitro; in contrast, PRUNE2 silencing or ectopic PCA3 expression increased cell proliferation and transformation (Figs. S7 and S8 A–C). Moreover, ectopic expression of PCA3 or antisense PCA3 (intron6-PRUNE2), which down-regulates PCA3, respectively decreased and increased endogenous, with no effect on exogenous mature PRUNE2 expression lacking complementarity with PCA3 (Fig. 1C and Figs. S2A and S8 D and E). Finally, we found that PRUNE2-deficient PC3 cells stably expressing ectopic PRUNE2 had lower levels of proliferation and transformation in vitro (Fig. S8 A and C). These results are consistent with the negative regulation of PRUNE2 by PCA3. We next investigated the downstream molecular mechanism(s) through which PRUNE2 suppresses tumor growth. PRUNE2 has three predicted functional domains (15): BCH, DHHA2, and PPX1 (Fig. S9A). BCH inhibits RhoA, a small GTPase that regulates the cytoskeleton, cell adhesion, and migration (16), whereas DHHA2 interacts with Nm23-H1, a metastasis suppressor (17). We found that endogenous PRUNE2 coimmunoprecipitates with RhoA and Nm23-H1 (Fig. S9 B–D). Consistent with an inhibitory role for PRUNE2 in RhoA signaling, PRUNE2 levels increased when LNCaP cells were grown in nonadherent culture conditions (Fig. S9E), and the distribution of PRUNE2 was inversely correlated with focal adhesion sites in LNCaP-derived spheroids (Fig. S9 C and F). In addition, we observed alterations in tumor cell adhesion and spreading, but no effect on apoptosis (Fig. S10 A–D). We also noted decreased adhesion, spreading, and migration of prostate cancer cells upon PRUNE2 expression and the opposite effect on ectopic expression of PCA3 or PRUNE2 silencing (Fig. S10 E–J). These results, along with the established functions of interacting proteins (16–18), suggest that PRUNE2 primarily decreases tumor growth by inhibiting cell proliferation but also affects adhesion, spreading, and migration. We subsequently extended these results to human tumor xenograft models; LNCaP prostate cancer cells stably expressing PRUNE2-shRNA, ectopic PCA3, PCA3-shRNA, or controls were s.c. administered into SCID mice. PRUNE2 silencing and ectopic PCA3 expression yielded markedly larger tumor xenografts than controls; in contrast, tumor growth was greatly diminished relative to controls when PCA3 was silenced (Fig. 4 A–C). Consistently, we observed increased serum prostate-specific antigen (PSA) concentrations in SCID mice that received LNCaP cells with ectopic PCA3 expression or PRUNE2 silencing compared to controls (Fig. 4D). In vitro, and also in tumor xenograft models, expression of antisense PCA3 (intron6-PRUNE2), which sequesters PCA3, decreased tumor growth in LNCaP but not in PC3 cells (Fig. 4 E and F and Fig. S8 A and B). Further, expression of ectopic PRUNE2 in LNCaP cells administered in SCID mice led to smaller tumors relative to controls (Fig. 4 G and H), illustrating the tumor suppressor activity of PRUNE2. Finally, silencing ADAR1, which increases PRUNE2 levels in LNCaP cells, reduced tumor cell proliferation in vitro and in vivo (Fig. 4 I and J and Fig. S11 A–C). These data show a functional role for the PRUNE2/PCA3 regulatory axis in prostate cancer. To explore the potential of clinical application of these findings, we specifically targeted the PCA3 sense strand with a modified siRNA (stealth RNAi-PCA3) serially administered to tumor-bearing mice with established prostate cancer xenografts. We observed tumor growth inhibition and serum PSA concentration reduction relative to scrambled siRNA control (Fig. 4 K–N). These results support the hypothesis that PRUNE2 expression has a functional tumor suppressive role in prostate cancer and suggest that the regulatory mechanism of PRUNE2 by PCA3 is a molecular target for intervention.
Fig. S7.

Function of PRUNE2/PCA3 in prostate cancer cells. (A) LNCaP cell proliferation in vitro after alteration of PCA3 and PRUNE2 levels or treatment with the androgen analog R1881. (B) Colony formation assay in soft agar medium of LNCaP cells transduced with ectopic PCA3 or PRUNE2, PCA3- or PRUNE2-shRNAs, or controls as indicated. In each experiment, mean ± SD is shown. *P < 0.05; **P < 0.01. (C) Cell cycle analysis by BrdU incorporation. (D) Cell-doubling time.
Fig. S8.
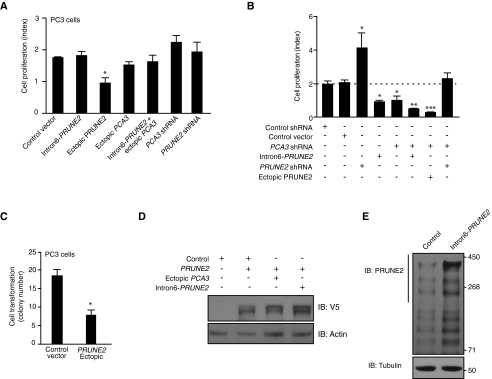
Epistasis analysis of the functional interplay between PRUNE2 and PCA3 in human prostate cancer cells. (A) Tumor cell growth analysis in PC3 cells stably expressing intron6-PRUNE2, ectopic PRUNE2, ectopic PCA3, intron6-PRUNE2 plus ectopic PCA3, PCA3-shRNA, PRUNE2-shRNA, or control shRNA constructs. (B) Tumor cell growth analysis in LNCaP cells stably expressing PCA3-shRNA, PRUNE2-shRNA, intron6-PRUNE2, V5-PRUNE2, or control constructs. (C) Anchorage-independent cell colony growth in soft agar medium of PC3 cells stably expressing V5-PRUNE2 or control vector. In each experiment, mean ± SD is shown. *P < 0.05, **P < 0.01, ***P < 0.001. (D) Immunoblots of extracts from LNCaP cells stably expressing PRUNE2-shRNA, PCA3-shRNA, ectopic PCA3, intron6-PRUNE2, or control shRNA constructs. (E) Immunoblots of whole extracts from LNCaP cells stably transduced with V5-PRUNE2, ectopic PCA3, intron6-PRUNE2, or control shRNA constructs. Antibodies against V5-tag or actin were used.
Fig. S9.
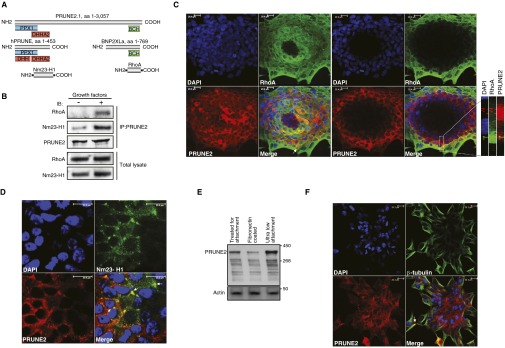
PRUNE2 domains, interactions, and colocalization with RhoA and Nm23-H1. (A) Predicted protein domains of PRUNE2 and putative interaction sites with RhoA and Nm23-H1. Conserved domains are indicated in blue (PPX1), red (DHH and DHHA2), and green (BCH), and protein sequences are indicated in gray. (B) PRUNE2 coimmunoprecipitation with RhoA and Nm23-H1. Starved LNCaP cell lysates were obtained after 10-min stimulation with a growth factor admixture (GF+; Materials and Methods) or BSA (GF−). Immunoprecipitation was performed with an anti-PRUNE2 antibody. Both RhoA and Nm23-H1 coprecipitated with PRUNE2 on growth factor stimulation. Total cell extracts before immunoprecipitation served as loading controls. (C and D) Immunostaining and confocal microscopy analysis of the entire LNCaP-derived spheroids were reconstructed by merging the full Z-section series. Colocalization of PRUNE2 (red) with RhoA (C); Nm23-H1 (D) is shown in green. DAPI, blue. Arrows indicate protein colocalization. (E) Immunoblots probed for PRUNE2 in LNCaP cell lysates grown in either nonadherent or adherent conditions as indicated. (F) Immunostaining and confocal microscopy analysis of the entire LNCaP-derived spheroids were reconstructed by merging the full Z-section series. Colocalization of PRUNE2 (red) and tubulin (green). DAPI, blue. Arrows indicate proteins colocalization.
Fig. S10.
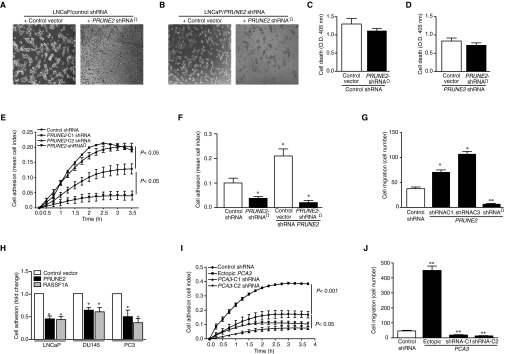
Biological function of PRUNE2 in prostate cancer cells. (A–D) PRUNE2 complementation assay in LNCaP cells stably expressing either control shRNA (A) or PRUNE2-shRNA (B) were transduced with an shRNA-resistant PRUNE2 (PRUNE2-shRNA) construct. In both cases, PRUNE2-shRNA expression resulted in a decrease in LNCaP cell adhesion and spreading. LNCaP cells stably expressing either control shRNA (C) or PRUNE2 shRNA (D) were transduced with PRUNE2-shRNA. In both cases, ectopic expression of PRUNE2-RNA did not result in significant changes in cell death. (E–J) Effects of PRUNE2 overexpression on prostate cancer cell adhesion and migration. (E–G) LNCaP cells stably expressing PRUNE2-shRNA, control shRNA, or two independent PRUNE2-shRNA constructs were evaluated for their capacity to adhere (E and F) and migrate (G) in the presence of RPMI containing 2.5% (vol/vol) FBS plus a growth factor admixture. (H) LNCaP, DU145, and PC3 prostate cancer cells were transfected with PRUNE2, an unrelated tumor suppressor gene RASSF1A (as a positive control), or vector alone as a negative control and analyzed for cell adhesion. (I and J) Effect of PCA3 overexpression on cell adhesion in LNCaP cells. Cells stably expressing PCA3, control shRNA, or two independent PCA3-shRNA constructs per gene were evaluated for their capacity to adhere (I) and migrate (J) in the presence of RPMI containing 2.5% (vol/vol) FBS plus a growth factor admixture.
Fig. 4.
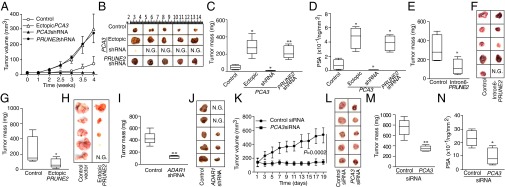
PRUNE2/PCA3 functions in tumor xenograft models of prostate cancer. (A–D) Male SCID mice received SC injection of 5 × 106 LNCaP cells stably expressing ectopic PCA3, PCA3-silenced, PRUNE2-silenced, or negative controls. Tumor growth was monitored and volume was measured (A). Tumors are shown after 4 wk (B); tumor mass (C) and serum PSA concentration (D) were determined. (E and F) Tumor growth in mice bearing LNCaP xenografts from cells stably expressing intron6-PRUNE2 (antisense sequence to PCA3) or controls. Tumor growth in SCID mice bearing PC3 xenografts of cells stably expressing either ectopic PRUNE2 or negative controls (G and H) and LNCaP cells stably expressing ADAR1-shRNA or negative controls (I and J). (K–N) PCA3 silencing in vivo by targeting SCID mice bearing LNCaP xenografts of cells stably expressing ectopic PCA3 constructs. Two cohorts of SCID mice with size-matched tumors (n = 10/group) received 8 μg of stealth chemically modified PCA3-siRNA or negative control-siRNA per dose; treatments and controls received a series of doses (n = 9) through alternating intratumoral or i.p. administration every other day. Tumor volumes, measured before each administration, were plotted over time (K), and representative tumors at the experimental end point are shown (L). Tumor xenograft mass (M) and serum PSA concentration (N) were determined at the end point. In each experiment, 6–10 mice per group were treated. N.G., no growth. Mean ± SD is shown. *P < 0.05, **P < 0.01.
Fig. S11.

Effects of ADAR1-silencing in prostate cancer cells. (A–C) Cell growth of LNCaP cells stably expressing ADAR1-shRNA or control shRNA. Cell proliferation (A), cell doubling time (B), and anchorage-independent colony formation in soft agar (C) are shown. In each experiment, mean ± SD is shown. *P < 0.05, **P < 0.01.
Levels of PCA3 and PRUNE2 Inversely Correlate in Human Prostate Cancer Specimens.
To determine the clinical relevance of our findings, we examined the expression of PCA3 and PRUNE2 in human prostate cancer. First, we performed qRT-PCR analysis on tumor RNA samples from prostate cancer patients (n = 48) and nonmalignant areas of the prostate (n = 9). PRUNE2 mRNA expression was detected more often in non–tumor-containing compared with the tumor-containing areas of the prostate (Fig. 5A). In contrast, PCA3 mRNA levels showed the opposite pattern, with high expression levels more frequently detected in tumors relative to nontumors, consistent with its role in the negative regulation of PRUNE2. To independently validate these clinical findings in silico, PRUNE2 and PCA3 expression levels were evaluated through Oncomine (19) in a large sample subset (n = 144) of primary nontreated prostate malignant tumors (n = 115) and nonmalignant prostate tissue (n = 29) (20). Although no statistically significant correlation with survival could be readily identified in this online dataset (20), a larger ongoing study is planned to fully address this question. Notably, to minimize variation, samples from prostate cancer-derived cell lines, metastatic lesions, and patients that received neoadjuvant therapy were excluded from the analysis. We next used The Cancer Genome Atlas (TCGA) as another unrelated large dataset (n = 50 nonmalignant control prostate samples; n = 333 prostate cancer samples) to validate the opposed expression between PRUNE2 and PCA3. We found that low PCA3 levels correlated with high PRUNE2 levels in nonmalignant control prostate samples and vice versa in prostate cancer samples (Fig. 5 B and C). Finally, we also analyzed the protein expression pattern of PRUNE2 in a large series of clinically annotated primary prostate cancer specimens (n = 145), matched to adjacent histologically normal prostate tissue (n = 145). In each case, immunohistochemical (IHC) staining was compared between the epithelial and stromal cells within tumors to the nonmalignant epithelial and stromal cells from adjacent nonmalignant areas of the same specimen (Fig. 5 D–F). We found a higher abundance of PRUNE2 in nonmalignant vs. malignant areas. The inverse correlation between the native expression of PRUNE2 and PCA3 mRNA in clinical samples again supports a meaningful role for their coregulation and tumor suppression in human prostate cancer.
Fig. 5.

PRUNE2/PCA3 expression in prostate cancer patient samples. (A) Analysis of PRUNE2 and PCA3 mRNA levels in human prostate cancer samples (n = 48) vs. nonmalignant prostate tissue (n = 9). Malignant (M) vs. nonmalignant (NM) control is indicated. PSA served as a positive control for the qRT-PCR. In each case, P values are for M vs. NM. (B) PCA3 and PRUNE2 expression levels of cDNA microarrays from Oncomine. Blue, nonmalignant gland (n = 29); red, prostate tumor (n = 115). Box-and-whisker plots with the data are presented with the horizontal lines within boxes representing median signal intensity. Black lines depict the calculated slopes linking to average intensity values. (C) RSEM. Normalized RNA-Seq data from TCGA for PCA3 and PRUNE2 mRNA in NM (blue) or M (red) from human prostate specimens are shown. Box-and-whisker plots with the corresponding data are presented with the horizontal lines in the boxes representing the median signal intensity. Black lines depict the calculated slopes linking to average intensity values. (D) Human TMA of prostate cancer samples showing high-abundance of PRUNE2 in NM adjacent prostate tissue control compared with M. In each case, IHC staining (i.e., % extent of expression in cells) was analyzed. Magnification, 20×. (E and F) PRUNE2 estimated expression in the epithelium (E) or stromal (F) component of the tumor samples (n = 145) with low-grade (n = 50) and high-grade (n = 95) vs. NM adjacent control tissues (n = 145) in human prostate cancer specimens. Mean ± SD is shown. **P < 0.01; ***P < 0.001.
RNA Editing of PCA3 and PRUNE2 in Human Prostate Cancer Patients.
We ultimately analyzed specimens obtained from index prostate cancer patients through RNA capture followed by next-generation sequencing and detected the presence of RNA editing (Fig. 6A), which was subsequently confirmed by classic Sanger sequencing (Fig. 6B) of genomic and cDNA clones from the same index patients. Bioinformatics demonstrated A > G/T > C alterations as the most frequent substitutions and data indicative of A-to-I editing in both PCA3 and PRUNE2 pre-mRNA strands, with no clear editing hotspots identified in human tumor samples (Fig. 6C). The editing maps provided for all patients show a similar distribution of alterations for both RNA strands, suggesting the interaction of the pre-mRNAs of both PCA3 and PRUNE2 transcripts (Fig. 6D).
Fig. 6.

RNA editing in specimens from prostate cancer patients. (A) All possible alterations, including putative editing sites, were determined as described for LNCaP cells and shown for each tumor sample. (B) A subset of editing sites suggested by large-scale sequencing was confirmed by PCR (with gDNA or cDNA as templates) followed by Sanger-sequencing. (C) Intersections of the putative edited sites among each independent patient sample are depicted. (D–F) Individual RNA editing maps of three prostate cancer patients are shown: Distribution of A > G (green)/T > C (red) sites over PCA3 and intron6-PRUNE2 pre-mRNA. Each square represents one individual base from the PCA3 locus (23,112 nt). Black borders delimit the bases of the four annotated exons (3,923 nt). Repeats (RepeatMasker): gray.
Discussion
lncRNAs have recently emerged as central regulators of gene expression in various biological settings, but only a few have known functional roles in human prostate cancer (1, 4, 5, 21–24). Here we present extensive data that are consistent with an antisense intronic lncRNA (i.e., PCA3) that acts by an ADAR-mediated RNA editing mechanism to down-regulate its target gene (i.e., PRUNE2). In this study, we establish the functional attributes of PCA3 as a transdominant negative oncogene that inactivates the unrecognized tumor suppressor gene PRUNE2 at the RNA level through an ADAR-mediated mechanism; such a remarkable regulatory unit located in a single genetic locus appears unique to human mammalian cells. Notably, the genomic region encompassing PRUNE2 contains several alternatively spliced isoforms (25–28), one of which is ∼3 kb shorter than the PRUNE2 full-length sequence identified here (presumably the canonical gene) and is found in human adult nerve cells (25), with a related mouse brain-specific isoform (26); thus, other tissue-specific isoforms with different functions may perhaps exist. Tumor suppressor genes have long been shown to affect cancer growth in the classic two-hit hypothesis (29, 30). More recently, it became clear that even partial inactivation of tumor suppressors contribute critically to tumorigenesis (31), as illustrated here. In sum, we show a striking function for the clinically well-established PCA3 marker that will lead to translational applications in human prostate cancer.
Materials and Methods
Details can be found in SI Materials and Methods. PCA3 and PRUNE2 sequence analyses were evaluated from cDNA microarray with Oncomine (19) or RNA-seq data from TCGA; expression was calculated by RNA-Seq by expectation maximization (RSEM) (32). Cell fractionation, nuclear RNA analysis, and immunoprecipitation/immunoblot were performed as previously described (13). siRNA and shRNA were custom-ordered against PCA3 or PRUNE2 (Table S1), respectively, and transfected into tumor cells (Ambion). Custom-ordered siRNAs against PCA3 (Tables S1 and S2) were transfected into tumor cells with the NeoFX transfection reagent (Ambion). RNA FISH and confocal microscopy RNAs were performed to detect PCA3 and PRUNE2. Cell culture and functional assays (cell proliferation, viability, adhesion, migration, soft agar colony formation, and tumor cell-derived spheroids) were performed. Tumor-bearing mouse models are described elsewhere (11). All animal experimentation was reviewed and approved by the Institutional Animal Care and Use Committee of the University of Texas M.D. Anderson Cancer Center (MDACC). Experiments with human samples were reviewed and approved by the Clinical Research Committee and by the institutional review board (IRB) at MDACC. All human specimens were obtained after the patients provided written informed consent under an IRB-approved experimental protocol. Total RNA samples purified from tumors from human prostate cancer patients were also obtained from the Tumor Bank at A.C. Camargo Cancer Center after IRB approval.
SI Materials and Methods
Reagents.
Anti-BrdU (Millipore), anti−β-actin, anti−β-tubulin (ECM Biosciences), ChIP grade anti-RED1, anti−Nm23-H1, anti-RhoA (Abcam), anti-PRUNE2 (ProteinTech), anti-AKT, anti-pAKT1, anti-pERK1/2, anti-p44/42 MAP kinase, ßanti-S6RP (Cell Signaling Technology), anti-ADAR1 (Sigma or Abnova), and anti-ADAR2 (Sigma) were commercially obtained. VEGF, basic FGF (bFGF), EGF, and insulin-like growth factor (IGF) were purchased from R&D Systems. An admixture (i.e., 10 ng EGF, 10 ng bFGF, 10 ng IGF, and 20 ng VEGF) supplemented with heparin (5 U/mL) to a final concentration of 50 ng/mL served for growth factor stimulation unless otherwise specified. Methyltrienolone (R1881; Perkin-Elmer) was used for androgenic stimulation in steroid-deprived conditions as indicated. RNaseA and RNase III (Invitrogen), DNaseI-RNase free (NEB), P54NRB, AR, pAR, and charcoal-stripped FBS (Invitrogen) were commercially obtained. Secondary antibodies were purchased from Jackson ImmunoResearch or Invitrogen.
Cell Lines and Tissue Culture.
Human tumor cell lines used (HeLa, LNCaP, PC3, DU145, SF-268, SF-539, SNB-75, U-87, BT-549, Hs587T, MCF-7, NCI-ADR-RES, NCI-H322M, A549K, EKVX, NCI-H266, SK-MEL-28, UACC-257, OVCAR-8, SK-OV-3, ACHN, HEK293, TK-10, KS1767, and COLO205) were grown in RPMI containing 5% (vol/vol) FBS. Human epithelial (PrEC) and stromal (PRSC) primary prostate cells, epithelial (RWPE-1 and RWPE-2) and stromal (WPMY-1) transformed prostate cells, and prostate cancer cells (VCaP and 22Rv) were cultured in optimized medium (ATCC).
Bioinformatics and Sequence Analysis.
Chromosomal locations, annotated transcripts, spliced expressed sequence tags, and sequence mapping were visualized on the Genome Browser from the University of California-Santa Cruz (https://genome.ucsc.edu), by using the latest version of the human genome assembly (hg19) available. Conserved domain analyses were performed through the Conserved Domain Database (www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), and sequence alignments were made with either ClustalW (www.ebi.ac.uk/Tools/msa/) or CLC bio (www.clcbio.com). PCA3 and PRUNE2 expressions were evaluated from cDNA microarray data of prostate cancer using Oncomine (1) or RNA-Seq data from the TCGA Research Network (cancergenome.nih.gov); for the latter, data were downloaded and expression values were calculated through RSEM (2) using all available samples.
Cloning and cDNA Generation.
Total RNAs from tumor cell lines or xenografts were isolated through the RNeasy kit (Qiagen), the All-in-One kit (Norgen Biotek), or the TRIzol reagent (Life Technologies). Total RNA samples from human normal tissues (prostate, brain, liver, kidney, breast, lung, pancreas, spleen, and testis) were commercially obtained (Stratagene). cDNAs were synthesized by using the SuperScript III reverse transcriptase (Invitrogen or Promega) from total RNA, with N15 random pentadecamers, oligo dT primers, or specific oligonucleotides as indicated (Table S1). PRUNE2 and PCA3 were amplified by RT-PCR with KAPA HiFi DNA polymerase (KAPA Biosystems), cloned into pENTR directional TOPO (Invitrogen), and fully sequenced. Verified coding sequences were reamplified and subcloned into a pcDNA-DEST40 expression vector (Invitrogen). shRNA-resistant PRUNE2 (PRUNE2 Ω shRNA) was created by site-directed mutagenesis.
siRNA and shRNA.
Custom-ordered siRNAs against PCA3 (Table S1) were transfected into tumor cells using the NeoFX transfection reagent (Ambion). PCA3-silencing experiments were performed with retroviral pLKO.1 and human GIPZ vectors from the RNAi Consortium (TRC) lentiviral shRNA library (Open Biosystems) expressing specific shRNAs for human PRUNE2 (oligonucleotide ID TRCN0000121740, referred to as PRUNE2-C1 and oligonucleotide ID TRCN0000144868, referred to as PRUNE2-C2), human PCA3 (oligonucleotide ID V2LHS_ 24225, referred to as PCA3-C1; and oligonucleotide ID V2LHS_24226, referred to as PCA3-C2), human ADAR1 (oligonucleotide ID TRCN0000050788, referred to as ADAR1-C1; and oligonucleotide ID TRCN0000050790, referred to as ADAR1-C2 (Sigma), human GIPZ ADAR2 (#RHS4287), and human TRIPZ lentiviral inducible shRNAmir (#RHS4740; Open Biosystems). Lentivirus particles for P54NRB-shRNA were purchased (TRCN0000074558 referred to as P54NRB-C1 and TRCN0000074559 referred to as P54NRB-C2; Sigma). Stable clones were maintained under puromycin selection. Validated nontargeting siRNA (Ambion) and shRNA (Open Biosystems) sequences served as negative controls. Customized Stealth chemically modified, HPLC-purified RNAi sequences against PCA3 or scrambled controls were purchased from Invitrogen.
Lentivirus Preparation.
Lentiviral vectors (pCCLsin.PTT.PGK.EGFP.Wpre, pMDLg/pRRE, pRSV-Rev, and pMD2.VSVG) were used. Briefly, 293FT cells were transiently transfected (Lipofectamine 2000; Invitrogen) for 16 h, after which the lentiviruses were harvested 24 and 48 h later and filtered through 0.22-µm pore cellulose acetate filters. Recombinant lentiviruses were concentrated by ultracentrifugation for 2 h at 50,000 × g. Lentiviral vector viability was confirmed by reporter gene expression and drug selection. Cells were transfected with the FuGeneHD reagent (Roche), and transgene expression was analyzed at 24, 36, 48, or 100 h after transfection. Corresponding empty plasmids served as negative controls.
qRT-PCR and Northern Blotting.
qRT-PCR analyses were performed with SYBR-green in a 7500 Fast Real-Time PCR system (Applied Biosystems). Gene expression levels were normalized against the average Ct of three standard endogenous controls (P0 large ribosomal protein, β-glucuronidase, and TATA box-binding protein), and the results were analyzed according to a standard ΔΔCt method. Data were reported as fold induction; samples were normalized onto their internal housekeeping genes followed by normalization of each sample to its control. For Northern blotting, customized LNA oligonucleotides (Exiqon) were used for PCA3 and PRUNE2 (Table S1). For retro-transcription, we designed specific antisense primers for PCA3 or PRUNE2 that allowed generation of specific cDNAs from corresponding pre-mRNA overlapped regions that enabled the generation of strand-specific cDNAs and PCR products for either PCA3 or PRUNE2. Amplified products were confirmed by sequencing.
Cell Fractionation and Nuclear RNA Analysis.
Nuclear/cytoplasmic RNA fractionation was performed (3). Tumor cells were grown in fibronectin-coated plates. At 70% confluence, cells were harvested, centrifuged, and rinsed with ice-cold PBS. In brief, cell pellets were resuspended by gentle pipetting in 200 µL lysis buffer A [10 mM Tris (pH 8.0), 140 mM NaCl, 1.5 mM MgCl2, 0.1% octylphenoxy poly(ethyleneoxy)ethanol (IGEPAL), and 2 mM vanadyl ribonucleoside complex] and were incubated on ice for 5 min. The same lysate sample served for total RNA extraction by the TRIzol reagent (Invitrogen) and for centrifugation (1,000 × g for 3 min at 4 °C), as well as to isolate the cytoplasmic fraction and pellet the nuclei. Cell equivalent amounts of cytoplasmic and nuclear RNA samples were used for nuclear retention analysis. Nuclear/cytoplasmic ratios were normalized to GAPDH, HYOU1, or SON RNA controls.
RNA FISH and Confocal Microscopy.
To detect PCA3 and PRUNE2 RNAs, cells were fixed in 3.6% (vol/vol) formaldehyde for 3 min at room temperature, followed by acetone:methanol 1:1 (vol/vol) for 5 min at −20 °C. Cells were permeabilized in PBS containing 0.39 Triton X-100 and 5 mM vanadyl ribonucleoside complex (Invitrogen) on ice for 5 min; vanadyl ribonucleoside complex (an RNase inhibitor) was omitted if the RNase enzymatic activity was to be determined. Cells were washed three times in PBS for 10 min and rinsed once in 2× saline sodium citrate (SSC) buffer before hybridization. Hybridization was carried out using labeled cy3, cy5, and 488 nm DNA-oligonucleotide probes in a moist chamber at 37 °C overnight (ON). For colocalization studies after in situ hybridization, cells were fixed for 5 min in PBS-containing 2% (vol/vol) formaldehyde. Standard immunofluorescence and imaging were performed.
Immunoprecipitation and Immunoblot Analysis.
Immunoprecipitation assays were performed as previously described (3). Briefly, a total of 3 × 106 subconfluent cells were starved for 36 h in RPMI containing 0.25% BSA and 0.05% FBS. Cells were stimulated for 15 min at 37 °C with a growth factor admixture described above. After washes with ice-cold PBS containing 0.1 mM sodium orthovanadate, cells were solubilized in 1 mL lysis buffer [50 mM Tris(hydroxymethyl) aminomethane HCI (pH 7.4), 150 mM NaCl, 1% Nonidet P-40 (Nonidet P-40), 0.25% sodium deoxycholate, 1 mM EGTA, 1 mM EDTA, 2.5 mM sodium pyrophosphate, 1 mM sodium fluoride, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate (Na3VO4) (pH 10.0), anti-protease, and anti-phosphatase mixture; (Sigma)], collected, and incubated on ice for 10 min. Cell lysates were centrifuged at 10,000 × g for 15 min, and supernatants were precleared for 1 h at 4 °C by incubation either with 15 µL protein A- or protein G-agarose (Roche). Precleared lysates were subsequently used for immunoprecipitation with specific antibodies as indicated. After incubation, the solution was centrifuged at 1,000 × g for 4 min and washed three times with 0.5 mL lysis buffer and once with ice-cold PBS containing 1 mM Na3VO4. Immunoprecipitates were separated by 3–8%, 4–12%, or 4% bis-Tris NuPAGE (Invitrogen) as indicated, transferred to nitrocellulose membranes, and immunoblotted with specified antibodies.
RNA–Protein Complex Immunoprecipitation.
Tumor cells at 70% confluence were rinsed twice and scrapped into ice-cold PBS. Cell pellets were resuspended in immunoprecipitation buffer [50 mM Tris⋅HCl (pH 7.4), 150 mM NaCl, 0.05% IGEPAL, 1 mM PMSF, and proteinase inhibitor mixture (Sigma)], subjected to two rounds of gentle sonication, and centrifuged to obtain cell extracts. For RNaseA treatment, 200 µg/mL RNaseA was added to cell extracts, and the admixture was incubated at 37 °C for 30 min. For immunoprecipitation, RNaseA-treated and nontreated cell extracts were precleared with 40 µL protein-A- plus protein-G-agarose beads (Roche) and 2.5 µg mouse anti-ADAR1 antibody or irrelevant isotype control antibody at 4 °C for 2 h, followed by 40 µL protein-A- plus protein-G-agarose beads for 30 min at room temperature.
UV Cross-Linking and ChIP.
Subconfluent cells cultured in complete RPMI for 24 h underwent UV-mediated cross-linking, extract preparation, and SDS/PAGE or standard immunoprecipitation. For Western blot oligo-hybridization, specific 3′-biotinylated oligonucleotides (Table S1) were used.
Cell Proliferation, Cell Viability, and Cell Death Assays.
Cells were seeded in 200 µL growth medium at a density of 5,000–10,000 cells per well onto E-Plates 96 (Roche). Cell attachment and growth were monitored every 15 min for 48–72 h with real-time cell electronic sensing (RT-CES) technology (Roche). The assay system expresses impedance in arbitrary cell index (CI) units. The CI at each time point is defined as (Rn-Rb)/15; where Rn is the cell-electrode impedance of the well when it contains cells, and Rb is the background impedance of the well with the medium alone. Cell proliferation comparable data were measured 72 h later with the WST-1 cell proliferation reagent (Roche). Cell viability was evaluated by the Trypan blue-exclusion methodology. Cell death assays were performed through a standardized cell death detection ELISA kit (Roche). In brief, cells were cultured in complete RPMI medium for 24 h. After cell lysis, the cytoplasmic fraction was prediluted to 1:10 (vol/vol) with incubation buffer and tested for nucleosomes in the immunoassay substrate reaction.
Cell Adhesion and Migration Assays.
Cells were seeded in 200 µL RPMI medium supplemented with 2.5% (vol/vol) FBS plus 50 ng of a growth factor admixture (described above) at a density of 5,000–10,000 cells per well onto E-Plates 96 (Roche). Cell adhesion was monitored every 15 min, during 4 h through RT-CES technology (Roche). A 24-well in colorimetric format (CytoSelect; Cell Biolabs) was used for cell migration assays. Briefly, a cell suspension (1,000 cells) was placed in the upper chamber, and RPMI medium containing 2.5% (vol/vol) FBS plus the growth factor admixture was placed in the lower chamber and incubated at 37 °C and 5% CO2 for 4 h. Cell migration was subsequently quantified.
Cell Cycle Analysis.
Cells were synchronized in RPMI containing 0.2% BSA and stimulated with 10% (vol/vol) FBS for 24 h. Proliferating cells were labeled by administration of BrdU at 10 µM for 1 h (Sigma-Aldrich). Cells were harvested by trypsinization, washed in ice-cold PBS, and fixed in 100% ethanol for 30 min on ice. DNA denaturation was achieved by incubation of fixed cells in 2 N HCl containing 0.05% Triton X-100 for 30 min at room temperature. Residual acid was neutralized with 0.1 M sodium borate (pH 8.5). Samples were incubated with an anti-BrdU monoclonal antibody, followed by cy-3−conjugated secondary antibody anti-mouse IgG (Jackson ImmunoResearch Laboratories). Total DNA content was measured with 50 µM propidium iodide (PI). Flow cytometry analysis was performed in a FACS Canto II System using the FACS Diva software (Becton-Dickinson).
Soft Agar Colony Formation Assay.
Standard analysis of anchorage-independent cell growth and colony formation was performed. In brief, cells transduced with the indicated constructs were suspended at either 5 × 103 or 105 cells per well in 2 mL of 0.35% low-melting agarose in 35-mm culture dishes containing 0.7% agarose base. Triplicates were prepared and evaluated for each construct. Colonies were allowed to form at 37 °C under standard tissue culture conditions for 21–28 d. After incubation, staining of the formed colonies with 0.005% crystal violet (CV) enabled visual inspection, photographs, and optical density measurements after CV solubilization.
Confocal Analysis of Tumor Cell-Derived Spheroids.
Tumor spheroids were prepared by growing 50–200 LNCaP cells in nonadherent 96-microwell culture dishes for 18 h in RPMI containing 10% (vol/vol) FBS. Spheroids were removed and cultured on fibronectin-coated slides in RPMI containing 2.5% (vol/vol) FBS plus a growth factor admixture (described above). Six hours later, spheroids were fixed, immunostained with the appropriate antibody, and 3D analyzed for PRUNE2, RhoA, Nm23-H1, and β-tubulin localization.
RNA-Capture Library Preparation and Large-Scale Sequencing Analysis.
RNA molecules derived from the PRUNE2/PCA3 locus were captured and sequenced in large scale for a comprehensive analysis of A-to-I editing. For the library preparation, mature and immature RNA molecules derived from the PRUNE2 locus (∼300 kb) were captured by using 120 nucleotide (nt) probes designed for a 2× tiling coverage (eArray; Agilent). Captured RNAs were used as templates for the construction of libraries through the SureSelect RNA Target Enrichment for Illumina Paired-end Multiplexed Sequencing kit and protocol (Agilent). Libraries were sequenced by using a MiSeq instrument (Illumina) generating >3 million 150-nt reads per sample (i.e., ∼450 million bases sequenced per sample). Reads were aligned to the human reference genome (hg19) with the Burrows-Wheeler Aligner (BWA-0.6.1). Repetitive regions from RepeatMasker were masked, and >400,000 on-target reads for each sample remained. After removing variants present in dbSNP (release 137), all discrepancies between the sample reads and hg19 (confirmed by at least three independent sequence reads) were considered and further analyzed through the CLC Genomics Workbench (version 6.0.3). A detailed analysis of putative ADAR1- and ADAR2-mediated editing (A > G/ T > C substitutions) was performed over the locus of interest, including the mapping of putative editing sites, the determination of editing frequency (i.e., putative edits per kilobase), and editing density (i.e., percentage of reads showing the alteration).
RNA-Editing Analysis by Sanger Sequencing.
Nuclear or total RNA samples were isolated and subjected to three pulses of sonication (300 Hz) to produce RNA fragments of 1,000–2,000 nt, followed by DNaseI treatment. RNA samples were added to two different minilibraries of specific primers (designed to cover intronic, as well as exonic regions of PCA3 and the corresponding intron6-PRUNE2), and the admixtures were heated to 99 °C for 10 min and ice-chilled for 5 min. Specific cDNAs for PCA3 and PRUNE2 were produced by reverse transcription with SuperScript II/III (Invitrogen) by using primers designed to bind intronic and exonic regions of PCA3 and the corresponding intron6-PRUNE2. Resultant cDNAs were amplified by PCR (850-1,000 bp) and subcloned in the TOPO vector, and individual single colonies were picked and amplified. The A-to-I editing (reflected by A > G/T > C changes) frequency was estimated by Sanger sequencing of >100 individual clones containing the expected inserts. As evidence of RNA editing, we considered only non-dbSNP alterations located outside of repetitive elements (including Alus) and only those represented by at least three distinct reads.
Tumor-Bearing Mouse Assays.
PC3 and LNCaP cell lines were established, each stably expressing ectopic PRUNE2, ectopic PCA3, control vector, PCA3 silenced, PRUNE2 silenced, and control-shRNA. In each case, stably expressing pool transduced cells and their corresponding controls were allowed to grow for 48 h to reach 85% confluence. Afterward, paired test and control tumor cells were counted, washed in serum-free medium, and resuspended to a final concentration of 5 × 107/mL in serum- and phenol-free basic RPMI medium. Cells were subsequently mixed in 50% volume of phenol-free Matrigel (Becton Dickinson), and cell suspensions (final volume of 200 µL) were administered s.c. in the right flanks of male 6-wk-old SCID mice (Charles River). Tumor xenograft growth was monitored serially over time. The MDA-PCa-133 patient-derived xenograft (PDX) has been reported elsewhere (4). Total RNA was obtained from an early tumor passage and used to clone PRUNE2. All animal experimentation was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Texas M.D. Anderson Cancer Center (MDACC).
Tissue Microarray and Immunohistochemistry.
A human tissue microarray (TMA) was constructed from prostate cancer patients (n = 145) and consisted of intermediate- or high-risk tumors (Gleason score ≥ 7, locally advanced ≥ pT2, peripheral zone tumors; n = 95) and low-risk tumors (Gleason score 6, peripheral zone tumors, previously untreated, who underwent prostatectomy; n = 50). Areas representative of all histologic tumor patterns of the Gleason grades were selected from the individual specimens. The TMA consisted of 1,500 cores, and each individual patient was represented by a set of 0.6-mm-diameter cores (median, 12; range, 18–53). For IHC analysis, images in each core of the TMA were acquired by the use of a BLISS imaging system (Bacus Laboratories). A standard percentage system was used for assessment of involvement (percentage of tumor cells exhibiting detectable staining). The extent of PRUNE2 protein expression was determined in tumor epithelium vs. adjacent stromal tissue; TMA slides were stained with an anti-PRUNE2/BMCC1 rabbit polyclonal antibody (ProteinTech Group) at a 1:70 dilution. The intensity of staining was scored as absent, low, or high. An automated stainer (DAKO) and standard 3,3-diaminobenzidine were used. Tumor samples were clinically annotated and selected from a serum and tissue bank supported by the National Cancer Institute (NCI) Specialized Program of Research Excellence (SPORE) in prostate cancer at MDACC. All human experimentation was reviewed and approved by the Clinical Research Committee (CRC) and by the institutional review board (IRB) at MDACC. All human specimens were obtained after the patients provided written informed consent under an IRB-approved experimental protocol. Total RNA samples purified from tumors from human prostate cancer patients were also obtained from the Tumor Bank at A.C. Camargo Cancer Center after its IRB approval.
Statistics.
We summarized PRUNE2 expression in the samples by the use of standard descriptive statistics for continuous variables or tabulations for categorical variables. The primary analysis was based on the involvement score (extent of staining) alone, which was treated as a continuous variable. Statistical significance was determined by the appropriate tests. The nonparametric Wilcoxon–Mann–Whitney test served to assess differences in expression between high-grade, low-grade, and bone metastatic cases and stromal and epithelial compartments. The Student t test or Fisher’s exact test was used in the data analysis for categorical variables as appropriate. To incorporate repeated measurements (e.g., TMA cores) from an individual patient, mixed-effects models were fitted to allow estimates of variability either within or among patients. The P values and SDs were reported in the data analysis with independently repeated experiments. All reported P values are two-sided, and P < 0.05 was considered statistically significant. Analyses were done with SAS for Windows (1999–2000; SAS Institute, release 8.1) and S-PLUS 2000 (1988–2000; Insightful Corporation, release 3).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants CA90270 (to R.P. and W.A.) and CA95616 (W.K.C.), AngelWorks, Gilson-Longenbaugh Foundation, Prostate Cancer Foundation (W.A. and R.P.), Fundação de Amparo à Pesquisa do Estado de São Paulo, and Associação Beneficente Alzira Denise Hertzog Da Silva (E.D-N.).
Footnotes
Conflict of interest statement: The University of New Mexico has filed patents on the technology and intellectual property reported here. If licensing or commercialization occurs, the researchers (A.S., A.K.L., D.N.N., E.D.-N., R.P., and W.A.) are entitled to standard royalties.
This article is a PNAS Direct Submission.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. FJ808772, FJ808773, and AF103907).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1507882112/-/DCSupplemental.
References
- 1.Walsh AL, Tuzova AV, Bolton EM, Lynch TH, Perry AS. Long noncoding RNAs and prostate carcinogenesis: The missing ‘linc’? Trends Mol Med. 2014;20(8):428–436. doi: 10.1016/j.molmed.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Bussemakers MJG, et al. DD3: A new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 1999;59(23):5975–5979. [PubMed] [Google Scholar]
- 3.Wei JT, et al. Can urinary PCA3 supplement PSA in the early detection of prostate cancer? J Clin Oncol. 2014;32(36):4066–4072. doi: 10.1200/JCO.2013.52.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 5.Fatica A, Bozzoni I. Long non-coding RNAs: New players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 6.Geisler S, Coller J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14(11):699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Machida T, et al. Increased expression of proapoptotic BMCC1, a novel gene with the BNIP2 and Cdc42GAP homology (BCH) domain, is associated with favorable prognosis in human neuroblastomas. Oncogene. 2006;25(13):1931–1942. doi: 10.1038/sj.onc.1209225. [DOI] [PubMed] [Google Scholar]
- 8.Clarke RA, et al. New genomic structure for prostate cancer specific gene PCA3 within BMCC1: Implications for prostate cancer detection and progression. PLoS ONE. 2009;4(3):e4995. doi: 10.1371/journal.pone.0004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavin MF, Clarke R, Gardiner RA. Differential expression of PCA3 and BMCC1 in prostate cancer. Prostate. 2009;69(16):1713–1714, author reply 1715. doi: 10.1002/pros.21068. [DOI] [PubMed] [Google Scholar]
- 10.Salagierski M, et al. Differential expression of PCA3 and its overlapping PRUNE2 transcript in prostate cancer. Prostate. 2010;70(1):70–78. doi: 10.1002/pros.21040. [DOI] [PubMed] [Google Scholar]
- 11.Lee YC, et al. BMP4 promotes prostate tumor growth in bone through osteogenesis. Cancer Res. 2011;71(15):5194–5203. doi: 10.1158/0008-5472.CAN-10-4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L-L, DeCerbo JN, Carmichael GG. Alu element-mediated gene silencing. EMBO J. 2008;27(12):1694–1705. doi: 10.1038/emboj.2008.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mallela A, Nishikura K. A-to-I editing of protein coding and noncoding RNAs. Crit Rev Biochem Mol Biol. 2012;47(6):493–501. doi: 10.3109/10409238.2012.714350. [DOI] [PubMed] [Google Scholar]
- 15.Ferreira LB, et al. PCA3 noncoding RNA is involved in the control of prostate-cancer cell survival and modulates androgen receptor signaling. BMC Cancer. 2012;12:507. doi: 10.1186/1471-2407-12-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soh UJ, Low BC. BNIP2 extra long inhibits RhoA and cellular transformation by Lbc RhoGEF via its BCH domain. J Cell Sci. 2008;121(Pt 10):1739–1749. doi: 10.1242/jcs.021774. [DOI] [PubMed] [Google Scholar]
- 17.Galasso A, Zollo M. The Nm23-H1-h-Prune complex in cellular physiology: A ‘tip of the iceberg’ protein network perspective. Mol Cell Biochem. 2009;329(1-2):149–159. doi: 10.1007/s11010-009-0115-4. [DOI] [PubMed] [Google Scholar]
- 18.Basile JR, Gavard J, Gutkind JS. Plexin-B1 utilizes RhoA and Rho kinase to promote the integrin-dependent activation of Akt and ERK and endothelial cell motility. J Biol Chem. 2007;282(48):34888–34895. doi: 10.1074/jbc.M705467200. [DOI] [PubMed] [Google Scholar]
- 19.Rhodes DR, et al. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Guttman M, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477(7364):295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: Insights into functions. Nat Rev Genet. 2009;10(3):155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 24.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Iwama E, et al. Cancer-related PRUNE2 protein is associated with nucleotides and is highly expressed in mature nerve tissues. J Mol Neurosci. 2011;44(2):103–114. doi: 10.1007/s12031-010-9490-2. [DOI] [PubMed] [Google Scholar]
- 26.Arama J, et al. Bmcc1s, a novel brain-isoform of Bmcc1, affects cell morphology by regulating MAP6/STOP functions. PLoS ONE. 2012;7(4):e35488. doi: 10.1371/journal.pone.0035488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris JL, et al. BMCC1 is an AP-2 associated endosomal protein in prostate cancer cells. PLoS ONE. 2013;8(9):e73880. doi: 10.1371/journal.pone.0073880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan CQ, Low BC. Functional plasticity of the BNIP-2 and Cdc42GAP Homology (BCH) domain in cell signaling and cell dynamics. FEBS Lett. 2012;586(17):2674–2691. doi: 10.1016/j.febslet.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 29.Knudson AG., Jr Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cavenee WK, et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature. 1983;305(5937):779–784. doi: 10.1038/305779a0. [DOI] [PubMed] [Google Scholar]
- 31.Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011;476(7359):163–169. doi: 10.1038/nature10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]