

Significance
Functional classification of DNA polymerases (DNAPs) usually divides them into replicative faithful replicases and error-prone enzymes devoted to DNA repair and DNA damage tolerance through translesion synthesis (TLS). When we analyzed the biochemical properties of phage Bam35 replicative DNAP, we found it to be a highly faithful DNAP that can couple strand displacement to processive DNA synthesis, suitable for rolling circle amplification of plasmidic DNA. Interestingly, it is also endowed with intrinsic TLS capacity opposite abasic sites and processive primer extension beyond the lesion. These features configure a versatile enzyme for accurate maintenance of viral genomic information over generations and, besides, to deal with DNA lesions, which suggest a possible application of Bam35 DNAP for the amplification of damaged or ancient DNA.
Keywords: protein-primed DNA polymerase, Bam35, abasic sites, translesion synthesis, isothermal DNA amplification
Abstract
DNA polymerases (DNAPs) responsible for genome replication are highly faithful enzymes that nonetheless cannot deal with damaged DNA. In contrast, translesion synthesis (TLS) DNAPs are suitable for replicating modified template bases, although resulting in very low-fidelity products. Here we report the biochemical characterization of the temperate bacteriophage Bam35 DNA polymerase (B35DNAP), which belongs to the protein-primed subgroup of family B DNAPs, along with phage Φ29 and other viral and mobile element polymerases. B35DNAP is a highly faithful DNAP that can couple strand displacement to processive DNA synthesis. These properties allow it to perform multiple displacement amplification of plasmid DNA with a very low error rate. Despite its fidelity and proofreading activity, B35DNAP was able to successfully perform abasic site TLS without template realignment and inserting preferably an A opposite the abasic site (A rule). Moreover, deletion of the TPR2 subdomain, required for processivity, impaired primer extension beyond the abasic site. Taken together, these findings suggest that B35DNAP may perform faithful and processive genome replication in vivo and, when required, TLS of abasic sites.
Replicative DNA polymerases (DNAPs) from A and B families, collectively termed replicases, exhibit a “tight fit” for their DNA and dNTP substrates and are wondrously adapted to form correct Watson–Crick base pairs, resulting in very pronounced fidelity (1, 2). This strict preference to produce A:T and G:C base pairs is also the Achilles heel of faithful DNA polymerases, however, because they are strongly inhibited by modified nucleotides present at sites of DNA damage, leading to the stalling of replication fork and eventually to replicative stress and cell death (3). At the stalled replication fork, the DNA polymerase may be exchanged by a translesion synthesis (TLS) polymerase, generally belonging to the Y family. These enzymes possess looser solvent-exposed active sites, which allows them to deal with aberrant DNA features much better, although with a very low polymerization accuracy with the risk of the accumulation of mutations and genetic instability (4, 5). Alternatively, nonbulky modified bases, such as uracil and 8-oxo-deoxyguanosine (8oxoG), can be bypassed by replicases, with faithful or mutagenic outcomes that can be modified by the sequence context and dNTP availability (6, 7).
Abasic or apurinic/apyrimidinic (AP) sites are the most common DNA lesions arising in cells when the N-glycosydic bond between the sugar moiety and the nucleobase is broken, either spontaneously or by a DNA glycosylase reaction product in the base excision repair pathway (8, 9). Unrepaired abasic sites are highly blocking lesions for replicative DNA polymerases (10), although mutant polymerases with impaired proofreading activity or with mutations in the polymerization active site residues that affect the incoming nucleotide selection have been shown to have enhanced AP site bypass capacity (11–15).
Among family B DNAPs, protein-primed polymerases constitute a heterogeneous group with an apparent monophyletic origin that can be found in various prokaryotic and eukaryotic viruses, mitochondrial plasmids, and eukaryotic mobile elements (16–19). This designation refers to their capacity, apparent for adenoviruses as well as different bacteriophage enzymes (20, 21), to perform genome replication primed by a specific protein, termed terminal protein (TP), that becomes covalently linked to the 5′ DNA ends. Identification and annotation of these polymerases in databases is based on the presence of two specific amino acid insertions, TPR1 and TPR2, involved in the interaction with TP and in processivity and strand displacement capacity, respectively (22, 23).
Bacillus subtilis bacteriophage Φ29 DNA polymerase (Φ29DNAP) is the paradigm of protein-primed DNAPs, with well-understood biochemical and structural properties (reviewed in refs. 18, 20, 24). Φ29DNAP is a highly faithful enzyme (25, 26) able to generate very long DNA molecules (27), coupling DNA synthesis and strand displacement. Φ29, along with other protein-primed genome replication phages, such as PRD1 or Cp-1 (28, 29), can undergo a lytic cycle only after infection of the host cell occurs. In contrast, phage Bam35, which infects Bacillus thuringiensis and related tectiviruses infecting Bacillus cereus sensu lato group (30, 31), are temperate viruses that can self-replicate as linear episomes within lysogenic cells.
In this work, we describe the biochemical properties of Bam35 DNA polymerase (B35DNAP) as a faithful, processive DNAP endowed with intrinsic strand displacement activity. Surprisingly, we also found that, despite its high fidelity, it can elude to some extent the tight quality check of proofreading activity, allowing the enzyme to processively bypass abasic sites in DNA. Furthermore, deletion of the TPR2 subdomain does not substantially reduce the insertion of nucleotides opposite the abasic site, but does impair its further extension. We discuss the potential implications of these findings for the bacteriophage replication cycle and possible applications.
Results and Discussion
Exonuclease and Polymerization Activities of Bam35 DNA Polymerase Are Coordinated.
B35DNAP shares 44% similarity and 29% identity with Φ29DNAP and contains most conserved catalytic residues, as well as the TPR1 and TPR2 insertions (Fig. S1). To evaluate both 3′-5′ degradative and 5′-3′ synthetic activities of B35DNAP, we analyzed the degradation and extension of full base-paired (Fig. 1A) and one 3′-mismatched nucleotide (Fig. 1B) primer/template substrates. In both cases, only degradation products could be detected in the absence of dNTP and with the addition of increasing amounts of dNTP, exonucleolysis was replaced by polymerization activity, requiring the same threshold of dNTP concentration (60 nM; lane 6 in Fig. 1 A and B) to replicate the full-length template with both primer/template substrates. This finding suggests that proofreading activity is able to remove the mismatched nucleotide at the 3′-primer terminus very efficiently and thereby extend the resulting matched primer. In support of this, when an exonuclease-deficient mutant (B35DNAPexo−; Materials and Methods) was used, mismatch extension required approximately 1,000-fold more nucleotide concentration than needed with correct base pairs (Fig. 1 C and D). Also, insertion of the correct nucleotide could be detected with approximately 100-fold less concentration compared with the incorrect dNTP (Fig. S2). Therefore, as expected for a replicative DNA polymerase, the B35DNAP polymerization active site is highly specific for insertion of correct base pairs, and eventual mistakes can be efficiently eliminated by proofreading 3′-5′ exonuclease activity.
Fig. S1.
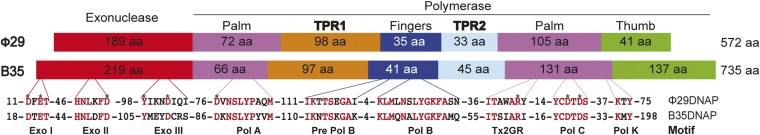
Schematic representation of Φ29 and Bam35 DNA polymerase sequences. Conserved domains and subdomains are represented as colored boxes, and their length is indicated. Identical residues are highlighted in red, and residues involved in DNA binding and catalysis are indicated with an asterisk.
Fig. 1.

Bam35 DNA polymerase synthetic and degradative activities. Shown is denaturing PAGE analysis of primer extension assays with a matched (A and C) or mismatched (B and D) 3′-terminus as depicted above the gels. The assays were carried out for 10 min at 37 °C in the presence of 10 nM of either WT (B35DNAP, A and B) or exonuclease-deficient (B35DNAPexo—, C and D) DNAPs and the indicated concentration of dNTPs. Positions of the 15-mer substrate and 33-mer product bands are indicated on the right.
Fig. S2.

B35DNAP nucleotide insertion fidelity. Shown is nucleotide insertion preference by the B35DNAPexo— mutant using the 3′-GTT template sequence context, in the presence of increasing amounts of each dNTP as indicated. Reactions were incubated for 10 min at 37 °C.
Bam35 DNA Polymerase Can Couple Strand Displacement to Polymerization During Faithful Processive DNA Synthesis.
To analyze the capacity of B35DNAP to couple polymerization and displacement of the nontemplate strand, we performed primed single-stranded M13 DNA rolling circle replication assays (Materials and Methods). As shown in Fig. 2A (lanes 1–9), B35DNAP was able to replicate M13 DNA and gave rise to a replication product larger than full-length M13 DNA (7.25 kb) with a length similar to that produced by Φ29DNAP (lane 11), indicating an intrinsic capacity of B35DNAP to efficiently couple polymerization and strand displacement. Moreover, B35DNAP is able to carry out DNA replication in a processive manner; the size of the replication products remained invariable on dilution of the enzyme up to 40-fold (lane 1 vs. lane 9).
Fig. 2.

Bam35 DNA polymerase can couple strand displacement and processive polymerization during rolling circle DNA replication. (A) Replication of primed M13 DNA was carried out as described in Materials and Methods in the presence of 40 μM each of the four dNTPs and 2.25–91 nM of B35DNAP (lanes 1–9) or 50 nM of Φ29DNAP (lane 11). After incubation at 37 °C for 20 min, the length of the synthesized DNA was analyzed by alkaline 0.7% agarose gel electrophoresis alongside a λ DNA ladder (lane 10) and autoradiography. M13 ssDNA unit length is indicated as well. (B) Multiple-displacement amplification of plasmidic DNA by B35DNAP. The assay was performed as described in Materials and Methods, in the presence of 0–10 ng of pUC19 plasmid DNA as input and 50 nM of B35DNAP (lanes 2–9). Linearized pUC19 plasmid (100 ng) was loaded in lane 1, and linear DNA fragments obtained after digesting Φ29 DNA with HindIII, used as DNA length markers (lane 10), are indicated on the right.
We also carried out isothermal multiple displacement amplification (MDA) of pUC19 plasmid DNA (32). As shown in Fig. 2B, B35DNAP can use random primers to successfully amplify circular dsDNA; more than 80% of the amplified DNA was linearized after digestion with the endonuclease EcoRI, rendering a band of identical size as the original digested plasmid (2.7 kbp; lane 1). This result indicates that most of the amplification products were dsDNA tandem repeats of the original plasmid. Importantly, B35DNAP was able to amplify a minimal DNA input of 1–2.5 ng (lanes 6 and 7), similar to that routinely used for Φ29DNAP-mediated MDA of plasmid DNA (26, 32). To determine the accuracy of DNA amplification by B35DNAP, we circularized digested samples from MDA experiments and subjected them to white/blue colony screening (Materials and Methods). As shown in Table 1, the average error rate was 5.1 ± 1.7 × 10−6, similar to that of Φ29DNAP and commercially available PCR polymerases (26, 33, 34). We also sequenced several mutant (white) clones to determine which type of mistake was the most frequent. The most common changes detected in the mutant clones were base insertions in homopolymeric runs (frequency of 2.7 × 10−3 among the mutant clones), with two marked hotspots, 5′-GGGAAAACCC and 5′-CCCCGGG (Fig. 3). Only one deletion was found, and substitutions were very scarce as well, with only four C:G→T:A transitions and one C:G→G:C transversion detected. In line with these results, available sequence stability data suggest that phage genome replication is likely very faithful in vivo, as shown for a short highly variable region found in a number of Bam35 closely-related viruses, which maintain a constant sequence after serially propagating several phage clones over a 50-d period (35).
Table 1.
Determination of error rate values for B35DNAP during MDA
| Experiment | Total colonies | f | f0 | d | Error rate |
| 1 | 1,171 | 1.8 × 10−2 | 5.7 × 10−3 | 11.2 | 3.2 × 10−6 |
| 2 | 10,605 | 2.7 × 10−2 | 4.6 × 10−3 | 9.9 | 6.5 × 10−6 |
| 3 | 10,682 | 2.1 × 10−2 | 2.2 × 10−3 | 10 | 5.6 × 10−6 |
| Average ± SD | 1.9 ± 0.2 × 10−2 | 4.2 ± 1.6 × 10−3 | 10.4 ± 0.9 | 5.1 ± 1.7 × 10−6 |
Mutant frequencies (f) were determined by dividing the total number of white plaques by the total number of plaques. f0 represents the background frequency in plasmid DNA purified from bacteria, digested and religated in the same conditions (Materials and Methods). Template doublings (d) were calculated using the equation d = (ng product)/(ng input). Error rate was calculated using the equation ER = (f-f0)/bp × d, where bp is the number of detectable sites in the lacZ gene (342 bp). Data from three independent experiments, and average ± SD values are shown.
Fig. 3.

Spectra of single base changes by B35DNAP. Bases are colored in red (A), blue (C), green (T), and orange (G). Base insertions, deletions, and substitutions are indicated above the sequence with inverted triangles (▼), Greek delta (Δ), and the mutated base, respectively, maintaining the same color code. Position 1 is the first transcribed nucleotide of the lacZ gene.
Similar analyses with other polymerases have shown that error rates are usually much higher for substitutions than for insertion/deletions (InDels, 4), except in the case of A-family human DNA polymerase θ (POLQ) (36, 37), which has a higher error rate than replicative polymerases (on the order of 10−3–10−4), owing mainly to nucleotide insertions (3.3 × 10−3) and deletions (1.4 × 10−3), particularly in homopolymeric DNA tracks. InDels are the signature for misaligned primer templates containing an extra base in the template strand (for deletions) or in the primer strand (for additions), stabilized by a number of correct base pairs that separate the extra base from the primer end at the catalytic site (38). Therefore, the relatively higher frequency of insertions compared with deletions produced by B35DNAP would indicate the possibility of misaligned primer–template intermediates containing an extra base in the primer strand, but not in the template strand (38). The average POLQ error rate is higher, however, and instead of replication, it has been involved in TLS and in an alternative end-joining pathway (39). We conclude that B35DNAP is able to amplify circular DNA by MDA with high yield at very low error rates, but the presence of homopolymeric DNA sequence tracks might be a source of frameshift (insertions) mutation accumulation.
Proficient Bypass of Abasic Sites by B35DNAP.
The bypass of abasic sites by replicative DNA polymerases is very low, depending on the sequence context and counteracted by the proofreading activity (12, 13, 40, 41). Fig. 4A shows a primer extension assay using two different template sequence contexts containing a tetrahydrofuran (THF) moiety, a stable analog of an abasic site, in the first template nucleotide and either a T (3′-GFT template; lanes 4–6) or an A (3′-GFA; lanes 10–12) in the second position, as well as the corresponding undamaged controls (3′-GTT, lanes 1–3; 3′-GTA, lanes 7–9). In the absence of damage, B35DNAP, like Φ29DNAP, used as a control, can extend the primer processively (lanes 3 and 9 vs. lanes 2 and 8); however, when they encounter an abasic site, Φ29DNAP is unable to replicate the template under these conditions (lanes 6 and 12), whereas B35DNAP is able to insert the first nucleotide and extend the primer beyond the THF, being more proficient in the 3′-GFT context than in the 3′-GFA context (lanes 5 and 11). Using both substrates, B35DNAP gave rise to a partial product accumulation at the lesion site (16 mer), suggesting that extension of the dNMP/THF partial mismatch is a limiting step. Interestingly, replication of 3′-GFT and 3′-GTT templates is equally processive, given that enzyme dilution with an excess of primer/template substrate does not modify the proportion of insertion (16-mer) and full-length TLS (33-mer) products (Fig. 4B). These results indicate that although some B35DNAP molecules may be unable to successfully extend the THF-containing mismatch or eventually drop, the polymerase is able to insert the nucleotide opposite the abasic site and subsequently extend the mismatch processively without dissociation from the template/primer substrate.
Fig. 4.

Abasic site TLS by B35DNAP. (A) Denaturing PAGE analysis of primer extension products by B35DNAP and Φ29DNAP on two different sequence contexts (lanes 1–6 vs. 7–12) and in the absence (3′-GTT, lanes 1–3; 3′-GTA, lanes 7–9) or presence (3′-GFT, lanes 4–6; 3′-GFA, lanes 10–12) of a THF (F) abasic site analog. The reactions, containing 1 nM of labeled duplex substrate, 10 nM of each enzyme, and either 100 nM or 100 μM of each dNTP for templates without or with an abasic site, respectively, were incubated for 30 min at 37 °C (Materials and Methods). Schematic representations of each template/primer substrate are depicted above. Positions of the 15-mer substrate and 16- and 33-mer products are indicated on the right. (B) Processive replication of primer/template substrates by B35DNAP. Assays were performed with excess (900 nM) of primer/template 3′-GTT (lanes 1–9) or 3′-GFT (lanes 10–18) substrates and decreasing concentrations of B35DNAP. Reactions were incubated for 30 min at 37 °C and loaded into denaturing PAGE. Positions of the 15-mer substrate and 16- and 33-mer products are indicated on the right.
TLS by DNA polymerases may occur via a misalignment mechanism, resulting in a one or two nucleotide deletion during extension and a DNA product shorter than that seen with nondamaged DNA (42, 43). Fig. 4 shows that the final product synthesized by B35DNAP reached full template length using both damaged and undamaged substrates, suggesting that, instead of a misalignment mechanism, the polymerase can insert and further elongate a nucleotide opposite the THF. Moreover, misalignments in the template strand that may lead to deletions are not favored during B35DNAP polymerization, because only one nucleotide deletion was found within all of the mutant clones of MDA products.
To further confirm this mechanism, we analyzed the incorporation preference opposite the damage. Using the mutant B35DNAPexo−, we found that B35DNAP preferentially inserts purines over pyrimidines (Fig. S3), in the preference order A>G>T>C, in agreement with the so-called “A rule” previously described for most DNA polymerases (44). This preference is the same either in the 3′-GFT sequence or the 3′-GFA sequence context (Fig. S3 A and B), ruling out the possibility that the polymerase is using the next nucleotide as a template.
Fig. S3.

Abasic site TLS by B35DNAP follows the A rule. Shown is nucleotide insertion preference opposite the THF by the B35DNAPexo— mutant using the 3′-GFT (A) or 3′-GFA (B) template sequence context, in the presence of increasing amounts of each dNTP as indicated. Reactions were incubated for 10 min at 37 °C.
To further characterize the TLS capacity of B35DNAP, we performed time course kinetics, with a 90:1 substrate:enzyme ratio, to favor single-hit polymerization (Fig. 5). Under these conditions, insertion probability (blue line), bypass (i.e., full-length replication) probability (red line), and relative bypass efficiency of the THF substrate (black line) compared with nondamaged template (gray line) were calculated (45). As shown in Fig. 5A, for the 3′-CTT and 3′-CFT templates, insertion probability kinetics was identical in the presence (blue line) or absence (gray line) of the damage, with extension of the primer beyond the THF (bypass probability; red line) as the limiting step, which led to a relative CFT/CTT full-length replication (black line) of approximately 60%. However, in the 3′-CFA template, the insertion probability opposite the THF (blue line) was lower than that in the nondamaged template 3′-CTA (gray line) (Fig. 5B), which, together with the limited elongation (red line), resulted in a maximum relative bypass below 20% (black line). These results indicate that the influence of the sequence context affects both insertion opposite the THF and extension beyond the mismatch, as has been shown for other polymerases (45, 46). Importantly, the bypass efficiency of B35DNAP is higher than that of other replicative polymerases, such as yeast polε (6–17%) (46), and comparable to previously reported values for Y family polymerases, including human DNA polymerase η (10–13%), Sulfolobus solfataricus Dpo4 (13–30%), and Escherichia coli DNA polymerase IV (25–40%) (45, 47, 48).
Fig. 5.

Time course analysis of abasic site TLS by B35DNAP. Assays were made with an excess of substrate (see text for details) and two alternative sequence contexts, 3′-CTT and 3′-CFT (A) or 3′-CTA and 3′-CFA (B), and with a 90:1 substrate:enzyme ratio. Under these conditions, maximum template utilization (at 30 min) was between 30--40%. Shown are mean and SE of three independent experiments of full-length replicated product in the absence of damage (gray line), or insertion probability (blue line) and bypass probability (red line), as well as relative full-length replication of the THF-containing template (black line).
B35DNAP TLS Is Counteracted by Proofreading Activity Up to 5 nt Beyond the Abasic Site.
To gain further insight into the B35DNAP TLS mechanism on abasic sites, we performed replication assays in the presence of dCTP to minimize primer terminus exonuclease degradation and A, A+T, or A+T+G, to allow partial primer extension (Fig. 6A). Under these conditions, in the absence of damage (3′-GTT template) the stepwise addition of incoming dNTPs gave rise to the corresponding correct base pairs, with an elongation of +2 nucleotides in the presence of A for a 3′-TT template (lanes 2–4), +4 (19-mer) with A+T for a 3′-TTAT sequence in the template (lanes 5–7), and full-length replication in the presence of the four dNTPs (lanes 8–10). In contrast, with the 3′-GFT template, the addition of A (even at a concentration of 1,000×) gave rise to only a +1 elongation (opposite the abasic THF), but this mismatch, rather than being elongated, because the next template base was a T, was somewhat degraded (lanes 12–14). Elongation of this mismatch was not observed with A+T (lanes 15–17), and only with the four dNTPs (lanes 18–20) could extension beyond the abasic site be detected, suggesting that when the A is inserted opposite the THF, the A:THF base pairs can be detected as a mismatch and likely undergo a futile exonuclease/polymerase cycle that is overcome when the four dNTPs are available and eventually several nucleotides are inserted. This was confirmed when we performed the assay with the B35DNAPexo— (Fig. 6B), which was able to extend beyond the THF to an extent similar to that in the absence of damage. Nevertheless, an accumulation of the A:THF was still detected, underscoring the difficult elongation of a THF-containing mismatch by the polymerization active site. Taken together, these results indicate that extension of a THF-containing mismatch is a limiting step for B35DNAP and, moreover, is strongly counteracted by the 3′-5′ exonuclease activity.
Fig. 6.

Exonuclease activity counteracts primer extension beyond the THF. (A) Detailed template strand sequence as well as the alternative primers used are depicted above, where “X” stands for either T or THF nucleotide and the sequence context 3′-GXT is highlighted. Polymerization activity by B35DNAP on a primer/template substrate without (lanes 1–10) or with THF (F) in the first template position (lanes 11–20) by sequential addition of 100 nM (lanes 1–10) or 100 μM (lanes 11–20) of the indicated dNTPs. (B) Similar primer extension assay by the B35DNAPexo— in the presence of 50 nM (lanes 1–13) or 1 μM (lanes 14–26) of the indicated dNTPs. The effects of dNTP concentration on polymerization on undamaged and damaged templates by WT and exonuclease-deficient B35DNAP are shown in Fig. 1 and Fig. S4, respectively. Positions of the 15-mer substrate and the 19-, 22-, and 33-mer products are indicated.
To analyze this quality check step in more detail, we performed primer extension assays with duplex substrates containing the primer terminus at different positions downstream of the damage and providing a high concentration (100 µM) of the first complementary dNTP (and the 3′ end nucleotide of the primer to reduce degradation). In the absence of damage, the correct nucleotide was inserted in all substrates and, owing to the high nucleotide concentration, partial spurious extension was also observed in some cases (Fig. 7A, lanes 8–14). However, with the 3′-GFT template, which has a THF at position 16 of the template strand, when the primer had 15–21 nucleotides (with THF at positions +1, 0, −3, and −5 from the primer terminus; lanes 15–18), partial degradation rather than polymerization was observed, indicating that the THF-containing mismatch could be detected and that the primer switched to the exonuclease active site. When the 3′ end was six nucleotides downstream, the THF (22-mer primer) or beyond (lanes 19–21), extension product was detected, suggesting that B35DNAP is able to detect the mismatch on the THF up to position −5, but polymerization will proceed beyond this point.
Fig. 7.

Short-term memory of abasic sites containing mismatch by B35DNAP. (A) Detailed template strand sequence as well as the alternative primers used are depicted above the gel, where “X” represents either T or THF nucleotide. The relative distance of the X with respect to the primer size is also indicated on the right. Reactions were performed for 10 min at 37 °C in the presence of 10 nM B35DNAP and 100 μM dATP (lanes 9, 11, 16, and 18), dATP and dCTP (lanes 8, 12, 14, 15, 19, and 21), dATP and dGTP (lanes 10 and 17), or dCTP and dGTP (lanes 13 and 20). (B) B35DNAP replication of the same primer/template substrates but with 10 µM of the four dNTPs. Primer length is specified above the gel and positions of the 15- and 22-mer substrates and the 16- and 33-mer products are indicated on the right.
To further understand how B35DNAP senses the abasic site-containing mismatch during polymerization, we performed the elongation assay with different primer sizes in the presence of all dNTPs (Fig. 7B), at a concentration (10 µM) that allows insertion in front of the THF, but elongation beyond that position is not detected (Fig. S4A). Under these conditions, undamaged primer/template substrates are fully replicated independent of primer length (lanes 8–14). Using a THF-containing template, we detected insertion of a single nucleotide opposite the abasic site only when the THF was at +1 position (lane 15). Analogously, when the primer terminus was already opposite the THF, the primer was not extended beyond this position (lane 16). Furthermore, even when the lesion was three nucleotides downstream of the 3′ end (lane 17), only a negligible extension was detected. In contrast, when the damage was at position −5 (lane 18) or, more clearly, at position −6 or beyond (lanes 19–21) downstream of the primer terminus, full-length replication was detected, indicating that B35DNAP is able to detect the mismatch on the THF up to position −4 or −5.
Fig. S4.

Effect of dNTP concentration on B35DNAP abasic site TLS. Primer extension assays of the 3′-GFT sequence context are shown. The assays were carried out for 10 min at 37 °C in the presence of 10 nM of either WT (A) or exonuclease mutant (B) B35DNAP and the indicated concentration of all dNTPs. Positions of the 15-mer substrate and 33-mer product bands are indicated on the right.
Interestingly, exonuclease degradation products up to the elimination of the abasic site-containing mismatch (15-mer) were detected with all of the substrates except that containing the longer primer (lane 21), suggesting that when the mismatch is nine nucleotides downstream of the 3′ primer end, it is completely out of the polymerase and thus unperceived for this quality check. Taken together, these results confirm that B35DNAP is able to sense the damage at a distance up to at least five nucleotides downstream of the primer terminus, and thus successful TLS requires processive DNA synthesis that gets away from the lesion very quickly.
Previous reports have shown that primer extension capacity of replicative polymerases can be modulated by both DNA damage and mismatches in positions −1 to −5 with respect to the primer termini (49–51). This mismatch-induced stalling, often referred to as “short-term memory,” is a consequence of long-range distortions in the DNA that affect the conformation of the protein active site, mainly by template strand-mediated distortions (51). In positions −3 and −4, the template base moves into the minor groove in a conformation that is stabilized by direct contacts between the enzyme and the DNA backbone and solvent-mediated contacts in the minor groove; however, minor groove interactions on Φ29DNAP:DNA complex are stabilized by conserved residues in the thumb subdomain (52, 53), thanks to interactions with the primer strand bases or sugar-phosphate backbone up to the −5 position (52). Similarly, the thumb subdomain of B35DNAP would provide interactions with the mismatched wobble up to one helix turn, providing a “memory” during stepwise polymerization. This short-term memory seems to be looser during processive DNA synthesis, given that full-length molecules are produced in the presence of all four dNTPs (Figs. 4 and 6), suggesting that TLS may escape from the proofreading activity during fast and processive genome replication.
Deletion of the TPR2 Subdomain Does Not Impair Nucleotide Insertion Opposite the Abasic Site, but Abrogates Primer Extension Beyond the Lesion.
As mentioned in the introductory section, protein-primed DNA polymerases contain two insertions, TPR1 and TPR2, which in the case of Φ29DNAP are involved in interactions with the TP and in processivity and strand displacement, respectively (22, 23). Recent work has suggested that TPR2 also may be required for TLS capacity in a transposon-derived protein-primed DNA polymerase (54). Analogously, abasic site bypass capacity of human POLQ depends on unique sequence inserts that are also required for polymerization proficiency and processivity (55). Thus, we constructed a TPR2 deletion mutant of B35DNAP (ΔTPR2; Materials and Methods), which, as was foreseen for all protein-primed DNA polymerases (23), has impaired activities (Fig. S5), with a distributive DNA polymerization (lanes 6–10) and poor exonucleolytic activity (lane 5).
Fig. S5.

Exonuclease and polymerase activities of B35DNAP ΔTPR2 mutant. Shown is the polymerization activity of the B35DNAP ΔTPR2 mutant (200 nM) on primer/template substrate in the absence (lane 5) or presence (lanes 6–10) of increasing concentrations of dNTPs. WT B35DNAP (10 nM) in the absence (lane 2) or presence (lane 3) of 100 nM dNTPs served as a control. Positions of the 15-mer substrate and 33-mer product bands are indicated on the right.
We compared B35DNAP ΔTPR2 mutant polymerization activity over 3′-GTT and 3′-GFT template contexts (Fig. 8) and found that whereas polymerization proceeded opposite the undamaged sequence (lanes 5–7), when ΔTPR2 encountered an AP site at the first nucleotide in the template strand, the insertion of one nucleotide was quite efficient, with more than 25% of substrate at position +1 after 10 min (lane 11), but elongation beyond this position was not detected, even at longer reaction times (lanes 11–13), whereas wild type (WT) B35DNAP exhibited full-length primer extension (lanes 8–10). Using a primer that already contained an A opposite the THF (16-mer), full-length elongation by the WT B35DNAP could be detected after 10 min (lane 15), whereas the ΔTPR2 mutant was unable to extend the primer beyond the AP site, and degradation of the primer at this position (from 16-mer to 15-mer) could be detected only after 1 h postreaction (lanes 18–20). These results suggest that the diminished TLS capacity of the ΔTPR2 mutant is related to its processivity impairment, but it is not essential for dNTP insertion opposite the abasic site. Thus, we conclude that B35DNAP processivity contributes to the ability of TLS by facilitating the reaching of a mismatch distance (5 nt) that cannot be detected by the polymerase short-term memory and performing successful elongation beyond the damage.
Fig 8.

The B35DNAP TPR2 motif is required for extension beyond the abasic site. Polymerization activity of WT (10 nM) and ΔTPR2 (200 nM) B35DNAP on primer/template substrates with or without a THF lesion, as depicted above. Reactions were performed for the indicated times at 37 °C in the presence of 100 μM dNTPs. Positions of the 15-mer substrate and the 16- and 33-mer products are indicated on the right.
Conclusions
Replication of genomic DNA entails processivity and accuracy, which usually requires large multiprotein complexes (1). In contrast, Φ29DNAP is able to perform highly faithful DNA replication by itself, thanks to intrinsic high processivity and strand displacement capacities (20). We show here that DNA polymerase from the temperate phage Bam35 has similar features that will allow replication of the viral genome. In addition, B35DNAP has efficient abasic site TLS that seems to be counteracted by a quality check mechanism that switches the primer termini to the exonuclease domain with a “memory” for abasic sites up to 5 nt upstream. As mentioned above, replication of damaged DNA requires a polymerase with either mismatch formation tolerance, like Y-family enzymes, or the capacity to generate primer misalignments with the template strand, leading to a −1 or −2 frameshift mutagenesis, as in the case of B family Pol II (43, 56). Here we show a possible alternative way in a faithful DNA polymerase that can tolerate THF-containing mismatches during processive DNA synthesis, which, although leading to mutation accumulation, will ultimately favor the maintenance of genome. In fact, during the lysogenic cycle, Bam35 and related phages can self-replicate for countless generations inside the host and, like the host genome, are subjected to endogenous and exogenous genotoxic agents, which might lead to the accumulation of DNA damage, with abasic sites being the most frequent in bacteria (8). Indeed, B. thuringiensis has been shown to be highly sensitive to alkylating DNA damage compared with other Bacillus species (57). In this possible scenario, B35DNAP would have evolved to safeguard the viral genome integrity in vivo, by providing faithful and processive DNA replication on the one hand and TLS capacity, when required, on the other hand.
Based in the B35DNAP features reported here, it is tempting to suggest a potential application for in vitro amplification of damaged or ancient DNA, which remains an important challenge for forensic or paleontologic applications (58, 59).
Materials and Methods
Nucleotides and DNAs.
DNA oligonucleotides (Table S1) were purchased from Sigma-Aldrich, except OL33GFT, OL33GFA and OL33GTA, which were purchased from Isogen. Unlabeled dNTPs were purchased from GE Healthcare. [γ-32P]ATP (3,000 Ci/mmol; 1 Ci = 37 GBq) and [α-32P]dATP (3,000 Ci/mmol) were supplied by PerkinElmer.
Table S1.
Sequences of oligonucleotides used in this work
| Oligonucleotide | Sequence |
| OL15 (15 mer) | 5′-GATCACAGTGAGTAC-3′ |
| OL15MM (15 mer) | 5′-GATCACAGTGAGTAG-3′ |
| OL16 (16 mer) | 5′-GATCACAGTGAGTACA-3′ |
| OL19 (19 mer) | 5′-GATCACAGTGAGTACAATA-3′ |
| OL21 (21 mer) | 5′-GATCACAGTGAGTACAATAGA-3′ |
| OL22 (22 mer) | 5′-GATCACAGTGAGTACAATAGAA-3′ |
| OL23 (23 mer) | 5′-GATCACAGTGAGTACAATAGAAC-3′ |
| OL25 (25 mer) | 5′-GATCACAGTGAGTACAATAGAACGA-3′ |
| OLC33GTT (33 mer) | 5′-ACTGGCCGTCGTTCTATTGTACTCACTGTGATC-3′ |
| OLC33GFT (33 mer) | 5′-ACTGGCCGTCGTTCTATFGTACTCACTGTGATC-3′ |
| OLC33GTA (33 mer) | 5′-ACTGGCCGTCGTTCTAATGTACTCACTGTGATC-3′ |
| OLC33GFA (33 mer) | 5′-ACTGGCCGTCGTTCTAAFGTACTCACTGTGATC-3′ |
| B35pol-fw | 5′-CGCGAAGCTTAAAGGAGGAAGCATATGATGACTACTACTAATAGAAAAAAGCGT-3′ |
| B35pol-rv | 5′-CCGGATCCTTATTATAAGAAACTTAATTCACCTAATAGTTCTTCAT-3′ |
| Exo-fw | 5′-CTTTACGCTAGCTACTGCAACACGTGGTTTGGACGGCGCCGTGTTTCGAA-3′ |
| Exo-rv | 5′-TTCGAAACACGGCGCCGTCCAAACCACGTGTTGCAGTAGCTAGCGTAAAG-3′ |
| ΔTPR2-fw | 5′-TGGAAAATTTGCAATGCAGCGGGAATATGCTAAGGCTGAATATATTCAAC-3′ |
| ΔTPR2-rv | 5′-GTTGAATATATTCAGCCTTAGCATATTCCCGCTGCATTGCAAATTTTCCA-3′ |
| LacZFW | 5′-AGCTTGTCTGTAAGCGGATGCCG-3′ |
F represents the THF abasic site analog. Names of complementary 33-mer oligonucleotides include three letters corresponding to the -1, +1, and +2 template bases in the direction of the polymerization.
Bam35 genomic DNA, kindly provided by Jaana Bamford (University of Jyväskylä, Jyväskylä, Finland), was used as a template to amplify gene 5 by PCR using B35pol-fw and B35pol-rv primers and Kapa Hi-Fi DNA polymerase in HiFi buffer (Kapa Biosystems). The PCR product was cloned into the BamHI and HindIII sites of vector pT7-4 (60). The B35DNAP 3′-5′ exonuclease-deficient (B35DNAPexo−) and TPR2 deletion (ΔTPR2) mutants were obtained using the QuikChange site-directed mutagenesis kit (Agilent Technologies), using plasmid pT7-4::B35DNAP as a template. Oligonucleotides Exo-fw and Exo-rv (Table S1) were designed to generate the mutations D19A and E21A, and ΔTPR2-fw and ΔTPR2-rv were designed to delete the region coding for residues E418 to Y458, obtaining B35DNAPexo− and ΔTPR2 mutants, respectively. In all cases, the complete B35DNAP gene was sequenced.
Proteins.
Restriction enzymes, T4 DNA ligase, and T4 polynucleotide kinase were purchased from New England Biolabs. WT Φ29DNAP was obtained from the laboratory stock (61).
B35DNAP WT and mutant proteins were expressed in E. coli Bl21(DE3) harboring the corresponding expression vector in ZYM-5052 autoinduction medium (62) with 100 mg/L ampicillin. Cultures were grown for 16 h at 27 °C, after which collected cells were disrupted by grinding with alumina and suspended in buffer A [50 mM Tris⋅HCl pH 7.5, 1 mM EDTA, 7 mM β-mercaptoethanol, and 5% (vol/vol) glycerol] containing 0.5 M NaCl. Alumina and cell debris were removed by centrifugation, and absorbance at 260 nm was adjusted to 120 U/mL before DNA precipitation with 0.3% polyethyleneimine and 1 M NaCl. After centrifugation at 20,000 × g for 20 min, ammonium sulfate was added to the supernatant to 69% saturation, followed by centrifugation at 20,000 × g for 30 min. The pellets containing B35DNAP (WT and mutants) were resuspended in buffer A and applied to a Q-Sepharose fast-flow column (GE Healthcare) at an ionic strength of ∼0.2 M NaCl. DNA polymerases were eluted with 0.25 M NaCl in buffer A and applied to a phosphocellulose column (P11; Whatman). After extensive washing with 0.25, 0.4, and 0.6 M NaCl in buffer A, purified DNA polymerases were eluted with 1 M NaCl. Proteins were maintained in the presence of 0.05% Tween 20 and 50% glycerol at −20 °C, or at −70 °C for long-term storage. Final purity of the proteins was estimated as >90% by SDS/PAGE, followed by Coomassie blue staining.
Replication of Primed M13 DNA.
Genomic M13mp18 single-stranded circular DNA (laboratory stock) was annealed to the universal M13 primer in a buffer containing 0.2 M NaCl and 60 mM Tris⋅HCl pH 7.5, to analyze processive DNA polymerization coupled to strand displacement by B35DNAP. The reaction mixture contained, in a final volume of 25 μL, 50 mM Tris⋅HCl pH 7.5, 1 mM DTT, 4% (vol/vol) glycerol, 0.1 mg/mL BSA, 0.05% Tween 20, 40 μM dNTPs, 0.5 μCi [α-32P]dATP, 3.2 nM primed M13mp18 ssDNA, 10 mM MgCl2, and the indicated concentrations of each DNA polymerase. After incubation for 20 min at 37 °C, the reactions were stopped by adding 30 mM EDTA, 0.5% SDS, and 0.3 M NaCl. The lambda DNA ladder used as a size marker was labeled by filling in with Klenow fragment (New England Biolabs) in the presence of [α-32P]dATP (63). DNA replication products were analyzed by electrophoresis in alkaline 0.7% agarose, and the gel was stained with ethidum bromide, dried, and autoradiographed.
Multiple Displacement Amplification of Plasmid DNA and Fidelity Analysis.
The amplification of pUC19 (Thermo Scientific), a plasmid that contains the lacZα indicator gene, was carried out as described by Nelson et al. (26), with minor modifications. In brief, the incubation mixture (20 μL) contained 40 mM Tris⋅HCl, pH 7.5, 50 mM KCl, 0.025% Tween 20, 10 mM MgCl2, 500 µM dNTPs, 25 µM 3′-protected hexamer random primers (Integrated DNA Technologies), 50 nM B35DNAP, and the indicated ammounts of pUC19 plasmid. The reactions were incubated for 16 h at 37 °C, after which 2 μL of each amplification product was digested with EcoRI-HF for 2 h at 37 °C and subjected to electrophoresis in 0.7% agarose gel.
To analyze the fidelity of B35DNAP, we treated a fraction of amplified DNA product with EcoRI-HF and DpnI simultaneously for 2 h at 37 °C. The digested products were purified using a PCR clean-up kit (Qiagen), and 170 ng of pUC19 linear plasmid was circularized with T4 DNA ligase in a final volume of 100 μL. To analyze mutations in the lacZα gene, we transformed 20 ng of ligated DNA into the E. coli XL-1 strain for blue/white colony screening. Several LacZα mutants (white colonies) were further analyzed by plasmid DNA sequencing with primer LacZFW (Table S1). Given the variability reported for the number of mutants that may be generated during plasmid replication or repair in different bacterial strains (64), the background mutation frequency (f0) for each experiment was determined with the original plasmid digested with EcoRI-HF, purified, and religated under the same conditions.
Primer Extension Assays.
Primer oligonucleotides OL15, OL15MM, OL16, OL19, OL21, OL22, OL23, and OL25 (Table S1) were purified electrophoretically on 8 M urea-20% polyacrylamide gels and 5′-labeled with [γ-32P]ATP using T4 polynucleotide kinase. Labeled oligonucleotides were hybridized to 1.2-fold molar excess of complementary unlabeled template oligonucleotides (OLC33s) in the presence of 50 mM NaCl and 50 mM Tris⋅HCl pH 7.5, resulting in a primer–template duplex substrate. Assays were performed in a final volume of 20 µL containing 50 mM Tris⋅HCl pH 7.5, 1 mM DTT, 4% (vol/vol) glycerol, 0.1 mg/mL BSA, and, unless stated otherwise, 1 nM of the indicated 5′-labeled primer/template duplex, 10 nM DNA polymerase, and the indicated dNTPs concentration. Reactions were triggered by the addition of 10 mM MgCl2. After incubation for the indicated times at 37 °C, the reactions were stopped by adding 10 μL of formamide loading buffer (98% formamide, 20 mM EDTA, 0.5% bromophenol blue, and 0.5% xylene cyanol). Samples were analyzed by 8 M urea-20% polyacrylamide gel electrophoresis in 1× Tris-borate-EDTA buffer. Gel bands were detected by autoradiography, and phosphorimages (Fuji BAS1000) were used for quantitation purposes with ImageJ software.
Supplementary Material
Acknowledgments
We thank Dr. J. K. Bamford for providing DNA of the Bam35 phage. Funding for this work was provided by the Spanish Ministry of Economy and Competitiveness (Grants BFU2011-23645 and BFU2014-52656, to M.S. and Doctoral Fellowship BES-2012-052228, to M.B.-O.) and the Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1510280112/-/DCSupplemental.
References
- 1.O’Donnell M, Langston L, Stillman B. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb Perspect Biol. 2013;5(7):a010108. doi: 10.1101/cshperspect.a010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johansson E, Dixon N. Replicative DNA polymerases. Cold Spring Harb Perspect Biol. 2013;5(6):a012799. doi: 10.1101/cshperspect.a012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11(3):208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 4.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18(1):148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang W. An overview of Y-family DNA polymerases and a case study of human DNA polymerase η. Biochemistry. 2014;53(17):2793–2803. doi: 10.1021/bi500019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCulloch SD, Kokoska RJ, Garg P, Burgers PM, Kunkel TA. The efficiency and fidelity of 8-oxo-guanine bypass by DNA polymerases delta and eta. Nucleic Acids Res. 2009;37(9):2830–2840. doi: 10.1093/nar/gkp103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Vega M, Salas M. A highly conserved tyrosine residue of family B DNA polymerases contributes to dictate translesion synthesis past 8-oxo-7,8-dihydro-2′-deoxyguanosine. Nucleic Acids Res. 2007;35(15):5096–5107. doi: 10.1093/nar/gkm545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362(6422):709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura J, Swenberg JA. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999;59(11):2522–2526. [PubMed] [Google Scholar]
- 10.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 11.Niimi A, et al. Palm mutants in DNA polymerases alpha and eta alter DNA replication fidelity and translesion activity. Mol Cell Biol. 2004;24(7):2734–2746. doi: 10.1128/MCB.24.7.2734-2746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanguy Le Gac N, Delagoutte E, Germain M, Villani G. Inactivation of the 3′-5′ exonuclease of the replicative T4 DNA polymerase allows translesion DNA synthesis at an abasic site. J Mol Biol. 2004;336(5):1023–1034. doi: 10.1016/j.jmb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Zhu Y, Song L, Stroud J, Parris DS. Mechanisms by which herpes simplex virus DNA polymerase limits translesion synthesis through abasic sites. DNA Repair (Amst) 2008;7(1):95–107. doi: 10.1016/j.dnarep.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong X, Pedersen LC, Kunkel TA. Characterization of a replicative DNA polymerase mutant with reduced fidelity and increased translesion synthesis capacity. Nucleic Acids Res. 2008;36(12):3892–3904. doi: 10.1093/nar/gkn312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel PH, Kawate H, Adman E, Ashbach M, Loeb LA. A single highly mutable catalytic site amino acid is critical for DNA polymerase fidelity. J Biol Chem. 2001;276(7):5044–5051. doi: 10.1074/jbc.M008701200. [DOI] [PubMed] [Google Scholar]
- 16.Filée J, Forterre P, Sen-Lin T, Laurent J. Evolution of DNA polymerase families: Evidences for multiple gene exchange between cellular and viral proteins. J Mol Evol. 2002;54(6):763–773. doi: 10.1007/s00239-001-0078-x. [DOI] [PubMed] [Google Scholar]
- 17.Koonin EV. Temporal order of evolution of DNA replication systems inferred by comparison of cellular and viral DNA polymerases. Biol Direct. 2006;1:39. doi: 10.1186/1745-6150-1-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salas M. Protein-priming of DNA replication. Annu Rev Biochem. 1991;60:39–71. doi: 10.1146/annurev.bi.60.070191.000351. [DOI] [PubMed] [Google Scholar]
- 19.Kapitonov VV, Jurka J. Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci USA. 2006;103(12):4540–4545. doi: 10.1073/pnas.0600833103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Vega M, Salas M. Protein-primed replication of bacteriophage Φ29 DNA. In: Kusic-Tisma J, editor. DNA Replication and Related Cellular Processes. InTech; Rijeka, Croatia: 2011. [Google Scholar]
- 21.Hoeben RC, Uil TG. Adenovirus DNA replication. Cold Spring Harb Perspect Biol. 2013;5(3):a013003. doi: 10.1101/cshperspect.a013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dufour E, et al. An aspartic acid residue in TPR-1, a specific region of protein-priming DNA polymerases, is required for the functional interaction with primer terminal protein. J Mol Biol. 2000;304(3):289–300. doi: 10.1006/jmbi.2000.4216. [DOI] [PubMed] [Google Scholar]
- 23.Rodríguez I, et al. A specific subdomain in Φ29 DNA polymerase confers both processivity and strand-displacement capacity. Proc Natl Acad Sci USA. 2005;102(18):6407–6412. [Google Scholar]
- 24.Blanco L, Salas M. Relating structure to function in Φ29 DNA polymerase. J Biol Chem. 1996;271(15):8509–8512. doi: 10.1074/jbc.271.15.8509. [DOI] [PubMed] [Google Scholar]
- 25.Esteban JA, Salas M, Blanco L. Fidelity of Φ29 DNA polymerase: Comparison between protein-primed initiation and DNA polymerization. J Biol Chem. 1993;268(4):2719–2726. [PubMed] [Google Scholar]
- 26.Nelson JR, et al. TempliPhi, Φ29 DNA polymerase-based rolling circle amplification of templates for DNA sequencing. Biotechniques. 2002;(Suppl):44–47. [PubMed] [Google Scholar]
- 27.Blanco L, et al. Highly efficient DNA synthesis by the phage Φ29 DNA polymerase: Symmetrical mode of DNA replication. J Biol Chem. 1989;264(15):8935–8940. [PubMed] [Google Scholar]
- 28.Caldentey J, Blanco L, Savilahti H, Bamford DH, Salas M. In vitro replication of bacteriophage PRD1 DNA: Metal activation of protein-primed initiation and DNA elongation. Nucleic Acids Res. 1992;20(15):3971–3976. doi: 10.1093/nar/20.15.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.García P, et al. Formation of a covalent complex between the terminal protein of pneumococcal bacteriophage Cp-1 and 5′-dAMP. J Virol. 1986;58(1):31–35. doi: 10.1128/jvi.58.1.31-35.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ravantti JJ, Gaidelyte A, Bamford DH, Bamford JK. Comparative analysis of bacterial viruses Bam35, infecting a gram-positive host, and PRD1, infecting gram-negative hosts, demonstrates a viral lineage. Virology. 2003;313(2):401–414. doi: 10.1016/s0042-6822(03)00295-2. [DOI] [PubMed] [Google Scholar]
- 31.Gillis A, Mahillon J. Phages preying on Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis: Past, present and future. Viruses. 2014;6(7):2623–2672. doi: 10.3390/v6072623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson JR. Random-primed, Phi29 DNA polymerase-based whole- genome amplification. Curr Protoc Mol Biol. 2014;6(105):Unit 15.13. doi: 10.1002/0471142727.mb1513s105. [DOI] [PubMed] [Google Scholar]
- 33.Cline J, Braman JC, Hogrefe HH. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996;24(18):3546–3551. doi: 10.1093/nar/24.18.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McInerney P, Adams P, Hadi MZ. Error rate comparison during polymerase chain reaction by DNA polymerase. Mol Biol Int. 2014;2014:287430. doi: 10.1155/2014/287430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jalasvuori M, et al. Identification of five novel tectiviruses in Bacillus strains: Analysis of a highly variable region generating genetic diversity. Res Microbiol. 2013;164(2):118–126. doi: 10.1016/j.resmic.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 36.Arana ME, Seki M, Wood RD, Rogozin IB, Kunkel TA. Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res. 2008;36(11):3847–3856. doi: 10.1093/nar/gkn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maga G, Shevelev I, Ramadan K, Spadari S, Hübscher U. DNA polymerase theta purified from human cells is a high-fidelity enzyme. J Mol Biol. 2002;319(2):359–369. doi: 10.1016/S0022-2836(02)00325-X. [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Diaz M, Kunkel TA. Mechanism of a genetic glissando: Structural biology of indel mutations. Trends Biochem Sci. 2006;31(4):206–214. doi: 10.1016/j.tibs.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Yousefzadeh MJ, et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 2014;10(10):e1004654. doi: 10.1371/journal.pgen.1004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hogg M, Wallace SS, Doublié S. Crystallographic snapshots of a replicative DNA polymerase encountering an abasic site. EMBO J. 2004;23(7):1483–1493. doi: 10.1038/sj.emboj.7600150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi JY, Lim S, Kim EJ, Jo A, Guengerich FP. Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases α, δ, η, ι, κ, and REV1. J Mol Biol. 2010;404(1):34–44. doi: 10.1016/j.jmb.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohashi E, et al. Error-prone bypass of certain DNA lesions by the human DNA polymerase kappa. Genes Dev. 2000;14(13):1589–1594. [PMC free article] [PubMed] [Google Scholar]
- 43.Wang F, Yang W. Structural insight into translesion synthesis by DNA Pol II. Cell. 2009;139(7):1279–1289. doi: 10.1016/j.cell.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strauss BS. The “A” rule revisited: Polymerases as determinants of mutational specificity. DNA Repair (Amst) 2002;1(2):125–135. doi: 10.1016/s1568-7864(01)00014-3. [DOI] [PubMed] [Google Scholar]
- 45.Kokoska RJ, McCulloch SD, Kunkel TA. The efficiency and specificity of apurinic/apyrimidinic site bypass by human DNA polymerase eta and Sulfolobus solfataricus Dpo4. J Biol Chem. 2003;278(50):50537–50545. doi: 10.1074/jbc.M308515200. [DOI] [PubMed] [Google Scholar]
- 46.Sabouri N, Johansson E. Translesion synthesis of abasic sites by yeast DNA polymerase epsilon. J Biol Chem. 2009;284(46):31555–31563. doi: 10.1074/jbc.M109.043927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stone JE, et al. Lesion bypass by S. cerevisiae Pol ζ alone. DNA Repair (Amst) 2011;10(8):826–834. doi: 10.1016/j.dnarep.2011.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maor-Shoshani A, Meira LB, Yang X, Samson LD. 3-Methyladenine DNA glycosylase is important for cellular resistance to psoralen interstrand cross-links. DNA Repair (Amst) 2008;7(8):1399–1406. doi: 10.1016/j.dnarep.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller H, Grollman AP. Kinetics of DNA polymerase I (Klenow fragment exo-) activity on damaged DNA templates: Effect of proximal and distal template damage on DNA synthesis. Biochemistry. 1997;36(49):15336–15342. doi: 10.1021/bi971927n. [DOI] [PubMed] [Google Scholar]
- 50.Ng L, Weiss SJ, Fisher PA. Recognition and binding of template primers containing defined abasic sites by Drosophila DNA polymerase alpha holoenzyme. J Biol Chem. 1989;264(22):13018–13023. [PubMed] [Google Scholar]
- 51.Johnson SJ, Beese LS. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116(6):803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- 52.Berman AJ, et al. Structures of phi29 DNA polymerase complexed with substrate: The mechanism of translocation in B-family polymerases. EMBO J. 2007;26(14):3494–3505. doi: 10.1038/sj.emboj.7601780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pérez-Arnaiz P, Lázaro JM, Salas M, de Vega M. Involvement of Φ29 DNA polymerase thumb subdomain in the proper coordination of synthesis and degradation during DNA replication. Nucleic Acids Res. 2006;34(10):3107–3115. doi: 10.1093/nar/gkl402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pastor-Palacios G, López-Ramírez V, Cardona-Felix CS, Brieba LG. A transposon-derived DNA polymerase from Entamoeba histolytica displays intrinsic strand displacement, processivity and lesion bypass. PLoS ONE. 2012;7(11):e49964. doi: 10.1371/journal.pone.0049964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hogg M, Seki M, Wood RD, Doublié S, Wallace SS. Lesion bypass activity of DNA polymerase θ (POLQ) is an intrinsic property of the pol domain and depends on unique sequence inserts. J Mol Biol. 2011;405(3):642–652. doi: 10.1016/j.jmb.2010.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodman MF, Woodgate R. Translesion DNA polymerases. Cold Spring Harb Perspect Biol. 2013;5(10):a010363. doi: 10.1101/cshperspect.a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morhoshi F, Munakata N. Diverse capacities for the adaptive response to DNA alkylation in Bacillus species and strains. Mutat Res. 1995;337(2):97–110. doi: 10.1016/0921-8777(95)00013-a. [DOI] [PubMed] [Google Scholar]
- 58.Höss M, Jaruga P, Zastawny TH, Dizdaroglu M, Pääbo S. DNA damage and DNA sequence retrieval from ancient tissues. Nucleic Acids Res. 1996;24(7):1304–1307. doi: 10.1093/nar/24.7.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.d’Abbadie M, et al. Molecular breeding of polymerases for amplification of ancient DNA. Nat Biotechnol. 2007;25(8):939–943. doi: 10.1038/nbt1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tabor S, Richardson CC. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci USA. 1985;82(4):1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lázaro JM, Blanco L, Salas M. Purification of bacteriophage Φ29 DNA polymerase. Methods Enzymol. 1995;262:42–49. doi: 10.1016/0076-6879(95)62007-9. [DOI] [PubMed] [Google Scholar]
- 62.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif. 2005;41(1):207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 63.Sambrook J, Russell D. Molecular Cloning: A Laboratory Manual. 4th Ed Cold Spring Harbor Lab Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 64.Jozwiakowski SK, Connolly BA. Plasmid-based lacZalpha assay for DNA polymerase fidelity: Application to archaeal family-B DNA polymerase. Nucleic Acids Res. 2009;37(15):e102. doi: 10.1093/nar/gkp494. [DOI] [PMC free article] [PubMed] [Google Scholar]