

Significance
Apoptosis, a type of programmed cell death, is critical for the removal of diseased or superfluous cells and is essential in maintaining tissue homeostasis. At the molecular level, apoptosis leads to the highly coordinated selective proteolysis of more than a thousand cellular proteins. Until now, determining the individual molecular consequence of proteolysis has been largely out of reach. Our article reports biotechnological advances that enable rapid and reliable posttranscriptional gene replacement for robust inducible and selective proteolysis of the replacement allele. We go on to further show the utility of our technology in deciphering synthetic lethality resulting from coordinated proteolysis of caspase substrates that control the apoptotic hallmark of chromatin fragmentation.
Keywords: DNA damage, apoptosis, ICAD, site-specific proteolysis, TEV protease
Abstract
Cellular demolition during apoptosis is completed by executioner caspases, that selectively cleave more than 1,500 proteins but whose individual roles are challenging to assess. Here, we used an optimized site-specific and inducible protease to examine the role of a classic apoptotic node, the caspase-activated DNase (CAD). CAD is activated when caspases cleave its endogenous inhibitor ICAD, resulting in the characteristic DNA laddering of apoptosis. We describe a posttranscriptional gene replacement (PTGR) approach where endogenous biallelic ICAD is knocked down and simultaneously replaced with an engineered allele that is susceptible to inducible cleavage by tobacco etch virus protease. Remarkably, selective activation of CAD alone does not induce cell death, although hallmarks of DNA damage are detected in human cancer cell lines. Our data strongly support that the highly cooperative action of CAD and inhibition of DNA repair systems are critical for the DNA laddering phenotype in apoptosis. Furthermore, the PTGR approach provides a general means for replacing wild-type protein function with a precisely engineered mutant at the transcriptional level that should be useful for cell engineering studies.
Apoptosis, programed cell death, is crucial during development and for maintaining tissue homeostasis in an adult organism (1–3). Dysregulation of apoptosis can have drastic disease consequences, such as cancer, autoimmunity, and neurodegeneration (4–6). Apoptosis is a tightly controlled cellular process that leads to the contained demolition of individual cells and preparation for their noninflammatory disposal through engulfment by phagocytic cells (7, 8). Induction of apoptosis causes prominent morphological changes, such as cellular blebbing and DNA laddering, resulting from chromatin cleavage (9, 10).
A great deal is known about the signaling events that initiate the hard-wired program of apoptosis that leads to caspase activation (11–13), cysteine-class aspartate-specific proteases that deconstruct the cell. The number of caspase substrates continue to grow every year, with over 1,500 substrates identified from several complementary global proteomic approaches (14–18). The substrate cleavage rates vary over 500-fold in cells (19). Although substrate identification and kinetics can begin to shed light on the proteolysis process, they do not address the role of specific cleavage events in apoptosis. Our understanding of how single substrates can drive apoptosis or how they depend on coordination with other substrates, thereby forming nodes of synthetic lethality, is lacking. Gene disruption and overexpression of cleaved substrates have suggested the importance of particular substrates in driving the apoptotic process (reviewed in ref. 20). However, overexpression approaches cannot mimic the temporal aspects or N- and C-terminal proteolytic fragments resulting from caspase proteolysis. Thus, there is a need to develop approaches for inducible site-specific proteolysis in cells to assess the role of specific substrates in apoptosis.
Recently, our group has developed an enzyme called the SNIPer (Single Nick in Proteome) that is capable of cleaving individual substrates in cells (21). The SNIPer is a split-protein version of NIa tobacco etch virus protease (TEVP), which relies on chemical induced dimerization of FRB (FKBP rapamycin binding) and FKBP in the presences of rapamycin for selective activation. The SNIPer was first applied to determine the role of selective proteolysis that activates individual caspases in apoptosis. By replacing the normal caspase cleavage sites in the executioner caspases-3, -6, and -7 with the unique TEVP recognition sequence [ENYLFQ/(G or S)], it was possible to generate caspaseTevS isoforms that could be cleaved and activated by TEVP upon small molecule addition. Interestingly, these experiments demonstrated that direct activation of a single substrate, caspase-3 or -7, lead to apoptosis, whereas caspase-6 does not (21). Can other individual substrates do the same?
DNA fragmentation is a catastrophic event for cells and a hallmark of apoptosis. DNA laddering is the result of double-stranded breaks at accessible regions of DNA between nucleosomes, thereby producing discrete chromosomal fragments (multiples of ∼180 bp) (22). The inhibitor caspase-activated DNase (ICAD)–CAD complex is believed to be the central apoptotic node for this fragmentation (23). Caspase cleavage of the regulator ICAD results in release, dimerization, and activation of CAD. Splicing of the ICAD transcript produces two variants, a cytosolic localized short (ICADS) isoform and the nuclear localized long (ICADL) isoform (24). Additionally, ICADL serves as a mandatory folding chaperone for CAD (24). Cells deficient in ICAD lack the CAD protein and DNA fragmentation, suggesting CAD is required for DNA fragmentation (25). Two types of apoptotic DNA fragmentation have been previously described: a high molecular weight and lower molecular weight cleavage. CAD has been implicated in generating either high molecular weight or lower molecular weight, or both, depending on the specific cell line and method used (26). Is CAD activation sufficient to cause DNA fragmentation and to kill cells?
Here, we present a system to test the cellular consequences for direct activation of CAD through site-specific proteolysis of ICAD using two small-molecule inducible TEVP strategies. This process required that we simultaneously deplete the endogenous transcripts for caspase sensitive ICAD and introduce TevS variants at physiological levels, an approach we call posttranscriptional gene replacement (PTGR). The recent advances in clustered regularly interspaced short palindromic repeats (CRISPR)-mediated genomic engineering are now routine for studies involving gene knockout and gene activation experiments; however, site-specific mutagenesis remains a substantial hurdle for efficient and robust Cas9 nickase-induced homology-directed repair (27). Using a single lentiviral vector for PTGR-mediated replacement and cellular engineering is a well-suited technology for experiments requiring replacement with site-specific mutations.
Combining selective and inducible proteolysis with substrate replacement, we discovered that CAD activation alone did not lead to substantial DNA fragmentation or cell death, but could only enhance fragmentation levels when combined with DNA-repair inhibitors or drug-induced chromatin relaxation. These data suggest that CAD cannot act alone in cancer cells, and that constitutive DNA damage response is sufficient to control CAD inflicted DNA damage (28). Moreover these studies show the power of combining a next generation of inducible TEVP strategies with a loss-of-function substrate through PTGR-mediated replacement for selective proteolysis of substrates in cells.
Results
Simultaneous Protein Knockdown and Replacement Using PTGR.
The CAD inhibitor, ICAD, is inactivated by caspase cleavage, resulting in a loss-of-function event. To place this event under proteolytic control of the SNIPer required concurrently reducing levels of the endogenous ICAD allele and introducing the TEVP sensitive (TevS) allele. The PTGR strategy allows simultaneous protein knockdown and replacement through the bicistronic expression of an engineered gene followed by a synthetic microRNA (miRNA) targeting the endogenous gene’s UTR or coding sequence (Fig. 1A and Fig. S1). The coexpression of each cassette results in miRNA reduction of the endogenous gene while simultaneously expressing the engineered substrate (Fig. 1B). The native caspase cleavage site is precisely replaced with a TEVP cleavage sequence, resulting in the orthogonal substrateTevS, which can no longer be cleaved by caspases. In addition, we have made a silent mutation in the replacement allele (substrateTevS) to make it resistant to the miRNA. For robust and broad specificity, the PTGR cassette is placed in a lentiviral backbone, pHR (Fig. 1C) (29). To achieve optimal tuned expression, we used a dual-selection strategy that relied on drug selection followed by stringent flow cytometry sorting of GFP+ cells.
Fig. 1.

Strategy for simultaneous protein knockdown and replacement using PTGR. (A) The PTGR vector uses a synthetic miRNA that is complementary to the endogenous target mRNA but is genetically orthogonal to the mutated region or UTR in the replacement allele. (B) The endogenous protein levels diminish as the orthogonal allele’s expression levels increase as a result of cistronic expression of the microRNA and the orthogonal allele. (C) The three components of the PTGR vector are inserted in a lentiviral backbone for targeting a wide variety of cell lines. Multisite Gateway cloning enables quick assembly of the final vector from donor plasmids, synthetic microRNA, and the orthogonal replacement allele. Donor plasmids are assembled and screened independently. The third component consists of an IRES sequence for cap-independent initiation of a combination selectable marker including both drug resistance and GFP fluorescence, separated by a “self-cleaving” T2A peptide sequence.
Fig. S1.

Three-component PTGR system and workflow. (A) Entry vectors harboring the replacement allele, in our case ICAD, are constructed by standard PCR techniques and BP-cloned into the pDONR-CMV-EGFP-P4-P1r. Endogenous allele-targeted synthetic miRNAs are ligated into PENTR-CMV-miR-L1-L2 using standard ligation of dsDNA (see SI Materials and Methods for design criteria). (B) Assembled pDONR and pENTR vectors can be immediately used to screen for miR knockdown efficiency by cotransfection of the two plasmids, using the loss of EGFP fluorescence as a read-out for functional transcript knockdown. A microRNA immune replacement allele can be quickly generated by overlap PCR extension, once the desired microRNA is selected. (C) The pDONR and pENTR vectors are combined with pHR-PTGR-GWR4-R2-EV using standard multisite Gateway cloning to generate the final PTGR lentiviral vector.
We tested functional knockdown and simultaneous replacement using a series of ICAD replacement alleles in HEK293 and HeLa cell lines (Fig. 2 and Fig. S2A). We observed a substantial reduction in ICAD protein levels in the ICAD knockdown cell line compared with empty vector HEK293 cells. As expected, there was decreased CAD expression in the absence of ICAD, consistent with its role as a folding chaperone. Upon ICAD replacement with engineered alleles, we observed expression of ICADTevS and corresponding rescue of CAD levels. Furthermore, CAD expression levels remained unchanged by PTGR-mediated replacement with WT or TevS alleles (Fig. 2). Taken together, these data suggest that (i) the ICADTevS allele was resistant to the orthogonal miR and (ii) both WT and LTevS alleles are able to act as chaperones for CAD folding during translation. We cross-validated the immunoblot results using quantitative RT-PCR (RT-qPCR) to directly monitor ICAD transcript levels (Fig. S2). The parental cell line expression level was set at 1.00, and mean value of expression of ICAD in PTGR ICAD knockdown cell line was 0.13 (+0.12/-0.05), corresponding to ∼85% reduction of ICAD expression.
Fig. 2.

Biochemical evidence that the PTGR strategy reduces the endogenous target while producing the replacement target. A series of stable cell lines were made using the PTGR vector and protein levels were analyzed by immunobloting against ICAD and CAD in HEK293 (A) and HeLa (B) cells. EV corresponds to cells engineered with an empty vector. ICAD knockdown (KD) was completed with a PTGR vector expressing Renilla in the position of the replacement ICAD allele. Three different ICAD replacement cells lines were created using ICAD long (L) and containing either WT caspase cleavage sequences or TevS sequences. In A, ICAD knockdown results in an absence of CAD protein, whereas PTGR mediated replacement rescues CAD levels. The addition of the larger TevS sequence at the two cleavage sites causes a slight upward molecular weight shift in the ICADLTevS allele. The short splice ICADSTevS allele is the clear lower molecular weight band in the ICAD blot (Top, far right). GAPDH was used as a protein loading control.
Fig. S2.

PTGR-mediated knockdown and replacement. (A) Additional immunoblotting panel of HeLa PTGR engineered cell lines, similar to Fig. 2B, with the addition of the third PTGR ICAD replacement allele (i.e., ICADSTevS). The short-splice ICADSTevS allele is the clear lower molecular-weight band in the ICAD blot. (B) Transcript reduction was quantitatively measured by RT-qPCR using specific Taqman probes to the endogenous ICAD gene (DFFA). Relative quantitation was performed using β-actin (ACTB) as control for total transcript level. Error bars represent SD for three biological replicates. The parental cell line was used to normalize expression levels. ICAD knockdown cells were engineered with a PTGR vector containing the coding sequence for Renilla in place of the ICAD replacement allele (same as Fig. 2A).
Induced and Selective Cleavage of ICADTevS.
We sought to preserve the natural pathway of proteolytic CAD activation and retain ICAD’s chaperone and inhibitor function (Fig. 3A). To complete this objective we combined our PTGR strategy with inducible and selective proteolysis of TEVP. Before combining PTGR and split-TEVP strategies, we next optimized the inducible protease approach. The split-TEVP, SNIPer, relies on the heterodimerization of FKBP and FRB in the presence of cell permeable rapamycin to assemble the N- and C-terminal halves of TEVP and activate the protease. A low level of TEVP activity is detectable with the SNIPer in the absence of rapamycin. Furthermore, split-TEVP has reduced activity upon rapamycin activation, likely a consequence incomplete assembly or instability of the split-protein (30). Although the lower activity of the SNIPer is adequate for testing apoptotic nodes undergoing positive feedback (such as caspase activation), we found it was less optimal for the current application where we wanted rapid and complete proteolysis of the ICADTevS substrate to ensure depletion of ICAD pools while outpacing substrate resynthesis rates.
Fig. 3.

Evidence of induced and selective cleavage of ICADLTevS. (A) ICAD is a mandatory folding chaperone and inhibitor of CAD. ICAD tightly inhibits CAD activation until it is cleaved which leads to dimerization and activation of CAD. To selectively activate CAD we used an orthogonal activation allele that that serves as a folding chaperone and is cleavable by TEVP (arrowheads). (B) Different strategies for inducible TEVP were constructed to maximize ease of use, expression levels, and protease activity. A dual vector version resulted in minimal decrease in full-length substrate, tagged with the FLAG epitope. The tetracycline inducible full-length TEVP results in near complete full-length substrate cleavage within 12 h. (C) Combining PTGR ICADLTevS and iTEVP in a stable cell line results in robust inducible expression of TEVP (Myc-tagged) and complete and sustained proteolysis of ICADLTevS.
As a result, we explored a variety of transcriptional and translational strategies to enhance inducible TEV proteolysis (Fig. 3B and Fig. S3). We first generated a single vector dual-expression system, using an internal ribosome entry site (IRES) sequence to drive the expression of the second, C-TEV, fragment; however, this suffered from low expression levels of the second fragment and poor activity upon dimerization. Next, we used a 2A “self-cleaving” peptide sequence to drive cistronic translation of N-TEV and C-TEV in two orientations (31). This 2A configuration yielded robust protein expression levels and inducible activity, with the C-Tev_T2A_N-Tev being the better of the two. However, background activity in the absence of rapamycin was still a concern as expression levels were increased (Fig. S3C). As a result of the limitations observed in the split-TEVP strategy, we explored tetracycline inducible promoters to drive full-length TEVP expression we call iTEVP. As expected, when ICADLTevS was expressed in cells containing either the SNIPer or iTEVP and activated with rapamycin or tetracycline, respectively, we observed a substantial decrease in full-length ICADLTevS. However, the iTEVP exhibited better inducible and selective cleavage of the target protein in the shortest period (Fig. 3C). We therefore moved forward with characterizing the biochemical consequences in cells engineered with the iTEVP system.
Fig. S3.

(A) Schematics of different coexpression strategies designed and tested to optimize for robust expression, inducibility, and activity of engineered TEVPs. (B) Each protease variant was cotransfected with mock substrate in 293FT cells and then treated for the indicated time with the inducer (10 nM rapamycin or 1 µg/mL doxycycline). Substrate cleavage was monitored by immunobloting for the full-length FLAG tagged substrate. (C) Characterization of SNIPer variant efficiencies by monitoring full-length and cleaved fragments of the TevS cleavage reporter, ECFP-TevS-YPET. HeLa cells were transiently cotransfected with proteolysis reporter and indicated SNIPer variants. Twenty-four hours posttransfection, cells were incubated for 6 h with 10 nM rapamycin or vehicle and then collected for immunobloting.
ICADLTevS is rapidly cleaved after doxycycline addition (Fig. 3C). To confirm our engineering approach results in an active CAD species, we undertook a series of in vitro CAD activity DNA fragmentation assays to confirm that our activation strategy worked in the engineered tissue culture cells. We expressed and purified recombinant heterodimers of ICAD:CAD and ICADTevS:CAD dimers and treated with activating protease, caspase-3 or TEVP, respectively. In both cases this resulted in an active CAD species (Fig. S4A). As expected, caspase-3 did not activate ICADTevS:CAD and neither did TEVP activate ICADWT:CAD, showing the orthogonality of the systems (Fig. S4B). We next determined if the ICADLTevS:CAD complex produced directly in HeLa cells could be activated by TEVP. Coimmunoprecipitation of the ICADLTevS:CAD complex from HeLa cells followed by TEVP treatment showed activity against both plasmid and chromatin substrates (Fig. S4 C and D). From these results we concluded that active CAD can be generated from an ICADLTevS:CAD complex by TEVP but not by caspase-3.
Fig. S4.

Functional activation of CAD. (A) Recombinantly coexpressed and purified ICADTevS/CAD duplex was activated by TEVP. CAD activity is measured by fragmentation of nuclear chromatin extracts following gel electrophoresis. (B) Recombinantly coexpressed ICADLWT/CAD and ICADLTevS/CAD is tested for orthogonality by addition of the indicated activating proteases and incubated for 1 h. Simultaneous protein status and CAD activity is monitored by SDS/PAGE (Lower) and plasmid migration in gel electrophoresis (Upper). (C) Immunoblot of coimmunoprecipitated ICAD:CAD. HA-CAD was transfected into order to increase pull down of the heterodimer complex in a FLAG coimmunoprecipitation. Input corresponds to 10% lysate. (D) Coimmunoprecipitated ICADTevS:CAD from mammalian cells is activated ex vivo by recombinant full-length TEVP and cleaves plasmid DNA and chromatin extracts.
Phenotypic Effects of Selective Activation of CAD by iTevP.
Following biochemical confirmation that the iTEVP system was optimized, we sought to understand the phenotypic consequences for specific activation of CAD in HeLa cells. Remarkably, we observed no significant change in cell viability or caspase activity upon doxycycline treatment for up to 48 h, as measured by Cell Titer Glo or Caspase-3/-7 Glo (Promega) (Fig. 4A and Fig. S5A). This came as an initial surprise to us because we believed that difficult to repair double-stranded DNA breaks would feedback and activate the apoptotic program. This finding indicated that a more detailed cellular characterization of the pathway downstream of CAD activation was required.
Fig. 4.

Phenotypic effects of selective CAD activation by iTEVP. (A) Cell viability, measured by Cell Titer Glo (detected by luciferase signal absorption at 405 nm) following iTEVP activation by addition of doxycycline in PTGR ICAD-engineered cells. (B) Cytoplasmic enrichment of free histone measured by ELISA following activation of iTEVP. (C) Phosphorylation of H2AX monitored by ELISA. All error bars in A–C represent SD for three biological replicates. Significance was determined by one-way ANOVA Dunnett correction. (D) Quantitation of TUNEL staining at 24 and 48 h following iTEVP activation (1 µg/mL Doxycycline) (red bar, mean; blue, SD). (E) Representative images from TUNEL staining following selective ICADLTevS cleavage. (F) Representative images from immunofluorescent staining for phosphorylation of serine-139 H2AX, following selective ICADLTevS cleavage induced by addition of doxycycline. (G) Representative images of single cell gel electrophoresis (comet assay) following addition of doxycycline. (Scale bars in E–G, 100 µm.) (H) Single cell high content image analysis of H2AXP levels plotted on a log scale; image intensity of entire population is shown (red bar, mean value; blue, SD; n = 2,870 cells). Significance was determined by one way ANOVA with Sidak correction. (I) Opencomet automated analysis of comet tail moment. Data are plotted on a Tukey boxplot (n = 43 cells per time point). *P ≤ 0.05; ***P ≤ 0.001.
Fig. S5.

Original data of Cell Titer Glo and Caspase Glo for vehicle/Dox/cotreatments. (A) Time-course monitoring cellular viability in engineered PTGR cell line (blue and red are HeLa-PTGR-ICADLTevS +iTevP; the remaining series are PTGR engineered Hek293 cells treated with 10 nM rapamycin) (see Fig. 2A). (B) Normalized changed in caspase-3/7 activity at 24 and 48 h following single and combination treatments. Normalized to inhibitor alone. No measured differences were found to significant P > 0.05 in an ANOVA t test. (C and D) Cellular viability with single and cotreatments with DNA repair inhibitors (C) and HDAC inhibitors (D). (E and F) Cellular caspase activity with single and cotreatments with DNA repair inhibitors (E) and HDAC inhibitors (F).
To determine the extent of cellular response to activation of CAD, we investigated the DNA damage-response pathway in greater detail, using ELISA-based detection strategies. Cytoplasmic nucleosomes were detected in cells treated with doxycycline (Cell Detection Plus, Roche) (Fig. 4B). Initially, immunoblotting for H2AX-Ser139 phosphorylation (H2AXP) as a marker for double-stranded DNA breaks, showed no change over an extended time course of treatment (Fig. S6). However, a sensitive ELISA-based detection system (Trevigen) revealed a marked increase in H2AXP levels following the addition of doxycycline in as little as 6 h (Fig. 4C).
Fig. S6.

Cell lysates from Fig. 3C were probed for H2AX phosphorylation at serine 139 and pan-H2AX. These bulk lysates show minimal response to DNA damage as a result of CAD activation.
To better understand the apparent discrepancy between the immunoblotting and ELISA-based detection methods, we hypothesized that the DNA damage signal might be coming from a subset of the cells exhibiting a strong response. To explore this idea, we looked for evidence of DNA damage and response at the single-cell level. Direct labeling of fragmented DNA by TUNEL staining showed only a small increase in TUNEL intensity of individual cells (Fig. 4 D and E). The TUNEL assay was significantly less sensitive in detecting early apoptotic cells as determined by a side-by-side comparison with H2AXP staining when using positive control DNA damage-inducing agents (Fig. S7). This is likely a result of the endogenous cellular amplification as the phosphorylation of H2AX spreads extensively around a site of DNA breakage while TUNEL directly labels a single DNA break (32, 33). High-content immunofluorescence quantitation of H2AXP levels showed clear enrichment after 12 h of doxycycline treatment over the vehicle control (Fig. 4F). Although the mean values were relatively consistent over time, a log plot of individual cell intensity of H2AXP staining demonstrates a time-dependent increase in H2AX phosphorylation (Fig. 4H). This finding indicates that over time CAD continues to cleave chromatin, thereby causing increased H2AXP signaling and we presume genotoxic stress in the cell.
Fig. S7.
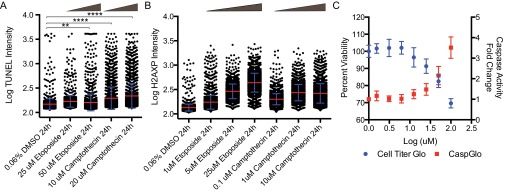
A comparison of the sensitivities of TUNEL assay (A) versus immunofluorescence of H2AX phosphorylation (B) staining using two standard DNA damage inducers, camptothecin and etoposide. High-content image analysis was used to identify and segment individual nuclei before pixel quantitation of TUNEL/H2AX phosphorylation staining (red bar, mean; blue, SD). ANOVA statistical testing showed all H2AXP conditions were P ≤ 0.0001 compared with DMSO control. (C) Correlation between cell viability and caspase-3/7 activity using twofold dose escalation of Etoposide (1.56–100 µM) at 24-h treatment time (error bars = SD). **P ≤ 0.01; ****P ≤ 0.0001.
To directly monitor DNA fragmentation following iTEVP induction, we used a single-cell gel electrophoresis (comet) assay. We observed an increase in comet tail moment in a small subset of treated cells compared with cells without iTEVP activation. The increase in comet tail moment indicates damaged chromatin migration following electrophoresis (Fig. 4G). The tail moment of individual cells is plotted using a box-and-whisker plot for each time point (Fig. 4I). Whereas the median remains nearly constant across the time course, there is a clear increase in heavily damaged cells starting at 6 h after iTEVP activation.
From these assays, it was apparent that although DNA damage signaling was taking place, many of the cells were still surviving. Two hypotheses emerged to explain the lack of robust apoptosis in the HeLa human cancer line upon selective CAD induction: (i) DNA repair machinery was so robust that it was quickly able to repair CAD-mediated damage and (ii) chromatin was only accessible to CAD in a subset of cells, possibly because of changes in chromatin accessibility known to vary during the cell cycle.
Caspase substrate profiling, both in our laboratory and others, previously demonstrated that many proteins involved in DNA repair are indeed targets of caspases, such as poly(ADP-ribose) polymerase (PARP), DNA-PK, and ATM/ATR among others (16, 34, 35). Moreover, a number of proteins involved in maintaining chromatin’s compact structure like the N-CoR/Smrt complex that contains histone deacetylase (HDAC) 3 and 7 are also targets (16). Therefore, we tested if available small-molecule inhibitors of these processes would synergize with CAD activation to impact cell death, caspase activation, H2AXP, and TUNEL staining. We thus tested the effects of combining sublethal doses of each of these inhibitors in the context of CAD activation, including: the DNA-PK inhibitor, Nu7441; the ATM inhibitor, Ku60019; the HDAC3 specific inhibitor, RGFP966; and the broad-spectrum HDAC inhibitor, TSA. Indeed we found each inhibitor, to varying degrees, resulted in a small increase in caspase activation (Fig. 5A) and TUNEL staining (Fig. 5B). However, there was no significant change in viability between inhibitor + doxycycline compared with inhibitor alone (Fig. S5B). There was a greater increase in phosphorylation of H2AX when cells were cotreated with doxycycline and various DNA pathway inhibitors (DNA repair and chromatin relaxation) (Fig. 5C and time-course detailed in Fig. S8B). These studies suggest that inhibiting a single arm of the DNA damage response synergizes with CAD activation, yet is still insufficient to trigger full-blown apoptosis.
Fig. 5.

Pharmacological sensitization of DNA damage with selective CAD activation. (A) Fold-change of caspase activity in engineered cells cotreated with doxycycline and DNA repair or HDAC inhibitors for 24 h. Significance determined by unpaired t test. (B) Single-cell quantitation of TUNEL staining with pharmacological inhibitors and cotreatments with 1 µg/mL doxycycline (48 h). The following inhibitors were used in treatments for samples for TUNEL and H2AXP staining: 1 µM Nu7441 for DNA-PK; 3 µM Ku60019 for ATM; 10 µM RGFP966 for HDAC3; 400 nM Trichostatin A (TSA) for HDAC. (C) Single-cell quantitation of H2AXP levels following treatment with 1 µg/mL doxycycline for 12 h. Significance determined by unpaired t test. (D) Single-cell quantitation of H2AXP levels with cotreatments with DNA repair inhibitors (Nu7441 and Ku60019) and doxycycline (12 h). Significance determined by unpaired t test. (E) Single-cell quantitation of H2AXP levels cotreated with HDAC inhibitors (RGFP966 and TSA) and doxycycline (12 h). Colored bars represents mean value and error bars are SD. Significance determined by unpaired t test. (F) Flow cytometry based assessment of apoptotic cells by PI/annexin V staining following S-phase synchronization induced by double thymidine block followed by 24-h treatment with doxycycline. Doxycycline-dependent increase is observed in synchronized cells from 2.3 to 5.0% annexin V positive and 8.3 to 18.5% annexin V and PI double-positive cells. (G) Cell cycle distribution of cells as measured by PI staining using flow cytometry. PTGR ICADLTevS cells were treated for an extended time with doxycycline to activate CAD. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Fig. S8.

(A) TUNEL staining from high content image analysis following 24-h treatment regimes with either pharmacological inhibitors of DNA repair (i) or chromatin condensation (ii). Vehicle and doxycycline treatment results are identical in A and B; treatment and staining was conducted simultaneously for all treatment conditions and separated for clarity in the figure. (B) H2AXP staining of cells over a time course of cotreatments with inhibitors (red bar, mean; blue, SD).
We hypothesized that chromatin accessibility to CAD should peak during S-phase, because DNA is stripped of many of its chromatin binding proteins. To test this idea, we synchronized a population of cells in early S-phase using a double thymidine block and released them following a 3 h preactivation of iTEVP with addition of doxycycline. This regime induced approximately twice the number of apoptotic cells, compared with synchronized cells in the absence of doxycycline as assessed by staining with annexin V and propidium iodide (PI) (Fig. 5E).
To assess the consequence of extended CAD activation in different stages of the cell cycle, we tracked the percentage of cells by PI staining using flow cytometry. We observed a marked arrest of cells in G2/M phase, a doubling of the percentage of cells (21–41%) in G2/M compared with cells without doxycycline (Fig. 5F). The arrest in G2/M was stable out to 72 h after addition of doxycycline. Overall, our data suggest that direct activation of CAD does lead to DNA cleavage, especially in S-phase, and that the DNA repair process once initiated can efficiently antagonize CAD activation.
Discussion
DNA fragmentation of is a classic hallmark of apoptosis. A key event in this process is believed to be the activation of CAD by caspase cleavage of its inhibitor and folding chaperone, ICAD. We engineered an inducible and selective proteolysis system to closely mimic caspase proteolysis of ICAD without parallel cleavage of over 1,500 other caspase substrates. The system preserved the critical role of ICAD as a CAD folding chaperone, by replacing the endogenous ICAD at physiological levels with one sensitive to TEVP. This process was enabled by designing the replacement ICAD allele to be resistant to the expressed synthetic miRNA, allowing the endogenous ICAD to be knocked down while the exogenous ICADTevS rescued endogenous function. This posttranscriptional gene-replacement strategy is simple, rapid to use, and genetically encoded. Recent advances in genome engineering using transcription activator-like endonucleases and CRISPR show great promise for gene ablation studies; however, the ability to use these techniques for site-specific biallelic replacement is not guaranteed and requires a large investment in reagents and screening time (36). We believe the PTGR strategy may be more broadly appropriate to many problems where one wishes to simultaneously replace endogenous biallelic expression with an engineered allele variant and screen for emergent functions.
We investigated two systems for inducing targeted proteolysis by TEVP, a split protein and full-length version. Although the SNIPer was able to cleave ICADTevS, we found the rate of proteolysis to be low and insufficient to deal with the steady-state levels of ICADTevS, probably because synthesis of new ICADTevS kept pace with split-TEVP proteolysis. Thus, we investigated a TetON full-length TEVP system for cleaving ICADTevS. Although there is a lag phase in TEVP activity as a result of transcriptional/translational delay in producing the full-length TEVP, the intact TEVP is about five-times more active than the split construct. Thus, we favored the iTEVP for the loss-of-function studies where rapid induction is not as important as complete proteolysis.
TEVP was capable of cleaving ICADTevS both in vitro and in cells and trigger hallmarks of DNA damage, such as H2AX phosphorylation. Chromatin fragmentation also significantly increased following selective ICADTevS cleavage, as demonstrated by increases in TUNEL staining, nucleosome release, and DNA tail moment in comet assays. Remarkably, we detected no significant change in cell death as measured by cellular ATP levels (Cell Titer-Glo) or caspase activity (Caspase-Glo).
In aggregate, our data suggest that DNA repair can largely out-compete DNA cleavage by CAD and preserve the cell. Among the many substrates that caspases cleave are proteins involved in DNA repair processes (Fig. 6). Indeed we observed increases in all DNA damage reporters upon pharmacological inhibition of DNA repair pathways. The accumulation of DNA damage in S-phase or with HDAC inhibition further suggests that exposed chromatin is more sensitive to CAD. Thus, our data suggest that the combined action of multiple caspase cleavages, DNA repair, and chromatin-modifying enzymes, work in coordination with CAD activation to facilitate DNA fragmentation in human cells during apoptosis.
Fig. 6.

Summary model of synthetic lethality and pharmacological sensitization through simultaneous activation CAD and inhibition of DNA repair. CAD activation through caspase or TEVP cleavage cleaves exposed regions of DNA (Center), which is opposed by either DNA repair (Right) or chromatin compaction (Left) promoted by HDACs. These opposing processes can be targeted by additional caspase cleavages (shown in red) or by pharmacological inhibitors (shown in light brown).
There have been reports that selective CAD activation can lead to DNA fragmentation and cell death in limited contexts, yet to our knowledge, our report is the first to achieve in situ activation in mammalian cells. Previous studies demonstrated the ability to activate CAD via addition of TEVP to ICADTevS/CAD in cytosolic extracts and utilization of a galactose-inducible TEVP in Saccharomyces cerevisiae (37, 38). Because yeast lack a traditional apoptotic pathway, caspases, and the ICAD/CAD node, the galactose inducible TEVP strategy only proved useful to study chromatin structure in this context. More recently, Samejima et al. described a cellular strategy that relied on an auxin-inducible degron system for inducing rapid and complete proteasomal degradation of huICAD in double-knockout DT40 chicken cells (39). It remains unclear how authentic their strategy is for selective CAD activation because it does not recapitulate both the spatial and endogenous site-specific mechanism of activation. Our approach of site-specific proteolysis closely mimics the endogenous mechanism. Our data are consistent with the view that apoptosis results from the synthetic lethality set up by coordinated caspase cleavage of multiple target pathways. Such a system provides useful safeguards for inadvertent triggering of cell death from a single node.
We believe the strategy of substrate replacement coupled with inducible and selective proteolysis provides a highly generalizable approach for assigning the functional consequences of selective cleavage of caspase substrates in cells. In addition, we envision the approach to be beneficial in characterizing the cross-talk between phosphorylation and apoptotic proteolysis by including either phosphorylation mimetic or nonphosphorylatable residues in the TevS allele series (40, 41). The approach outlined with the PTGR and SNIPer technology may prove particularly beneficial in profiling site-specific proteolysis throughout cell biology: for example, during cell-cycle progression mediated by separase during anaphase (42), differentiation, and intercellular signaling by Notch proteolysis (43), and mediating the unfolded protein response by ATF6 proteolysis (44).
As the proteomic-generated map of caspase substrates continues to grow, our understanding of the functional consequence resulting from individual cleavage events remains in its infancy. The present studies offer a strategy to precisely investigate substrate proteolysis and can provide new insight to the complexity and cooperativity between caspase cleavage events during apoptosis.
Materials and Methods
SNIPER, iTEVP, and the PTGR backbone was assembled from cassettes obtained from Kaveh Ashrafi (University of California, San Francisco, CA) and Addgene. PTGR- and iTEVP-engineered HeLa cells were plated in 96-well microclear plates (Greiner). Cells were allowed to attach and were treated for the indicated amount of time with the indicated small molecules: doxycycline (Sigma), Nu7441 (Torcis), Ku6009 (Torcis), RGFP966 (Selleckchem), TSA (Cells Signaling). Cells were fixed and permeabilized with 1% PFA in PBS +0.1% Triton X-100 for 10 min. H2AXP immunofluorescence protocol was developed from Zeitlin 2009 (45). High-content image analysis included cell identification by nuclear staining (DAPI), and segmentation was refined by watershed clump breaking and sieve algorithms to eliminate false targets. Using DAPI signal as a threshold area, H2AXP signal was recorded for each target cell in a field of view. Batch processing was then used to analyze >2,000 cells per well, across a 96-well plate. Target and pixel intensities were then exported to Microsoft Excel for numerical review and final graphical representation and statistical analysis using Prism (Graphpad). Statistics were performed using Prism as indicated in the figure legends. Additional experimental details are described in SI Materials and Methods. See Table S1 for selected primer sequences used.
Table S1.
Selected primer sequences
| Vector | Name | Sequence |
| TEVP | ||
| IRES Single vector |
N-Tev Rev | aggatcgaattctcaggtctggaagttggtgg |
| T2A-myc Fwd | aacatgcggtgacgtggaggagaatcccggccctgaacaaaagctgatctctgaggag | |
| BspEI T2A Fwd | gatccttccggaagcggagagggcagaggaagtcttctaacatgcggtgacgtggagg | |
| C-Tev EcoRv Rev | gatcctgatatctcacatgaacaccttatgtccgcc | |
| KpnI IRES Fwd | gatcctggtaccccattgtatgggatctga | |
| Myc IRES Rev | cctcagagatcagcttttgttccatggttgtggccatattatcatcgtgt | |
| Myc IRES Fwd | acacgatgataatatggccacaaccatggaacaaaagctgatctctgagg | |
| T2A | 2A Myc FRB N Fwd | gtcttctaacatgcggtgacgtggaggagaatcccggccctgcccccggcagcgaacaaaagctgatctctgaggagg |
| Single vector | ||
| C-Tev 2A FRB | gggcggacataaggtgttcatgtccggaagcggagagggcagaggaagtcttctaacatgcggtgacgtggaggagaatcccggccctgcccccggcagcgaacaaaagctgatctctgaggagg | |
| C-Tev 2A overlap | gggcggacataaggtgttcatgtccggaagcggagagggcagaggaagtcttctaacatgcggtgacgtg | |
| C-Tev Rev | catgaacaccttatgtccgccc | |
| XbaI Stop N Rev | gatccttctagatcaggtctggaagttggtgg | |
| NcoI Mycortho Fwd | gatcccatggagcagaagctgatcagcgaggaggacctgctggaggggggcggtatgggt | |
| XhoI FKBP Rev | aggatcctcgagtcaggtctggaagttggtggtcacc | |
| iTEVP | BamHI TEVP | gatcctggatccggcgagagccttttcaagggcc |
| NcoI Myc Fwd | gatcctccatggaacaaaagctgatctctgagg | |
| EcoRI TEVP Rev | aggatcgaattcttatcaaagctgagtggcttccttaacg | |
| PTGR | ||
| Backbone | CMV_EGFP_ApaI_Fwd | aaagggaaagggccccgatgtacgggccagatatacgcgttgacatt |
| CMV_EGFP_ApaI_Rev | aaagggaaagggccctttgctagctcacttgtacagctcgtccat | |
| F-AgeI-GW-attR4_2 | aaagggaaaaccggtcgccaagctcgaaattaaccctcactaaa | |
| R-AgeI-GW-attR4_2 | ccttgaagcgcatgaactccttgatgatggccatgtt | |
| AttB1-miR-F | ggggacaagtttgtacaaaaaagcaggctgctaagcacttcgtggccgtcgatcgtttaaagggaggtagtgagtcga | |
| AttB2-miR-R | ggggaccactttgtacaagaaagctgggttctagactcgagtgcggccagatctgggcca | |
| Lenti | ||
| GBEcoRIKONotIGW4Fwd | tccactagtccagtgtggtggaattcgcctaatgcaggccgccatag | |
| GBEcoRIKONotIGW4FwdII | gcctaatgcaggccgccatagtgactggatatg | |
| EcoRI CMRccdb | gatcctgaattcggctttacactttatgcttccggctc | |
| GW1Rev | ||
| GBNotICMVrev | gcctgcaggtcgactctagagtcgcggccgcgtccgttacataacttacggtaaatggcc | |
| GBXbaIXhoI CMV rev | gaaccgcgggccctctagactcgag | |
| mfeI pmeI BlastR Fwd | gatcctcaattgatcgtttaaacttattagttcctggtgtacttgagg | |
| pacI plocIRES rev | gatcctttaattaatgctagctaatgaaccgggcgc | |
| GBNotIBswIPacIIRES rev | tgcaggtcgactctagagtcgcggccgccgtacgcttaattaatgctagctaatgaacc | |
| ICAD Alleles | Fwd N FLAG ICAD att | ggggacaactttgtatagaaaagttgccaccatggattacaaggatgacgacgataaggaggtgaccggggacgccggg |
| RevTEV_1 | gccccgctctggaagtagagattttcgtctgtttcatctacatcaaaggactctt | |
| Fwd TEV1 | ctacttccagagcggggcaggattaaaatggaaaaacgtggccaggcagctgaaagaaga | |
| Rev TEV2 | accactctggaagtagagattttcgtctactgcatccacctcctcaccaa | |
| Fwd TEV2 | gacgaaaatctctacttccagagtggtacgggtatcagcagagagacctcctcgga | |
| Rev att ICADLong | ggggactgcttttttgtacaaacttgtcatgtgggatcctgtctggctcgctta | |
| Rev att ICADShort | ggggactgcttttttgtacaaacttgtcagtgaccctggtttccgcccacctccaaatcctgactagataagctcagctc | |
| petDuet | ||
| BamHi HuCAD F | gtgacaggatccgatgctccagaagcccagagcgtg | |
| SalI HuCAD R | gctgcagtcgactcactggcgtttccgcacaggc | |
| NdeI HuICAD F | cgacgtcatatggattacaaggatgacgacgataagg | |
| XhoI HuICAD R | tcatgcctcgagtcatgtgggatcctgtctggctcgc | |
| miR-155-ICAD#3 | Top | tgctgaaagtaatcgtcatcatccacgttttggccactgactgacgtggatgaacgattacttt |
| Bottom | cctgaaagtaatcgttcatccacgtcagtcagtggccaaaacgtggatgatgacgattactttc | |
| Mir155 neg.control | Top | tgctgaaatgtactgcgcgtggagacgttttggccactgactgacgtctccacgcagtacattt |
| Bottom | cctgaaatgtactgcgtggagacgtcagtcagtggccaaaacgtctccacgcgcagtacatttc |
SI Materials and Methods
Molecular Biology.
TEVP.
The second-generation SNIPer and iTEVP vectors were assembled using standard molecular biology techniques, including PCR, overlap extension PCR, restriction enzyme cloning, Gibson assembly, and Gateway recombination (Life Technologies). Briefly, the different split TEVP, SNIPer versions were cloned from the original SNIPer and either inserted around an existing IRES sequence or split and a 2A sequence was inserted via PCR overlap extensions. The full-length TEVP was inserted into the second generation pLenti TetON vector (26430, Addgene). The mycNLS tagged TEVP sequence was cloned into a pENTR vector at NcoI and EcoRI, before Gateway recombination mediated insertion into the empty destination lentiviral backbone.
pETDuet ICAD/CAD cloning.
Human ICAD and CAD sequences were subcloned into pETDuet (Novagen) to yield coexpression constructs for recombinant expression and purification. Briefly, human CAD coding sequence, generated by PCR from cDNA (Open Biosystems), was ligated in-between the BamHi and SalI sites of the empty pETDuet vector, thereby creating pETDuet–huCAD. ICADLTevS was generated by PCR overlap extension using ICADWT as a template. The two caspase cleavage sites (DETD-117 and DAVD-224) in ICAD were sequentially mutated to TEVP cleavable sequences (ENLYLFQ), following methods outlined in Gray et al. (21). ICADWT or ICADLTevS were then ligated into pETDuet–huCAD, between NedI and XhoI, thereby creating two expression vectors, pETDuet–huCAD–ICADWT and pETDuet–huCAD–ICADLTevS.
Posttranscriptional gene replacement.
The PTGR vector was assembled from elements from several different sources using the aforementioned standard molecular biology techniques. The basic MultiSite Vectors: pDONR-P4-P1r, pDNOR-P1-P2 (pDNOR221), and pKA452 were a kind gift of Brendan Mullaney and Kaveh Ashrafi (University of California, San Francisco, CA).To generate pDONR-CMV-EGFP-P4-P1r, 5′ and 3′ ApaI restriction sites were added to the CMV-EGFP segment from pEGFP-N1 (Clontech) by PCR and then subcloned into the unique ApaI site of pDNOR-P4-P1r. pENTR-CMV-miR-GWL1-L2 was constructed by first cloning the miR-155 scaffold by a PCR-based synthetic gene construction approach, a second round of PCR to add attB1 and attB2 sites, and the resulting product was Gateway BP (Invitrogen) cloned into pDONR221 to generate pENTR-attL1-miR155-EV-attL2. An ApaI and PvuII attL1-miR155-EV-attL2 fragment was then subcloned into the SmaI and HpaI sites of a modified pIRES2-dsRED vector (Clontech; the BsaI sites were previously removed by site-directed mutagenesis) to yield pENTR-CMV-miR-L1-L2. The acceptor cassette R4-R2 was generated by PCR amplifying the attR4-attR2 Gateway acceptor cassette from pKA452. An additional 5′ AgeI restriction site was added during PCR, and the PCR product was cloned into the unique AgeI site of pQCXI-mCherry. The IRES–GFPTurbo2A–BlastR fusion protein was generated by PCR amplification from pLoc GFPTurbo2A-BlastR, a kind gift from Justin Farlow and Zev Gartner (University of California, San Francisco, CA), the product was subcloned into pHR using homology based Gibson recombination to generate pHR–IRES–GFPTurbo2A–BlastR. To generate pHR–CMV–R4-R2–IRES–GFPTurbo2A–BlastR, the previously generated attR4-attR2 Gateway acceptor cassette with the CMV promoter (pQCXI) was amplified using PCR and ligated into the pHR vector between the EcoRI and NotI, to yield the final PTGR acceptor vector.
Control and ICAD-targeting miRNAs were designed on Invitrogen’s BLOCKIT web-server (rnaidesigner.lifetechnologies.com/rnaiexpress/setOption.do?designOption). miR vectors were constructed by linearizing pENTR-CMV-miR-GW-L1-L2 with BsaI and directly ligating annealed 65-mer oligonucleotides. Resulting miR vectors were validated by DNA sequencing. Two TevS sites were added by a two round overlapping PCR protocol using human ICAD-L/DFF45 cDNA (Open Biosystems); attB4 and attB1 sites were added in the second round of PCR, and PCR products were BP-cloned into pDONR-P4-P1r. Once a robust miR-ICAD was identified, the corresponding 21-nt region in the expressed ICAD alleles was subjected to site-directed mutagenesis in which five silent mutations were made (silent codon mutations in residues 70–74, VDDD). The EGFP–ICAD fusion construct was prepared by two rounds of overlapping PCR and cloned into the EcoRI and NotI sites of pcDNA3.1-Myc/His (Invitrogen). BP and LR cloning were performed by using BP Clonase II and LR Clonase II (Invitrogen) according to the manufacture’s recommendations. Acceptor vectors were propagated in One Shot ccdB Survival Escherichia coli and Gateway transformants were propagated in Stbl3. See Fig. S1 for a workflow diagram.
Cell Culture.
Viral production.
The 293FT cells were transfected with a mixture of second-generation lentiviral packaging plasmids at ∼80% confluences. Lipofectamine 2000 (Invitrogen) was used for lipid-based transfection of the plasmids using 4 µg DNA and 2.5 µL lipofectamine per well of a six-well plate. Transfected DNA for viral packaging was mixed at 1:1:1 (VSV-G:pCMVdelta8.91:Lenti) ratio. Media was changed to complete DMEM 10% FBS after 6 h of incubation with the lipid:DNA mixture. The supernatant was harvested and cleared by passing it through a 0.2-µm syringe at 48 h and 72 h posttransfection. The cleared supernatant was kept at 4 °C in-between collection points. The supernatant was concentrated using Clontech LentiX Concentrator according to the manufactures supplied protocol. Freshly adhered target HeLa cells (∼50% confluent), were then incubated with the viral supernatant + 8 µg/mL polybrene for a minimum of 6 h before the media was changed to fresh complete DMEM. Cells were expanded for a minimum of 48 h before they were grown in selection media. Drug selection for the stable cell line was started by the addition of 200 µg/mL Hygromycin B (InvivoGen) and 400 µg/mL G418 (Life Technologies) for TetR and iTEVP, respectively. Following the death of a negative control plate, Inducible expression of iTEVP was analyzed by immunoblot following addition of 1 µg/mL of doxycycline. The TetR-iTEVP cells were then engineered using the indicated lentiviral PTGR ICAD replacement construct. Cells with stable expression of the PTGR cassette were selected on 4 µg/mL Blasticidin (InvivoGen) following standard viral infection. Following the death of a negative control plate, cells were analyzed for expression of GFP markers by epifluorescence microscopy (Zeiss), flow cytometry (BD Calibur), and immunoblot for desired replacement phenotype.
Stable cell line selection and characterization.
GFP signal was very faint by conventional wide-field epifluorescence but was evident by the more sensitive FACS, laser excitation, and PMT detection using an AriaII (BD Biosciences). This is consistent with known low-level expression driven by cap-independent translation facilitated by the IRES sequence. Synthetic miRNA were cloned into the pDonr–miR backbone and used to screen for target knockdown. We assessed miR efficacy in several ways: (i) immunoblot detection of target protein with miR transfected cells, (ii) assessment of a loss of EGFP signal follow cotransfection of an EGFP-stop-target substrate and pDonr-miR, and (iii) RT-qPCR analysis of the target mRNA levels following 72 h of miR transfection (data available upon request).
Cellular treatments pharmacological inhibitors were conducted with the appropriate vehicle control, between 0.1% and 0.5% DMSO, for all cellular experiments. The concentration of DMSO was held constant across experimental sets and was dictated by compound solubility and desired treatment concentration. Next, 2 mM thymidine (Sigma Aldrich) was used for double thymidine block for cell synchronization experiments, 12-h on, 12-h off, followed by 12-h on.
Recombinant Protein Expression and Purification.
petDuet ICADWT:CAD and ICADLTevS:CAD were individually transformed into BL-21 Rosetta chemically competent cells for expression. Coexpression was induced with 1 mM isfopropyl-β-d-thiogalactopyranoside (IPTG) at an OD600 = 0.5, cultures were then grown overnight (∼16 h) at 28 °C. Bacterial cell pellets were lysed by several passages through a microfluidizer in binding buffer (50 mM Tris pH 7.5, 500 mM NaCl, 20 mM Imidazole, 5% glycerol) plus Leupeptin, Pepstatin, and PMSF protease inhibitors. The soluble protein fraction was collected after lysate clarification at 20,000 rpm at 4 °C for 30 min. The 6× histidine tag on the N terminus of CAD was used to purify the complex on a nickel column (HisTrap HP, GE Life Sciences) on an AKTA (GE Life Sciences). The column was washed with four to five column volumes of binding buffer before gradient elution in elution buffer (50 mM Tris pH 7.5, 500 mM NaCl, 250 mM Imidazole, 5% Glycerol). Eluted fractions were screened for 40 and 45 kDA by SDS/PAGE. Desired fractions were then pooled and concentrated (10 molecular weight cut-off) prior dilution into Buffer A (25 mM Tris pH 8.0, 5% glyercol) and loading on ion exchange column (HiTrap Q HP, GE Life Sciences). Elution of ICAD:CAD complex was done by gradient elution to 50% buffer B (25 mM Tris pH 8.0, 1 M NaCl, 5% glyercol). Eluted fractions were screened by SDS/PAGE and desired fractions pooled and concentrated prior making aliquots and snap freezing in liquid nitrogen. Protein concentration was measured using the Edelhoch method.
Immunofluorescence.
Treated cells were washed in PBS and then incubated with H2AXP (Millipore; 1:1,000 dilution) for 30 min in a humidified chamber at 37 °C. Wells were washed once with PBS and once with PBS+0.1%Triton then incubated with 1:100 Alexa Fluor 488 anti-mouse (Life Technologies) and counter-stained with DAPI and Alexa Fluor 546 phalloidin (Life Technologies) where indicated in the figure legends.
RT-qPCR.
Cells to CT (Taq-man gene expression, Life Technologies) was used for rapid mRNA isolation and quantification. DNA fragmentation factor A (DFFA) (ICAD)-specific Taqman probe (Hs00189336_m1) was obtained from Life Technologies and used in combination with Taqman Gene-expression Master Mix (4369016, AB). Biological triplicates were run on an ABI-7300 instrument. The manufacturers’ suggested isolation and cycling protocols were followed.
Comet Assay.
Cells were treated in 12-well plates (Corning #3512) for the indicated times and then collected by through standard trypsization and washed once in PBS, counted, and then 500 cells were added to low-melt agarose (Trevigen) and immediately spread on glass comet slides (Trevigen). The agarose slurry was allowed to solidify at 4 °C for 30 min before adding lysis buffer (Trevigen) for overnight incubation at 4 °C. The slides were transferred to a large electrophoresis chamber and 1 V/cm2 was applied across the slides for 45 min. The slides were washed in double-distilled H2O and then incubated with SYBR-Green I dye (Life Technologies) for 10 min, followed by another H2O wash and then allowed to dry before imaging. Slides were imaged using an inverted epifluorescence Zeiss microscope, 10× objective, GFP filter set. Saved images were analyzed using the free software plugin for ImageJ, opencomet (opencomet.org).
Cell Viability and Caspase Activity Measurements.
Cells were plated in 96-well plates (Corning #3596). Addition of drug-containing media was carried out media exchange with a 2× addition of small-molecule–containing media. Cells were lysed using the Cell Titer Glo reagent (Promega) 1:1 50:50 and allowed to incubate while shaking for >10 min before measurement on a spectramax M5. Cellular caspase activity was measured using Caspase Glo-3/7 reagent in a similar format as the cell titer glo except the incubation step was increased to 45 min to allow for sufficient amplification of the signal. All experiments were done in biological triplicate with the mean and SD calculated for each treatment set.
Western Blot.
Cells were trypsanized, spun down at 5,000 rpm at 4 °C, washed once with cold PBS, spun again, and then lysed in MPER (ThermoFisher) supplemented with Roche protease and phosphatase inhibitor mixture mixture and 150 mM NaCl for a more complete lysis of nuclei. Following cellular lysis with MPER, additional solubilization and shearing of DNA was accomplished with short 1-s bursts of a microtipped sonicator at 20% power. This process was then followed by clearing of insoluble material by centrifugation at 20,800 × g for 10 min. Cleared lysates were then flash frozen until further analysis by SDS/PAGE and immunoblotting. Protein loading was normalized by a standard BCA assay (Thermo Fisher). Proteins were separated on a 4–12% TrisGlycine gel (Bio-Rad) and transferred to a PVDF membrane. Membranes were blocked in 5% BSA in PBS+0.1% Tween-20 (PBST) for 30 min. Immunoblotting was conducted overnight at 4 °C. The following antibodies and their respective dilutions were used for immunoblotting: ICAD (611036, BD Biosciences) at 1:1,000, FLAG-M2 (F1804, Sigma-Aldrich) at 1:2,000, HA tag (Cell Signaling) at 1:1,000, GAPDH (14C10, Cell Signaling) 1:5,000, CAD (AB16926, Millipore), H2AXP (Ser-139) (JBW301, Millipore) at 1:2,500, H2AX (D17A3, Cell Signaling) at 1:2,500.
Immunoprecipitations.
Coimmunoprecipitations were performed following detergent lysis of cells as outlined above in Western blot methods. An N-terminal FLAG tag was used to imunnoprecipitate ICAD from cell lysates using an FLAG M2 Affinity Gel (A2220, Sigma-Aldrich). The manufacturer’s suggested protocol was followed for washing and 3×FLAG-tagged peptide was used for elution in 20% glycerol and CAD buffer. An N-terminal HA tag was used to facilitate CAD complex purification from cell lysates expressing exogenous CAD from transient transfection, anti-HA conjugated Sepharose beads (C29F4, Cell Signaling). The manufacturer’s suggested protocol was followed.
Flow Cytometry.
Cell viability.
Data from all flow cytometry experiments were collected on a Calibur instrument (BD Biosciences). Singlets were selected in the SSA vs. FSA plot before further gating of fluorescent signal from Annexin V-AlexaFluor488 and PI staining. All gates were drawn uniformly among all experiments and analyzed using FlowJoX. Exported values from FlowJo were visualized using Prism6.
Cell cycle analysis.
Cells were trypisinized and washed twice in cold PBS. Cells were fixed in 70% ethanol, washed in PBS, and then incubated overnight in FxCycle Pi/RNase staining solution (Life Technologies). Cells were gated on FSC-H SSC-H then single cells were gated based on FL-3-W vs. FL-A. Cellular DNA content was measured using the cell cycle analysis package in FlowJoX.
Statistics.
Statistics were performed using Prism6 (Graphpad). Data were imported into Prism and corresponding analysis was performed as indicated in the figure legends. When statistical testing was appropriate and indicated in the text, it was determined by unpaired t test between two populations with Welch’s correction for nonequal stand a one-way ANOVA, with Dunnett correction applied for multiple comparisons. If no significance is shown, then P > 0.05.
Supplementary Material
Acknowledgments
The authors thank Drs. J. T. Koerber, Jason Porter, Nick Agard, Sami Mahrus, Julie Zorn, and Profs. David Morgan and Charles Craik for their helpful insights during this research; and the Univerisity of California, San Francisco Laboratory for Cell Analysis and Small Molecule Discovery Center for use of their flow cytometry instruments and Incell2000. This study was supported in part by National Institute of General Medical Sciences Grants 1R01GM097316-01, 5R01GM081051-08, and R01CA136779, and a National Science Foundation Graduate Research fellowship (to C.W.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1504141112/-/DCSupplemental.
References
- 1.Renehan AG, Bach SP, Potten CS. The relevance of apoptosis for cellular homeostasis and tumorigenesis in the intestine. Can J Gastroenterol. 2001;15(3):166–176. doi: 10.1155/2001/164727. [DOI] [PubMed] [Google Scholar]
- 2.Vaux DL, Korsmeyer SJ. Cell death in development. Cell. 1999;96(2):245–254. doi: 10.1016/s0092-8674(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Xu M. Apoptotic DNA fragmentation and tissue homeostasis. Trends Cell Biol. 2002;12(2):84–89. doi: 10.1016/s0962-8924(01)02206-1. [DOI] [PubMed] [Google Scholar]
- 4.Los M, Wesselborg S, Schulze-Osthoff K. The role of caspases in development, immunity, and apoptotic signal transduction: Lessons from knockout mice. Immunity. 1999;10(6):629–639. doi: 10.1016/s1074-7613(00)80062-x. [DOI] [PubMed] [Google Scholar]
- 5.Mason KD, et al. Proapoptotic Bak and Bax guard against fatal systemic and organ-specific autoimmune disease. Proc Natl Acad Sci USA. 2013;110(7):2599–2604. doi: 10.1073/pnas.1215097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cotter TG. Apoptosis and cancer: The genesis of a research field. Nat Rev Cancer. 2009;9(7):501–507. doi: 10.1038/nrc2663. [DOI] [PubMed] [Google Scholar]
- 7.Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407(6805):784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 9.Enari M, et al. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391(6662):43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 10.Cotter TG, Lennon SV, Glynn JM, Green DR. Microfilament-disrupting agents prevent the formation of apoptotic bodies in tumor cells undergoing apoptosis. Cancer Res. 1992;52(4):997–1005. [PubMed] [Google Scholar]
- 11.Taylor RC, Cullen SP, Martin SJ. Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 12.Ashkenazi A, Salvesen G. Regulated cell death: Signaling and mechanisms. Annu Rev Cell Dev Biol. 2014;30:337–356. doi: 10.1146/annurev-cellbio-100913-013226. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27(48):6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 14.Van Damme P, et al. Caspase-specific and nonspecific in vivo protein processing during Fas-induced apoptosis. Nat Methods. 2005;2(10):771–777. doi: 10.1038/nmeth792. [DOI] [PubMed] [Google Scholar]
- 15.Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell. 2008;134(4):679–691. doi: 10.1016/j.cell.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahrus S, et al. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell. 2008;134(5):866–876. doi: 10.1016/j.cell.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timmer JC, Salvesen GS. N-terminomics: A high-content screen for protease substrates and their cleavage sites. Methods Mol Biol. 2011;753:243–255. doi: 10.1007/978-1-61779-148-2_16. [DOI] [PubMed] [Google Scholar]
- 18.Pham VC, et al. Complementary proteomic tools for the dissection of apoptotic proteolysis events. J Proteome Res. 2012;11(5):2947–2954. doi: 10.1021/pr300035k. [DOI] [PubMed] [Google Scholar]
- 19.Agard NJ, et al. Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proc Natl Acad Sci USA. 2012;109(6):1913–1918. doi: 10.1073/pnas.1117158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer U, Jänicke RU, Schulze-Osthoff K. Many cuts to ruin: A comprehensive update of caspase substrates. Cell Death Differ. 2003;10(1):76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gray DC, Mahrus S, Wells JA. Activation of specific apoptotic caspases with an engineered small-molecule-activated protease. Cell. 2010;142(4):637–646. doi: 10.1016/j.cell.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284(5756):555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 23.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391(6662):96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 24.Sakahira H, Enari M, Nagata S. Functional differences of two forms of the inhibitor of caspase-activated DNase, ICAD-L, and ICAD-S. J Biol Chem. 1999;274(22):15740–15744. doi: 10.1074/jbc.274.22.15740. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Wang X, Bove KE, Xu M. DNA fragmentation factor 45-deficient cells are more resistant to apoptosis and exhibit different dying morphology than wild-type control cells. J Biol Chem. 1999;274(52):37450–37454. doi: 10.1074/jbc.274.52.37450. [DOI] [PubMed] [Google Scholar]
- 26.Samejima K, Earnshaw WC. Trashing the genome: The role of nucleases during apoptosis. Nat Rev Mol Cell Biol. 2005;6(9):677–688. doi: 10.1038/nrm1715. [DOI] [PubMed] [Google Scholar]
- 27.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciccia A, Elledge SJ. The DNA damage response: Making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naldini L, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272(5259):263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 30.Magliery TJ, et al. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: Scope and mechanism. J Am Chem Soc. 2005;127(1):146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- 31.Yang S, et al. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther. 2008;15(21):1411–1423. doi: 10.1038/gt.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273(10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 33.Bewersdorf J, Bennett BT, Knight KL. H2AX chromatin structures and their response to DNA damage revealed by 4Pi microscopy. Proc Natl Acad Sci USA. 2006;103(48):18137–18142. doi: 10.1073/pnas.0608709103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crawford ED, et al. Conservation of caspase substrates across metazoans suggests hierarchical importance of signaling pathways over specific targets and cleavage site motifs in apoptosis. Cell Death Differ. 2012;19(12):2040–2048. doi: 10.1038/cdd.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lüthi AU, Martin SJ. The CASBAH: A searchable database of caspase substrates. Cell Death Differ. 2007;14(4):641–650. doi: 10.1038/sj.cdd.4402103. [DOI] [PubMed] [Google Scholar]
- 36.Miyaoka Y, et al. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods. 2014;11(3):291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ageichik AV, Samejima K, Kaufmann SH, Earnshaw WC. Genetic analysis of the short splice variant of the inhibitor of caspase-activated DNase (ICAD-S) in chicken DT40 cells. J Biol Chem. 2007;282(37):27374–27382. doi: 10.1074/jbc.M704307200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao F, Widlak P, Garrard WT. Engineered apoptotic nucleases for chromatin research. Nucleic Acids Res. 2007;35(13):e93. doi: 10.1093/nar/gkm486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samejima K, et al. Auxin-induced rapid degradation of inhibitor of caspase-activated DNase (ICAD) induces apoptotic DNA fragmentation, caspase activation, and cell death: A cell suicide module. J Biol Chem. 2014;289(45):31617–31623. doi: 10.1074/jbc.M114.583542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell. 2009;138(5):838–854. doi: 10.1016/j.cell.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dix MM, et al. Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell. 2012;150(2):426–440. doi: 10.1016/j.cell.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Queralt E, Lehane C, Novak B, Uhlmann F. Downregulation of PP2A(Cdc55) phosphatase by separase initiates mitotic exit in budding yeast. Cell. 2006;125(4):719–732. doi: 10.1016/j.cell.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 43.Selkoe D, Kopan R. Notch and Presenilin: Regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565–597. doi: 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- 44.Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell. 2000;100(4):391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 45.Zeitlin SG, et al. Double-strand DNA breaks recruit the centromeric histone CENP-A. Proc Natl Acad Sci USA. 2009;106(37):15762–15767. doi: 10.1073/pnas.0908233106. [DOI] [PMC free article] [PubMed] [Google Scholar]