

Significance
MyD88-dependent signaling is cluster of differentiation 14 (CD14)-dependent only at low LPS concentrations, whereas activation of the TIR-domain–containing adapter-inducing IFN-β (TRIF) pathway requires CD14 at all LPS concentrations, leading to internalization of the Toll-like receptor 4 (TLR4) complex into endosomes whereupon TRIF is recruited. Using alternative TLR4 agonists, or macrophages rendered tolerant to LPS, we dissociate TLR4 complex internalization from CD14 and TRIF-dependent signaling. In response to LPS, CD14 contributes to the formation of a TLR4/MD2 complex dimer that, in turn, promotes endocytosis and IRF3 activation.
Keywords: TLR4 endocytosis, agonistic antibody, small-molecule ligands, Eritoran, endotoxin tolerance
Abstract
Dimerization of Toll-like receptor 4 (TLR4)/myeloid differentiation factor 2 (MD2) heterodimers is critical for both MyD88- and TIR-domain–containing adapter-inducing IFN-β (TRIF)-mediated signaling pathways. Recently, Zanoni et al. [(2011) Cell 147(4):868–880] reported that cluster of differentiation 14 (CD14) is required for LPS-/Escherichia coli- induced TLR4 internalization into endosomes and activation of TRIF-mediated signaling in macrophages. We confirmed their findings with LPS but report here that CD14 is not required for receptor endocytosis and downstream signaling mediated by TLR4/MD2 agonistic antibody (UT12) and synthetic small-molecule TLR4 ligands (1Z105) in murine macrophages. CD14 deficiency completely ablated the LPS-induced TBK1/IRF3 signaling axis that mediates production of IFN-β in murine macrophages without affecting MyD88-mediated signaling, including NF-κB, MAPK activation, and TNF-α and IL-6 production. However, neither the MyD88- nor TRIF-signaling pathways and their associated cytokine profiles were altered in the absence of CD14 in UT12- or 1Z105-treated murine macrophages. Eritoran (E5564), a lipid A antagonist that binds the MD2 “pocket,” completely blocked LPS- and 1Z105-driven, but not UT12-induced, TLR4 dimerization and endocytosis. Furthermore, TLR4 endocytosis is induced in macrophages tolerized by exposure to either LPS or UT12 and is independent of CD14. These data indicate that TLR4 receptor endocytosis and the TRIF-signaling pathway are dissociable and that TLR4 internalization in macrophages can be induced by UT12, 1Z105, and during endotoxin tolerance in the absence of CD14.
Toll-like receptor 4 (TLR4) signaling plays a crucial role in host defense against Gram-negative bacteria by recognizing the outer membrane component, lipopolysaccharide (LPS) (1–3). TLR4 signaling is initiated by transfer of an LPS monomer from LPS binding protein (LBP) to cluster of differentiation 14 (CD14) (GPI-linked or soluble). In turn, CD14 transfers monomeric LPS to myeloid differentiation factor 2 (MD-2), a protein that associates noncovalently with TLR4 (4). Appropriate ligand binding to MD2 results in dimerization of two TLR4/MD2 complexes (4). TLR4 is unique in that it is the only TLR that activates both myeloid differentiation primary response 88 (MyD88) and TIR-domain–containing adapter-inducing IFN-β (TRIF)-dependent signaling pathways (5, 6). MyD88-mediated, TLR4 signaling occurs mainly at plasma membranes and involves IL-1R–associated kinases phosphorylation, association of TNF-receptor–associated factor 6, and downstream signaling that results in NF-κB activation and induction of proinflammatory mediators such as TNF-α and IL-6 (7). In contrast, TRIF-mediated signaling in response to LPS occurs at the endosomal membrane after internalization of the TLR4 that, in turn, activates IFN regulatory factor 3 (IRF3), resulting in production of IFN-β, IP-10, and other IRF-3–dependent genes, as well as delayed NF-κB activation (8). Recent studies have shown that the endocytosis of TLR4 is tightly controlled by several molecules. Rab11a, ARF6, and p120-catenin have been implicated in Escherichia coli/LPS-induced TLR4 endocytosis and IRF3 activation (9–11). Zanoni et al. showed that CD14 plays critical roles in translocation of TLR4 into endosomes and in activation of IRF3 that are dependent upon the enzymatic activities of PLCγ2 and Syk (12). However, CD14-independent TLR4 endocytosis and TRIF signaling have not been reported.
UT12 is a monoclonal antibody (MAb) with specificity for the mouse TLR4/MD2 complex and mediates LPS-like signaling (13). It has been shown that UT12 induces endotoxic shock-like symptoms in mice including augmentation of TNF-α and IL-6. Furthermore, UT12 induced long-term tolerance and protection against LPS-induced lethal shock in mice (14). However, the ability of UT12 to induce translocation of TLR4/MD2 into endosomes, as well as its potential for mediating TRIF-dependent signaling, has not been reported. Recently, a group of substituted pyrimido[5,4-b]indoles, synthetic ligands for TLR4 that activate NF-κB that act in a CD14-independent manner, were discovered by high-throughput screening (15). These synthetic ligands induced IL-6 and IP-10 in a TLR4/MD2-dependent, but CD14-independent manner (16). They, too, have not been tested for TLR4 endocytosis and TRIF-dependent intermediates.
In this study, we report, for the first time to our knowledge, CD14-independent translocation of TLR4 to endosomes and TRIF signaling by UT12 and small synthetic TLR4 ligands (1Z105). A TLR4 antagonist, Eritoran, that binds to a deep hydrophobic pocket in MD2 and blocks signaling induced by LPS, UT12, and 1Z105, blocked only TLR4 internalization and dimerization induced by LPS and 1Z105. Despite TLR4/MD2 internalization, endotoxin-tolerized macrophages fail to activate TRIF-mediated signaling. These findings reveal previously unidentified insights into the possible role of CD14 in LPS-mediated TLR4 endocytosis and signaling and demonstrate that TLR4 endocytosis and signaling are dissociable processes.
Results
UT12-Induced TLR4 Endocytosis and TRIF-Dependent Signaling Are CD14-Independent.
Zanoni et al. (12) reported the requirement for CD14 in TLR4 endocytosis and the production of IFN-β by LPS. We compared internalization of TLR4 induced by LPS to that induced by UT12, a MAb directed against a TLR4/MD2 epitope that acts as a TLR4 agonist (13, 17). In WT macrophages, both LPS and UT12 induced TLR4 internalization, whereas in CD14−/− macrophages only UT12 induced TLR4 endocytosis (Fig. S1A). An isotype-matched control antibody failed to induce TLR4 internalization in either WT or CD14−/− macrophages (Fig. S1A). The effect of UT12 on macrophages from FcR α-chain−/− and γ-chain−/− mice was evaluated to rule out activation of TLR4 internalization and TRIF signaling by FcR-mediated endocytosis. UT12 induced a similar level of TLR4 internalization in the absence of either FcR α- or γ-chains (Fig. S2). UT12-induced TLR4 internalization was time-dependent in both WT and CD14−/− macrophages (Fig. 1 A and B). LPS- and UT12-induced TLR4 internalization in WT macrophages were similarly time-dependent, but again, LPS, but not UT12, failed to induce TLR4 endocytosis in CD14−/− macrophages (Fig. 1 A and B).
Fig. S1.

Dose-dependent TLR4 internalization in WT and CD14−/− PMs. (A) WT and CD14−/− PMs were treated with medium only or with increasing doses of LPS (0.1–1,000 ng/mL), UT12 (1–1,000 ng/mL), UT12-istoype antibody (1,000 ng/mL), or (B) synthetic TLR4 ligands 1Z105 and 1Z204 (0.1–10 μM) and a control compound, 1Y88 (10 μM) for 90 min, and TLR4 surface expression was analyzed by flow cytometry and quantified by MFI at each dose.
Fig. S2.
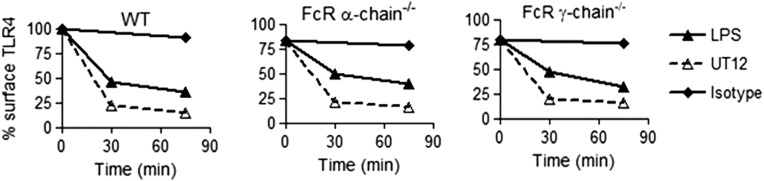
FcR α-chain and γ-chain deficiency does not affect UT12- and LPS-induced TLR4 internalization. WT and FcR α-chain−/− and γ-chain−/− BMDMs were untreated or treated with UT12 or LPS for indicated times, and TLR4 surface expression was analyzed by flow cytometry and quantified by MFI at each time point.
Fig. 1.

CD14 is not required for UT12-induced TLR4 endocytosis and downstream signaling in PMs. WT and CD14−/− mouse PMs were medium-treated or treated with LPS (100 ng/mL), UT12 (1,000 ng/mL), and UT12 istoype (1,000 ng/mL) for the indicated times, and TLR4 surface expression was analyzed by flow cytometry (A and B). A representative histogram was shown after 90 min of treatment (A), and TLR4 internalization was quantitated by mean fluorescence intensity (MFI) at each time point (B). Total cell lysates from WT and CD14−/− PMs were prepared at 30 min (A, Left) and 60 min (A, Right) after treating with LPS, UT12, and UT12-isotype control antibody and activating signaling molecules analyzed by Western blotting (C). For cytokine/chemokine secretion, WT and CD14−/− PMs were treated with LPS, UT12, and UT12-isotype control antibody for 16 h, and culture supernatants were analyzed by ELISA (D). CD14 expression in WT and CD14−/− PMs was analyzed by flow cytometry (E, Left). WT PMs were treated with medium only, LPS, UT12, and UT12-isotype control antibody for indicated times, and CD14 surface expression was analyzed by flow cytometry and quantified by MFI (E, Right). Data represent the mean ± SEM from two to three independent experiments. #P < 0.05, nontreated vs. treated groups; *P < 0.05, treated WT vs. treated CD14−/− groups. (NT, not treated; Iso, isotype).
Activation of signaling molecules required for the MyD88- and/or TRIF-dependent signaling pathways were compared in WT and CD14−/− macrophages stimulated by LPS and UT12. LPS (100 ng/mL) activated the TRIF-signaling intermediates TBK1 and IRF3 in WT macrophages by 30 min; however, LPS failed to activate TBK1 or IRF3 in CD14−/− macrophages, even after 60 min (Fig. 1C, Right). NF-κB activation by LPS was also defective in CD14−/− macrophages at 30 min (Fig.1C, Left), but became detectable at 60 min (Fig. 1C, Right). Similarly, MAPK activation by LPS was defective in CD14−/− macrophage at 30 min, but was increased slightly at 60 min (Fig. 1C), consistent with the previously reported role for CD14 in enhancing kinetics of MyD88-dependent gene induction (18). On the other hand, the activation of NF-κB, MAPK, and TBK1/IRF3 by UT12 was CD14-independent both at early and at late time points (Fig. 1C).
To measure the effect of CD14 on production of type I IFN and other proinflammatory cytokines induced by LPS or UT12, secreted cytokines/chemokines in culture supernatants of WT and CD14−/− macrophages were analyzed. Both LPS (100 ng/mL) and UT12 (1,000 ng/mL) elevated proinflammatory cytokine levels (e.g., TNF-α and IL-6) in WT and CD14−/− macrophages, although the level of secretion induced by LPS was consistently greater than that induced by UT12 (Fig. 1D). In contrast, LPS-induced IFN-β and IP-10 production was significantly blunted in CD14−/− macrophages (Fig. 1D) under conditions where UT12-induced IFN-β and IP-10 levels were CD14-independent (Fig. 1D).
Zanoni et al. (12) also reported that CD14 was internalized into endosomes with TLR4. Therefore, we investigated the endocytosis of CD14 in LPS- vs. UT12-stimulated WT macrophages. CD14−/− macrophages were used to confirm the specificity of the anti-CD14 antibody. CD14−/− macrophages failed to express CD14 protein whereas WT macrophages strongly expressed CD14 (Fig. 1E, Left). In contrast to TLR4, surface expression of CD14 was reduced minimally by LPS stimulation at 30 min, but not altered at 90 min (Fig. 1E, Right). However, UT12 did not induce CD14 internalization at early or late time points (Fig. 1E, Right). These data clearly suggest that TLR4 and CD14 endocytosis is dissociable.
To determine if UT12-induced TLR4 internalization and IRF3 activation involves the same signaling molecules reported for LPS (12), we tested the effect of the Syk and PLC-γ2 inhibitors, piceatannol and U73122, respectively. Both inhibitors blocked LPS- and UT12-induced TLR4 internalization and IRF3 activation similarly (Fig. S3). These data suggest that whereas both Syk and PLC-γ2 are required for UT12-mediated TLR4 internalization and IRF3 activation, CD14 is not.
Fig. S3.

Syk and PLC-γ2 inhibitors reversed LPS- and UT12-induced TLR4 endocytosis and IRF3 activation. PMs from WT mice were pretreated with either DMSO (0.1%) or the Syk inhibitor, piceatannol (75 μM), or the PLC-γ2 inhibitor, U73122 (5 μM) for 30 min and then treated with LPS (50 ng/mL) and UT12 (100 ng/mL) for 90 min. TLR4 internalization was analyzed by flow cytometry; a representative histogram is shown (A and B). TLR4 surface expression was quantified using MFI (C and D). For signaling studies, PMs were pretreated with piceatannol (E) or U73122 (F) for 30 min and stimulated with LPS or UT12 for 60 min as described above, and total cell lysates were subjected to Western blotting (E and F). Data represent mean ± SEM from three independent experiments. (NT, not treated; Iso, Isotype).
MyD88 and TRIF Deficiencies Do Not Affect TLR4 Internalization.
MyD88 and TRIF are adapter molecules responsible for NF-κB and IRF3 activation, respectively (5–7). As previously reported (12), TLR4 endocytosis induced by LPS was both MyD88- and TRIF-independent, and this was observed for UT12 stimulation as well (Fig. S4 A and C). Secretion of TNF-α and IL-6 induced by LPS or UT12 was significantly diminished in MyD88−/− macrophages, but IFN-β and IP-10 levels were either equivalent to or slightly less in MyD88−/− compared with WT macrophages (Fig. S4B), consistent with the classification of IFN-β and IP-10 as MyD88-independent (8, 19). In TRIF−/− macrophages, both MyD88-dependent (TNF-α and IL-6) and TRIF-dependent (IFN-β and IP-10) cytokines induced by LPS or UT12 were completely inhibited (Fig. S4D), consistent with previous reports that TRIF plays an important role in the production of both MyD88- and TRIF-dependent cytokines/chemokines (20, 21).
Fig. S4.

MyD88 and TRIF deficiency did not affect TLR4 internalization induced by LPS and UT12. WT, MyD88−/−, and TRIF−/− PMs were treated with medium only or with LPS (100 ng/mL), UT12 (1000 ng/mL), or 1Z105 (5 μM) for the indicated times, and TLR4 surface expression was analyzed by flow cytometry and quantified by MFI at each time point (A and C). Culture supernatants were collected after 16 h of treatment, and ELISAs were performed for secreted cytokines/chemokines (B and D).
TRIF-Dependent Cytokine/Chemokine Production by TLR3 Ligand Is CD14-Independent.
Because TLR3 is located in endosomes and its ligand, polyinosinic:polycytidylic acid (pI:C), strongly induces IFN-β, we assessed the involvement of CD14 in TLR3 signaling. Several reports have provided evidence for the requirement of CD14 in TLR3-mediated signaling (22, 23). Lee et al. claimed that CD14 physically interacts with pI:C and mediates TLR3 activation (23). pI:C treatment of macrophages did not affect surface expression of TLR4 or CD14 (Fig. S5 A and B). The production of TNF-α in pI:C-stimulated CD14−/− macrophages was greatly inhibited, in contrast to IL-6, IFN-β, and IP-10, which were minimally or not affected (Fig. S5C). Hence, CD14 is not absolutely required for TLR3-mediated, TRIF-dependent cytokines and chemokine secretion.
Fig. S5.

CD14 deficiency minimally affected pI:C-induced IFN-β. WT and CD14−/− PMs were treated with medium or with pI:C (20 μg/mL) for the indicted times, and surface expression of TLR4 and CD14 was analyzed by flow cytometry (A and B) (NT, not treated; Iso, Isotype). (A) A representative histogram of surface TLR4 at 3 h (Left). TLR4 internalization quantified by MFI at each time point (Right). (B) A representative histogram for surface CD14 is shown at the indicated times (Left). CD14 internalization quantified by MFI at each time point (Right). (C) Culture supernatants from cells analyzed in B were harvested at 16 h and analyzed for secreted cytokines/chemokines.
Synthetic Small-Molecule TLR4 Ligands Induce TLR4 Endocytosis and IRF3 Activation in a CD14-Independent Manner.
Recently, Hayashi et al. identified synthetic chemical ligands that activate TLR4 in a CD14-independent, MD2-dependent manner and resulted in the secretion of both MyD88- and TRIF-dependent cytokines/chemokines (16). To extend these findings, we investigated their effects on TLR4 internalization and endocytic signaling. Two agonists, 1Z105, and the less active ligand, 1Z204, dose-dependently induced TLR4 internalization in both WT and CD14−/− macrophages (Fig. S1B). An inactive control compound, 1Y88, did not induce TLR4 endocytosis in either WT or CD14−/− macrophages, even at higher concentrations (Fig. 2A and Fig. S1B). Similar to UT12, 1Z105 (5 μM) potently induced TLR4 endocytosis in a time-dependent fashion. The less active 1Z204 induced TLR4 endocytosis partially at 5 μM (Fig. 2A). The activation of signaling events by these compounds was also similar to the effect of UT12 (compare Fig. 2B to Fig. 1C). Furthermore, cytokine/chemokine production was also CD14-independent (Fig. 2C) and correlated with the degree of activation of NF-κB and TBK1/IRF3 axes (Fig. 2B).
Fig. 2.

TLR4 synthetic small-molecule ligands induce receptor endocytosis and related signaling in a CD14-independent manner. WT and CD14−/− PMs were medium-treated or treated with LPS (100 ng/mL) and different synthetic ligands (5 μM) for the indicated times, and TLR4 internalization was analyzed by flow cytometry and quantified by MFI at each time point (A) as described in the legend to Fig. 1. Total cell lysates from WT and CD14−/− PMs were prepared 60 min after treating cells with LPS or synthetic ligands, and activation of signaling molecules was analyzed by Western blotting (B). For cytokine/chemokine secretion, WT and CD14−/− PMs were treated with LPS and synthetic ligands for 16 h, and culture supernatants were analyzed by ELISA (C). Data represent the mean ± SEM from two to three independent experiments. #P < 0.05, nontreated vs. treated groups; *P < 0.05, treated WT vs. treated CD14−/− groups.
LPS-induced B7 costimulatory molecules (CD80 and CD86) are TRIF-TRAM–dependent (24, 25). Similar to previously published reports in TRIF-deficient macrophages (24) and TRAM-deficient B220-positive cells (25), LPS-induced up-regulation of CD80 and CD86 was perturbed in CD14−/− macrophages (Fig. S6). However, as expected, both UT12- and 1Z105-induced up-regulation of CD80 and CD86 was not affected in CD14−/− macrophages (Fig. S6). Overall, these data suggest that CD14 is not absolutely required for TLR4 endocytosis and its downstream signaling induced by UT12 and small-molecule TLR4 agonists. To rule out any differences in TLR4 internalization and TRIF signaling induced by UT12 and 1Z105 in primary peritoneal macrophages (PMs) vs. bone marrow-derived macrophages (BMDMs), we repeated our studies in BMDMs. BMDMs behaved very similarly to peritoneal macrophages with respect to TLR4 endocytosis, TRIF signaling, and cytokine/chemokine production induced by UT12 and 1Z105 (Fig. S7 A–D). Surface expression of CD14 was not modulated in BMDM upon stimulation with LPS, UT12, or 1Z105 (Fig. S7E), in contrast to the slight decrease in CD14 seen in LPS-stimulated PMs (Fig. 1E). Furthermore, we performed colocalization analysis of CD14 to the endosome in WT BMDMs using confocal microscopy. As shown in Fig. S8, CD14 (green) did not colocalize with endosomes (red) when BMDMs were treated with LPS, UT12, or 1Z105.
Fig. S6.

CD14-independent up-regulation of B7 costimulatory molecules by UT12 and 1Z105. WT and CD14−/− mouse PMs were medium-treated (NT) or treated with LPS (100 ng/mL), UT12 (1,000 ng/mL), or 1Z105 (5 μM) for 18 h, and surface expression of CD80 (A) and CD86 (B) was analyzed by flow cytometry. (A and B) A representative histogram after 90 min treatment. (C and D) CD80/CD86 up-regulation quantified by MFI. Data represent mean ± SEM from three independent experiments. (NT, not treated; Iso, Isotype).
Fig. S7.

CD14 is not required for UT12- and 1Z105-induced TLR4 endocytosis and downstream signaling in BMDMs. WT and CD14−/− BMDMs were medium-treated or treated with LPS (100 ng/mL), UT12 (1,000 ng/mL), or 1Z105 (5 μM) for the indicated times, and TLR4 surface expression was analyzed by flow cytometry (A and B) (NT, not treated; Iso, Isotype). A representative histogram is shown after 90 min treatment (A), and TLR4 internalization was quantified by MFI at each time point (B). Total cell lysates from WT and CD14−/− BMDMs were prepared at 60 min after treatment with medium only or with LPS, UT12, or 1Z105, and activation of signaling molecules was analyzed by Western blotting (C). For secreted cytokines/chemokines, WT and CD14−/− BMDMs were treated with LPS, UT12, or 1Z105 for 16 h, and culture supernatants were collected and analyzed by ELISA (D). WT BMDMs were treated with medium only, LPS, UT12, and 1Z105 for the indicated times, and CD14 surface expression was analyzed by flow cytometry and quantified by MFI (E). Data represent the mean ± SEM from two to three independent experiments.
Fig. S8.

UT12 and 1Z105 did not induce CD14 colocalization with endosome in BMDMs. WT BMDMs were treated with medium (Med), LPS (100 ng/mL), UT12 (1 μg/mL), and 1Z105 (5 μM) for 30 min. Cells were fixed, permeabilized, blocked, and stained with anti–CD14-Alexa488 (green), anti–EEA-Alexa647 (red), and DAPI (blue) and analyzed using confocal microscopy. Merged images of CD14, EEA1, and DAPI are shown. Original magnification: 40×. (Scale bar: 20 μm.)
The dynamin inhibitor, dynasore, prevented LPS-induced TLR4 internalization in WT macrophages (Fig. S9 A and C, Left) as previously reported (8). As expected, LPS did not induce TLR4 internalization in the presence or absence of dynasore in CD14−/− macrophages (Fig. S9 B and C, Right). 1Z105-induced TLR4 internalization was inhibited by dynasore in both WT and CD14−/− macrophages (Fig. S9 A–C), whereas UT12-induced internalization was not (Fig. S9 A–C). However, dynasore completely inhibited both 1Z105- and UT12-induced IRF3 activation in both WT and CD14−/− macrophages (Fig. S9D), resulting in inhibition of both MyD88-dependent (TNF-α) and TRIF-dependent (IFN-β) cytokines (Fig. S9E). This latter observation extends the findings of Kagan et al. who reported complete inhibition of LPS-induced IL-6 (MyD88-dependent) and RANTES (TRIF-dependent) by dynasore in WT macrophages (8). In fact, dynasore enhanced degradation of IκB-α in medium-treated WT and CD14−/− macrophages (Fig. S9D) yet did not induce NF-κB–dependent cytokines, suggesting that nuclear translocation of NF-κB did not occur in macrophages treated with dynasore alone.
Fig. S9.

Dynasore inhibited LPS- and 1Z105-driven not UT12-induced TLR4 endocytosis but inhibited LPS-, UT12-, and 1Z105-induced IRF3 activation. BMDMs from WT and CD14−/− mice were pretreated with either DMSO or dynasore (80 μM) for 60 min and then treated with medium only, LPS (50 ng/mL), UT12 (100 ng/mL), or 1Z105 (5 μM) for 90 min. TLR4 internalization was analyzed by flow cytometry; and a representative histogram is shown (A and B) (NT, not treated; Iso, Isotype). TLR4 surface expression was quantified by MFI (C). For signaling studies, BMDMs were stimulated with LPS or UT12 or 1Z105 in the absence or presence of dynasore for 60 min, and total cell lysates were subjected to Western analysis (D). Culture supernatants were collected after 16 h of treatment described above and cytokines were analyzed by ELISA (E). Data represent mean ± SEM from two experiments.
TLR4 Antagonist Eritoran Inhibits LPS and 1Z105, but Not UT12-Induced TLR4 Internalization.
Eritoran (E5564) is a synthetic lipid A analog that binds in the deep hydrophobic pocket of MD2 and competitively inhibits binding of lipid A to MD2 and thereby inhibits downstream signaling (26). Eritoran was also shown to bind to CD14 and block the transfer of lipid A to MD2 (27). Eritoran blocks LPS-induced MyD88- and TRIF-dependent cytokine production in human and murine macrophages (28). Therefore, we sought to determine if Eritoran would block TLR4 internalization induced by the various TLR4 agonists. Eritoran inhibited LPS- and 1Z105-induced TLR4 internalization in WT macrophages (Fig. 3A), consistent with the MD2 dependency of these two agonists. However, Eritoran failed to inhibit UT12-induced TLR4 internalization (Fig. 3A), suggesting that the TLR4/MD2 epitope recognized by UT12 is distinct from the MD2 hydrophobic pocket or is not modified by binding of Eritoran to the MD2 hydrophobic pocket. Because 1Z105 induces TLR4 internalization in a CD14-independent manner and was inhibited by Eritoran (Fig. 3A), we asked whether Eritoran would inhibit 1Z105-induced TLR4 internalization in CD14−/− macrophages. Eritoran inhibited 1Z105-induced TLR4 internalization in CD14−/− macrophages (Fig. 3B), suggesting that Eritoran is acting independently of CD14 despite the fact it is able to bind to CD14. Furthermore, we observed that Eritoran inhibited LPS-, UT12-, and 1Z105-induced activation of TBK1/IRF3, NF-κB, and MAPKs (Fig. 3C). R848, a TLR7/8 ligand, was used as control, and Eritoran did not inhibit any signaling induced by this agonist (Fig. 3C). Eritoran also inhibited LPS-, UT12-, and 1Z105-induced cytokine/chemokine gene expression (Fig. 3D).
Fig. 3.

The TLR4 antagonist E5564 failed to inhibit UT12-induced receptor internalization, but inhibited MyD88- and TRIF-dependent signaling. WT PMs were pretreated with 10 ng/mL E5564 for 60 min and then treated with LPS, UT12, or 1Z105 for the indicated times. TLR4 internalization was analyzed by flow cytometry, and surface expression was quantitated using MFI (A). TLR4 internalization induced by 1Z105 was analyzed after 90 min of treatment in CD14−/− PMs in the absence or presence of E5564 as described above (B). PMs were stimulated with LPS, 1Z105, UT12, or R848 in the absence or presence of E5564 (10 ng/mL) for 60 min, and total cell lysates were subjected to Western blotting (C). PMs were treated with different TLR4 ligands as described above, and RNA was isolated after 1 and 5 h. Gene expression was analyzed by quantitative real-time PCR (qRT-PCR) (D). HEK293T cells were treated with different TLR4 ligands in the absence or presence of Eritoran for 30 min, and induction of TLR4 dimerization was analyzed by immunoprecipitation followed by Western analysis (E). Data represent the mean ± SEM from two to three independent experiments. *P < 0.05, treated without vs. with E5564 groups.
To determine if Eritoran interfered in TLR4 dimerization, we compared the ability of LPS, UT12, and 1Z105 to induce TLR4 dimerization in the absence or presence of Eritoran. Eritoran blocked TLR4 dimerization induced by LPS and 1Z105 in TLR4-expressing HEK293T cells (Fig. 3E); however, it failed to block TLR4 dimerization induced by UT12 (Fig. 3E), consistent with the failure to block UT12-induced TLR4 internalization (Fig. 3A). These data clearly indicate that TLR4 dimerization is critical for its internalization induced by different ligands.
CD14-Independent Endocytosis of TLR4 in Macrophages Rendered Tolerant by LPS and UT12.
In mice and macrophages exposed to LPS, a transient period of LPS-hyporesponsiveness ensues, which has been referred to as “endotoxin tolerance,” which has been associated with epigenetic changes that result in differential gene expression (29–32). Because the expression of TRIF-dependent genes is strongly “tolerized,” we sought to determine the role of TLR4 internalization in this process. Cells were treated overnight with medium (M) only, LPS (L), or UT12 (U) and then restimulated the next day with M, L, or U. We observed decreased surface expression of TLR4 both in nontolerized (M/L, M/U) and tolerized (L/M, L/L, U/U) WT macrophages (Fig. 4A, Left). In CD14−/− macrophages, however, LPS treatment of mock-tolerized macrophages (M/L) did not reduce the surface expression of TLR4, consistent with our data in Figs. 1 and 2. Interestingly, macrophages rendered tolerant by overnight LPS treatment exhibited significant TLR4 internalization, even in the absence of CD14, and without or with LPS restimulation (Fig. 4A, L/M, L/L), although the TLR4 internalization seen in CD14−/− macrophages was somewhat less than that observed in WT macrophages. When UT12 was used as the TLR4 agonist, however, we found that nontolerized (M/U) and tolerized (U/U) macrophages exhibited similar levels of TLR4 endocytosis both in WT and CD14−/− macrophages (Fig. 4A). Heterotolerance to LPS or UT12 (U/L or L/U) also induced CD14-independent TLR4 internalization (Fig. 4B). Inhibition of LPS- or UT12-induced NF-κB, MAPK, and TBK1/IRF3 signaling was comparable in LPS- or UT12-tolerized WT and CD14−/− macrophages, thus dissociating TLR4 internalization and signaling (Fig. 4C). Both TNF-α and IL-6 levels were tolerized to the same extent in WT and CD14−/− macrophages (Fig. 4D). Although LPS (M/L) did not induce IFN-β in CD14−/− macrophages (as observed in Fig. 1D), production of IFN-β was completely blocked in tolerized macrophages (Fig. 4D), despite TLR4 internalization.
Fig. 4.

Endotoxin tolerance induced TLR4 endocytosis in CD14−/− PMs. WT and CD14−/− PMs were tolerized overnight for 18 h with LPS or UT12. Cells were washed to remove endotoxin or UT12 and restimulated with either medium or LPS or UT12 for indicated times. Surface expression of TLR4 (A and B) was analyzed by flow cytometry as described in Fig. 1. Similarly, WT and CD14−/− PMs were tolerized for 18 h as described above, and total cell lysate was prepared after 30 min of restimulation with medium only, LPS, or UT12; activation of signaling molecules was analyzed by Western blotting (C). For cytokine secretion, WT and CD14−/− PMs were tolerized for 18 h as described above and restimulated with LPS or UT12 for 16 h, and culture supernatants were analyzed for cytokine/chemokine level by ELISA (D). For each graph, data represent the mean ± SEM from two to three independent experiments. Percentage surface TLR4 in WT and CD14−/− was normalized using medium-treated WT macrophages as 100%. #P < 0.05, medium treated vs. nontolerized and tolerized groups; *P < 0.05, M/L (nontolerized) vs. L/L and U/L (tolerized); ‡P < 0.05, M/U (nontolerized) vs. U/U and L/U (tolerized).
Discussion
TLR4 endocytosis and trafficking to the endosomal compartment is important for the regulation of TRIF-mediated signaling induced by LPS (8, 33). This process is tightly regulated by dynamins, clathrin, and associated Rab proteins (9, 34). Kagan and coworkers reported that, upon LPS stimulation, TLR4 is recruited to the endosome from the plasma membrane where it interacts with TRAM and TRIF adaptor molecules, leading to activation of the IRF3 pathway (8). However, the specific mechanism by which TLR4 is transported to the endosome was incompletely defined. The small GTPase ADP ribosylation factor 6 (ARF6) and Rab family of GTPases have been investigated in controlling endocytic transport of receptors (10). Recently, Husebye et al. showed that Rab11a, a small GTPase, regulates recruitment of TLR4 and TRAM to E. coli phagosomes and controls both E. coli- and LPS-induced IRF3 activation (9). Zanoni and coworkers (12) reported the requirement for membrane-bound CD14 in controlling LPS- and E. coli-induced TLR4 endocytosis and TRIF-mediated signaling in macrophages. Furthermore, they reported that TLR4 internalization and IRF3 activation is mediated by PLC-γ2 and Syk (12). Very recently, a requirement for p120-catenin, a prototypic member of subfamily of armadillo repeat domain proteins, has been shown to regulate MyD88-dependent NF-κB and TRIF-dependent IRF3 activation reciprocally. Silencing of p120-catenin diminished LPS-induced TLR4 internalization and IRF3 activation while increasing NF-κB translocation (11).
We and others previously reported that CD14 is required for MyD88-dependent signaling at low, but not high, concentrations of LPS (12, 35). This suggests that CD14 is primarily responsible for the transfer of LPS to MD2, a necessary coreceptor for TLR4, when the concentration of LPS is limiting (36, 37). CD14 dependency for MyD88-dependent signaling is overcome at higher LPS doses, possibly due to a direct interaction of LPS monomers with MD2. In contrast, the CD14 dependency required for TRIF-mediated signaling cannot be overcome by increasing the LPS concentration (12). When LPS or E. coli are presented on beads to CD14-deficient dendritic cells, both TLR4 internalization and TRIF-dependent signaling are preserved (12). This implies that, in the case of soluble LPS, CD14 also regulates the trafficking of TLR4 into the endosome where it, in turn, recruits the downstream adapters TRAM and TRIF to the TIR domain of TLR4 dimer.
Our data confirm and significantly extend these findings. TLR4 endocytosis and TRIF-mediated signaling were induced by treatment of macrophages with UT12, a mouse antibody directed against an epitope formed by TLR4/MD2 interaction (13, 14), and small synthetic TLR4 ligands (1Z105 and 1Z204) that bind to MD2 and signal through both MyD88-dependent and TRIF-dependent pathways in the absence of CD14 (16). Although it is possible that the UT12 monoclonal antibody also activates internalization through FcγR-dependent uptake of UT12/TLR4/MD2 immune complexes, UT12 is a mouse IgG3 that has high affinity for FcRn and very low affinity/no affinity toward FcγRI, FcγRIIB, FcγRIII, and FcγRIV (38, 39). For all of these FcγRs, either FcR α- and/or γ-chains are required for activation (40). UT12-induced TLR4 internalization was not altered in macrophages derived from mice deficient in either FcR α- and γ-chains (Fig. S2), ruling out the possibility of FcR involvement in TLR4 internalization. Furthermore, the isotype control antibody for UT12 did not induce TLR4/MD2 internalization.
Moreover, LPS- and 1Z105-, but not UT12-induced TLR4 internalization was blocked by dynasore, and yet, TNF-α and IFN-β levels were completely blocked in UT12-treated macrophages. This suggests that either dynamin is acting further downstream in the TLR4-signaling pathway triggered by UT12 leading to gene expression or that dynasore has an off-target effect that underlies inhibition of MyD88-dependent cytokines. Zanoni et al. previously showed that Syk and PLC-γ2 were key signaling components for TLR4 internalization (12). Consistent with their findings, we found that Syk/PLC-γ2 inhibitors blocked both LPS- and UT12-induced TLR4 internalization, as well as IRF3 phosphorylation. The PLC-γ2 inhibitor partially prevented IκB-α degradation (Fig. S3E), supporting the notion that it may also act further downstream and/or have off-target effects on other mediators involved in MyD88-dependent signaling.
More interestingly, macrophages rendered hyporesponsive to LPS or UT12 by a standard “tolerance” regimen (31) retained the ability to internalize TLR4 in a CD14-independent fashion, yet exhibited decreased MyD88- and TRIF-dependent signaling and cytokine production. A correlation between endotoxin tolerance and a transient down-regulation of surface TLR4 after LPS stimulation has been reported by some groups (41), but was completely restored to normal levels at the time of LPS challenge in tolerized cells (30). Regardless, both TLR4 endocytosis by endotoxin tolerance and induction of tolerance are CD14-independent. We reported here that although surface expression TLR4 is endocytosed in endotoxin-tolerized cells, none of the signaling cascades are activated. Thus, the TLR4 endocytosis that occurs during endotoxin tolerance is completely dissociable from TRIF signaling (Fig. S10D).
Fig. S10.

Hypothetical model of CD14-dependent and -independent TLR4 internalization and TRIF-dependent signaling and their dissociation. Unlike LPS that requires CD14 for stabilization of the TLR4/MD2 complex, UT12 and/or 1Z105 bind directly to TLR4/MD2 and form a stable TLR4/MD2 dimer complex, even in the absence of CD14, that leads to NF-κB activation whereas TLR4/MD2 internalization leads to IRF3 activation (A). UT12-induced TLR4 endocytosis is not affected in the presence of E5564, but both NF-κB and IRF3 signaling are blocked (B). E5564 blocks 1Z105-induced TLR4/MD2 dimer complex formation that inhibits both TLR4 endocytosis and NF-κB and IRF3 signaling (C). In tolerized macrophages, TLR4 endocytosis is not affected, but both NF-κB and IRF3 signaling are blocked (D).
Because CD14 is required for LPS-induced TLR4 delivery to endosomes and is not required for MyD88-dependent signaling at higher LPS concentrations, we propose that CD14 not only assists in LPS transfer to MD2, but perhaps is also is required for stabilizing the TLR4/MD2 complex at the plasma membrane that, in turn, favors complex internalization (Fig. S10A). The involvement of CD14 in tight heterodimerization of TLR4/MD2 has been shown recently by Tanimura and coworkers (42). In the case of UT12 and 1Z105, CD14 is required neither for transfer of the ligand nor for internalization of the TLR4/MD2 complex (Fig. S10A), possibly due to their innate abilities to bind MD2 directly.
Furthermore, we demonstrated another surprising dissociation between TLR4 endocytosis and TRIF-dependent signaling using Eritoran (Fig. S10B), an inactive lipid A analog that binds to a MD2 hydrophobic pocket and competes for LPS binding, thereby blocking both MyD88-dependent and TRIF-dependent signaling. Eritoran treatment failed to induce TLR4 endocytosis, and we confirmed by a TLR4 dimerization assay that this is due to a failure to bring two TLR4/MD2 complexes into an active conformation. The inhibition of LPS- and 1Z105-induced TLR4 endocytosis by Eritoran mediated by preventing the formation of the TLR4/MD2 complex (Fig. S10C) confirmed that these molecules compete for the same MD2-binding site. Moreover, our findings that Eritoran did not inhibit UT12-induced endocytosis, but inhibited UT12-induced signaling, suggests the possibility that the presence of Eritoran in the endosome, along with the UT12-TLR4/MD2 complex, may cause a conformational change within the TIR domains of the TLR4 dimer that prevents its interaction with the adapter, TRIF adapter (Fig. S10B).
In summary, we have found that TLR4 endocytosis and TRIF signaling are dissociable. We propose that CD14 may help in the stabilization of TLR4/MD2 complex at plasma membrane that, in turn, leads to endocytosis and TRIF-dependent signaling.
Materials and Methods
Cell Culture.
PMs.
C57BL/6J (WT), CD14−/−, MyD88−/−, and TRIF−/− mice (6–8 wk) were injected with sterile thioglycollate (Remel) as described previously (43). Cells were obtained and treated as described in SI Materials and Methods.
BMDMs.
BMDMs were derived from C57BL/6 (WT), CD14, FcR α-chain, and γ-chain–deficient mice bone marrows. BMDMs were cultured and treated as described in SI Materials and Methods.
Cells stimulation and FACS staining.
Cells were stimulated with the indicated concentrations of TLR ligands and RNA was isolated for gene expression studies. Culture supernatants were collected for analyzing cytokine secretion, and cell lysates were prepared for Western analysis. For FACS analysis, PMs and BMDMs (2 × 105) were stimulated with the indicated concentrations of different TLR ligands as described in SI Materials and Methods.
Inhibition of TLR4 internalization by dynasore.
WT and CD14−/− BMDMs were pretreated with dynasore (80 μM) (Sigma-Aldrich) for 60 min in serum-free culture medium as described in SI Materials and Methods.
TLR4 dimerization assay.
Four × 105 HEK293T cells were plated per well in a six-well tissue culture plate. After 24 h, HEK293T cells were transfected with expression vectors for FLAG-TLR4 (100 ng/well), eCFP-TLR4 (100 ng/well), CD14 (75 ng/well), and MD2 (100 ng/well), and cells were treated as described in SI Materials and Methods.
Animal Assurances.
All animal studies were carried out with approval from the University of Maryland, Baltimore Institutional Animal Care and Use Committee.
Statistical Analysis.
One-way ANOVA with Tukey’s Multiple Comparison post hoc test was performed to assess statistical significance (P values <0.05) using GraphPad PRISM 4.0 (GraphPad Software).
Detailed experimental procedures are available in SI Materials and Methods.
SI Materials and Methods
Reagents and Mice.
Protein-free E. coli K235 LPS (<0.008% protein) was prepared as described previously (44). Purified UT12 was the kind gift of Robert Munford and Mingfang Lu (NIH, Bethesda, MD). Synthetic ligands for TLR4 were prepared as previously described (16). Phycoerythrin (PE)-labeled anti-mouse TLR4 (SA15-21), PE-labeled anti-mouse CD14 (Sa14-2), PE-labeled rat IgG2a, and anti-mouse CD16/32 (Fc blocking antibody) antibodies were purchased from Bio Legend. Antibodies directed against phospho-TBK1, phospho-IRF3, phospho-ERK1/2, phospho-JNK1/2, JNK, IκB-α, and β-actin were purchased from Cell Signaling Technology. Eritoran was kindly provided by Eisai Inc. under a Materials Transfer Agreement. WT C57BL/6J and CD14−/− mice (C57BL/6J background) were purchased from the Jackson Laboratory, and MyD88−/− and TRIF−/− mice (C57BL/6J background) were bred in-house at the University of Maryland Baltimore. Leg bones from FcR α-chain−/− and γ-chain−/− mice were kindly provided by Jeffrey Ravetch (The Rockefeller University, New York).
Cell Culture.
PMs.
C57BL/6J (WT), CD14−/−, MyD88−/−, and TRIF−/− mice (6–8 wk) were injected with sterile thioglycollate (Remel) as described previously (43). Four days later, cells were obtained by peritoneal lavage. Cells were plated and allowed to adhere overnight, followed by extensive washing to remove nonadherent cell types. Macrophages were cultured in RPMI 1640 supplemented with 2% (vol/vol) FBS, 2 mM glutamine, 1% (vol/vol) penicillin, and streptomycin as described previously.
BMDMs.
BMDMs were derived from C57BL/6 (WT), CD14 and FcR α-chain, and γ-chain–deficient mice bone marrows. BMDMs were cultured in DMEM supplemented with 25–30% (vol/vol) LADMAC (ATCC) conditioned medium as a source of macrophage colony-stimulating factor-1, 10% (vol/vol) FBS, 10 mM Hepes, 1 mM sodium pyruvate, 2 mM glutamine, 1% (vol/vol) penicillin, and streptomycin for 6 d. Cells were washed and stimulated with different ligands, as indicated, in the absence of conditioned medium.
Cells stimulation and FACS staining.
Cells were stimulated with the indicated concentrations of TLR ligands, and RNA was isolated for gene expression studies. Culture supernatants were collected for analyzing cytokine secretion, and cell lysates were prepared for Western analysis. For FACS analysis, PMs and BMDMs (2 × 105) were stimulated with the indicated concentrations of different TLR ligands. Cells were washed and blocked with anti-CD16/CD32 antibody (Fc blocking) for 20 min on ice and stained with PE-labeled anti-mouse TLR4, PE-labeled anti-mouse CD14 and PE-labeled rat IgG2a for 30 min on ice. Cells were washed twice to remove unbound antibodies and analyzed using BD FACSCanto II (Becton and Dickinson). Data were analyzed by FlowJo software. For inhibition studies, cells were pretreated with Eritoran (Eisai Inc.) for 60 min or with piceatannol/U73122 for 30 min, then treated with different ligands and analyzed as described above.
Inhibition of TLR4 internalization by dynasore.
WT and CD14−/− BMDMs were pretreated with dynasore (80 μM) (Sigma-Aldrich) for 60 min in serum-free culture medium. Then cells were stimulated with UT12 and 1Z105 in serum-free medium. For LPS treatment, cells were stimulated in culture medium containing a final concentration of 0.14% serum as a source of LBP. TLR4 internalization, signaling, and cytokines were analyzed as described above.
TLR4 dimerization assay.
HEK293T cells (4 × 105) were plated per well in a six-well tissue culture plate. After 24 h, HEK293T cells were transfected with expression vectors for FLAG-TLR4 (100 ng/well), eCFP-TLR4 (100 ng/well), CD14 (75 ng/well), and MD2 (100 ng/well). Following 24 h transfection, cells were pretreated without or with E5564 (20 ng/mL) for 30 min. Subsequently, cells were stimulated with media alone, LPS (1 μg/mL), 1Z105 (1.66 μM), or UT12 (1 μg/mL) for 30 min. Cells were washed one time with cold PBS and lysed in 1 mL B1 Lysis buffer [1% Triton × 100, 25 mM Hepes, 150 mM NaCl, with Complete Mini protease inhibitor mixture (Roche)]. Lysates were precleared with protein G slurry (Roche) for 30 min at 4 °C. Lysates were subsequently immunoprecipitated for 3 h at 4 °C using 1 μg of anti-eCFP monoclonal antibody (Origene) with protein G slurry. Immunoprecipitates were washed three times in ice-cold B1 lysis buffer, and immune-precipitated complexes were resolved on 10% (vol/vol) SDS/PAGE. Gels were transferred to PVDF and probed with anti-FLAG M2 monoclonal antibody (Sigma-Aldrich).
Induction of in vitro endotoxin tolerance.
Cells were stimulated without or with LPS (100 ng/mL) or UT12 (1,000 ng/mL) for 20 h, washed, and then “challenged” by treating the cells without or with LPS (100 ng/mL) or UT12 (1,000 ng/mL) for the indicated times. Macrophage treatments are designated as M/M (medium/medium), M/L (medium/LPS treated), M/U (medium/UT12 treated), L/L (LPS/LPS treated), and U/U (UT12/UT12 treated), corresponding to the initial/challenge treatments. FACS staining was carried out as described above. For cytokine ELISAs, culture supernatants were collected 16 h after rechallenge with LPS or UT12. For signaling, cell lysates were prepared using cell lysis buffer (Cell Signaling Technology) after rechallenge with LPS or UT12 for 30 min.
qRT-PCR.
Total RNA was isolated from macrophages using a High Pure RNA Isolation Kit (Roche) according to the manufacturer’s instructions. A total of 1 μg of RNA was reverse-transcribed using an oligo(dT) cDNA synthesis kit (Bio-Rad RT system). Differential gene expression was analyzed by qRT-PCR using SyBR green as per the manufacturer’s guidelines in 7900 HT Fast Real-time PCR system (Applied Biosystems). mRNA expression profiles were normalized to the levels of the housekeeping gene HPRT in each sample, and the fold change in expression was calculated by the 2-ΔΔCt method.
Western analysis.
Whole-cell lysates from PMs and BMDMs were prepared by the addition of lysis buffer (Cell Signaling Technology) and subsequent incubation at 4 °C. Cell lysates were separated by electrophoresis in a denaturing SDS/PAGE gel and transferred to a PVDF membrane at constant voltage (100 V). Blots were incubated overnight in relevant primary antibodies at 4 °C, washed four to five times with PBST, and then incubated with appropriate HRP-conjugated secondary antibody (Jackson ImmunoResearch). Blots were developed using ECL plus reagents (Amersham Bioscience).
ELISA.
Cytokine/chemokine levels in macrophage culture supernatants were analyzed by ELISA at the Cytokine Core Facility (University of Maryland Baltimore). IFN-β ELISA was performed according to the protocol established by Roberts et al. (45).
Immunofluorescence microscopy.
WT and CD14−/− BMDMs were stimulated with different ligands for 30 min in chamber slides, fixed in 4% paraformaldehyde (10 min at room temperature), permeabilized with 0.25% Triton-X100 (10 min at room temperature), and blocked with 5% (vol/vol) normal goat serum (30 min at room temperature). Cells were incubated with rabbit anti-EEA1 (Cell Signaling Technology; dilution 1:100) and rat anti-CD14 (eBioscience; dilution 1:50) for 1 h at room temperature. Alexa Fluor 488-conjugated goat anti-rat IgG and Alexa Fluor 647-conjugated goat anti-rabbit IgG (Molecular Probes) secondary antibodies were added at a dilution of 1:100. Samples were then analyzed with a LSM 510 Zeiss confocal microscope.
Supplementary Material
Acknowledgments
We thank Dr. Dennis Carson and Dr. Tomoko Hayashi (University of California at San Diego Moores Cancer Center) for providing synthetic small TLR4 ligands. This work was supported by National Institutes of Health Grant AI018797 (to S.N.V.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.C.K. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424980112/-/DCSupplemental.
References
- 1.Montminy SW, et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol. 2006;7(10):1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- 2.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 3.Shibata T, et al. Intracellular TLR4/MD-2 in macrophages senses Gram-negative bacteria and induces a unique set of LPS-dependent genes. Int Immunol. 2011;23(8):503–510. doi: 10.1093/intimm/dxr044. [DOI] [PubMed] [Google Scholar]
- 4.Gioannini TL, et al. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc Natl Acad Sci USA. 2004;101(12):4186–4191. doi: 10.1073/pnas.0306906101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300(5625):1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 6.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 7.Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125(5):943–955. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 8.Kagan JC, et al. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9(4):361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Husebye H, et al. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity. 2010;33(4):583–596. doi: 10.1016/j.immuni.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Acker T, et al. The small GTPase Arf6 is essential for the Tram/Trif pathway in TLR4 signaling. J Biol Chem. 2014;289(3):1364–1376. doi: 10.1074/jbc.M113.499194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Z, et al. Differential role for p120-catenin in regulation of TLR4 signaling in macrophages. J Immunol. 2014;193(4):1931–1941. doi: 10.4049/jimmunol.1302863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zanoni I, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147(4):868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahrun U, et al. Preparation and characterization of agonistic monoclonal antibodies against Toll-like receptor 4-MD-2 complex. Hybridoma (Larchmt) 2007;26(6):393–399. doi: 10.1089/hyb.2007.0523. [DOI] [PubMed] [Google Scholar]
- 14.Ohta S, et al. Induction of long-term lipopolysaccharide tolerance by an agonistic monoclonal antibody to the toll-like receptor 4/MD-2 complex. Clin Vaccine Immunol. 2006;13(10):1131–1136. doi: 10.1128/CVI.00173-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan M, et al. Identification of substituted pyrimido[5,4-b]indoles as selective Toll-like receptor 4 ligands. J Med Chem. 2013;56(11):4206–4223. doi: 10.1021/jm301694x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi T, et al. Novel synthetic toll-like receptor 4/MD2 ligands attenuate sterile inflammation. J Pharmacol Exp Ther. 2014;350(2):330–340. doi: 10.1124/jpet.114.214312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe. 2008;4(3):293–302. doi: 10.1016/j.chom.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perera PY, Vogel SN, Detore GR, Haziot A, Goyert SM. CD14-dependent and CD14-independent signaling pathways in murine macrophages from normal and CD14 knockout mice stimulated with lipopolysaccharide or taxol. J Immunol. 1997;158(9):4422–4429. [PubMed] [Google Scholar]
- 19.Palsson-McDermott EM, et al. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat Immunol. 2009;10(6):579–586. doi: 10.1038/ni.1727. [DOI] [PubMed] [Google Scholar]
- 20.Hoebe K, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424(6950):743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301(5633):640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 22.Baumann CL, et al. CD14 is a coreceptor of Toll-like receptors 7 and 9. J Exp Med. 2010;207(12):2689–2701. doi: 10.1084/jem.20101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee HK, Dunzendorfer S, Soldau K, Tobias PS. Double-stranded RNA-mediated TLR3 activation is enhanced by CD14. Immunity. 2006;24(2):153–163. doi: 10.1016/j.immuni.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 24.Hoebe K, et al. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol. 2003;4(12):1223–1229. doi: 10.1038/ni1010. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto M, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4(11):1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 26.Kim HM, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130(5):906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 27.Shirey KA, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. 2013;497(7450):498–502. doi: 10.1038/nature12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Y, Pearlman E. Inhibition of corneal inflammation by the TLR4 antagonist Eritoran tetrasodium (E5564) Invest Ophthalmol Vis Sci. 2009;50(3):1247–1254. doi: 10.1167/iovs.08-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henricson BE, Benjamin WR, Vogel SN. Differential cytokine induction by doses of lipopolysaccharide and monophosphoryl lipid A that result in equivalent early endotoxin tolerance. Infect Immun. 1990;58(8):2429–2437. doi: 10.1128/iai.58.8.2429-2437.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Medvedev AE, Lentschat A, Wahl LM, Golenbock DT, Vogel SN. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J Immunol. 2002;169(9):5209–5216. doi: 10.4049/jimmunol.169.9.5209. [DOI] [PubMed] [Google Scholar]
- 31.Rajaiah R, et al. Dissociation of endotoxin tolerance and differentiation of alternatively activated macrophages. J Immunol. 2013;190(9):4763–4772. doi: 10.4049/jimmunol.1202407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447(7147):972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 33.Latz E, et al. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J Biol Chem. 2002;277(49):47834–47843. doi: 10.1074/jbc.M207873200. [DOI] [PubMed] [Google Scholar]
- 34.Husebye H, et al. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006;25(4):683–692. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perera PY, et al. CD11b/CD18 acts in concert with CD14 and Toll-like receptor (TLR) 4 to elicit full lipopolysaccharide and taxol-inducible gene expression. J Immunol. 2001;166(1):574–581. doi: 10.4049/jimmunol.166.1.574. [DOI] [PubMed] [Google Scholar]
- 36.Akashi S, et al. Lipopolysaccharide interaction with cell surface Toll-like receptor 4-MD-2: Higher affinity than that with MD-2 or CD14. J Exp Med. 2003;198(7):1035–1042. doi: 10.1084/jem.20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes Infect. 2004;6(15):1361–1367. doi: 10.1016/j.micinf.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 38.Gavin AL, Barnes N, Dijstelbloem HM, Hogarth PM. Identification of the mouse IgG3 receptor: Implications for antibody effector function at the interface between innate and adaptive immunity. J Immunol. 1998;160(1):20–23. [PubMed] [Google Scholar]
- 39.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119(24):5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 40.Ravetch JV, Nimmerjahn F. 2013. Fundamental Immunology: Fc Receptors and their Role in Immune Regulation and Inflammation, ed Paul WE (Lippincott Williams and Wilkins, Philadelphia), pp 583–600.
- 41.Nomura F, et al. Cutting edge: Endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J Immunol. 2000;164(7):3476–3479. doi: 10.4049/jimmunol.164.7.3476. [DOI] [PubMed] [Google Scholar]
- 42.Tanimura N, et al. The attenuated inflammation of MPL is due to the lack of CD14-dependent tight dimerization of the TLR4/MD2 complex at the plasma membrane. Int Immunol. 2014;26(6):307–314. doi: 10.1093/intimm/dxt071. [DOI] [PubMed] [Google Scholar]
- 43.Dobrovolskaia MA, Vogel SN. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002;4(9):903–914. doi: 10.1016/s1286-4579(02)01613-1. [DOI] [PubMed] [Google Scholar]
- 44.McIntire FC, Sievert HW, Barlow GH, Finley RA, Lee AY. Chemical, physical, biological properties of a lipopolysaccharide from Escherichia coli K-235. Biochemistry. 1967;6(8):2363–2372. doi: 10.1021/bi00860a011. [DOI] [PubMed] [Google Scholar]
- 45.Roberts ZJ, et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J Exp Med. 2007;204(7):1559–1569. doi: 10.1084/jem.20061845. [DOI] [PMC free article] [PubMed] [Google Scholar]