

Significance
Although vimentin intermediate filaments (VIFs) are the most stable cytoskeletal component in motile cells, VIFs undergo dramatic reorganization during cell spreading, cell division, and motility. Here, we studied the first step of IF assembly using the vimentinY117L mutant, which forms oligomers called unit-length filaments (ULFs) but cannot assemble into mature VIFs. We discovered that ULFs, unlike VIFs, are extremely dynamic and rapidly exchange subunits with the soluble vimentin pool. Surprisingly, this process requires ATP but seems independent of the vimentin phosphorylation events previously shown to trigger filament disassembly. We believe that dynamic exchange of subunits could play a role in the regulation of ULF assembly and maintenance of a soluble vimentin pool during the reorganization of the filament network.
Keywords: vimentin, dynamic, photoconversion, intermediate filaments, cytoskeleton
Abstract
Intermediate filaments (IFs) are a component of the cytoskeleton capable of profound reorganization in response to specific physiological situations, such as differentiation, cell division, and motility. Various mechanisms were proposed to be responsible for this plasticity depending on the type of IF polymer and the biological context. For example, recent studies suggest that mature vimentin IFs (VIFs) undergo rearrangement by severing and reannealing, but direct subunit exchange within the filament plays little role in filament dynamics at steady state. Here, we studied the dynamics of subunit exchange in VIF precursors, called unit-length filaments (ULFs), formed by the lateral association of eight vimentin tetramers. To block vimentin assembly at the ULF stage, we used the Y117L vimentin mutant (vimentinY117L). By tagging vimentinY117L with a photoconvertible protein mEos3.2 and photoconverting ULFs in a limited area of the cytoplasm, we found that ULFs, unlike mature filaments, were highly dynamic. Subunit exchange among ULFs occurred within seconds and was limited by the diffusion of soluble subunits in the cytoplasm rather than by the association and dissociation of subunits from ULFs. Our data demonstrate that cells expressing vimentinY117L contained a large pool of soluble vimentin tetramers that was in rapid equilibrium with ULFs. Furthermore, vimentin exchange in ULFs required ATP, and ATP depletion caused a dramatic reduction of the soluble tetramer pool. We believe that the dynamic exchange of subunits plays a role in the regulation of ULF assembly and the maintenance of a soluble vimentin pool during the reorganization of filament networks.
Cell shape, mechanical properties, and motile behavior are determined by the cytoskeleton composed of three interconnected polymers: actin microfilaments, microtubules, and intermediates filaments (IFs). Whereas the structural polarity of microfilaments and microtubules is essential for their functions, IFs are apolar structures that contribute to the mechanical properties of the cell (reviewed in ref. 1). This important function is perfectly adapted for each cell type because about 70 different human IF proteins are expressed in a tissue- and differentiation-specific manner. The common feature of all IF proteins is the presence of a central coiled-coiled domain, which favors the formation of a very stable polymer.
In vitro, the spontaneous self-assembly of vimentin IFs occurs in three steps that can be initiated by increasing the salt concentration (reviewed in ref. 2). During the first step of assembly, eight vimentin tetramers, the smallest stable polymer in vitro, anneal laterally to form the so-called unit-length filament (ULF) with an approximate length of 65 nm (3). During the second phase, ULFs and longer filaments elongate by longitudinal annealing via end-to-end fusion (4). Finally, during the third phase of assembly, a radial compaction of the filaments occurs, resulting in the maturation of the 10-nm-wide IF (5, 6). The resulting mature IFs are very stable in vitro. For example, it takes 5–9 h before IFs labeled with a fluorescent dye integrate segments from IFs labeled with a second fluorescent dye (7, 8).
In cells, IFs are also extraordinarily stable at steady state compared with actin microfilaments or microtubules. Nevertheless, at least three major forms of vimentin can be observed in cells: the most abundant mature filaments that radiate from the juxtanuclear region to the cell periphery, the short filaments of different sizes that are not yet connected to the filament network, and nonfilamentous particles that might be aggregates of ULFs (9). In addition, the presence of a small soluble pool of tetrameric vimentin was also reported in live cells (10). The hierarchical assembly of keratin IFs from particles at the cell periphery to mature filaments at the cell center was also nicely revealed by live-cell imaging (11). As opposed to the quasi-unidirectional assembly of IFs in vitro, it is now well accepted that IF networks in cells undergo dynamic reorganization, which is a prerequisite for the cell-protective functions of IFs in response to several stress-related signaling events. For example, it was shown that the vimentin IF (VIF) network is reorganized during cell spreading and motility (12). Several mechanisms were proposed to be involved in IF reorganization, depending on the type of IF protein as well as the biological context. For example, it has been shown that keratin IFs can exchange subunits (13). Keratin subunits are released near the cell center, whereas the assembly of filaments occurs preferentially at the cell periphery (11, 14). Recent studies using photoactivation and photoconversion demonstrate that reorganization of VIFs and neurofilaments mainly occur via a different mechanism. Instead of releasing soluble subunits, these filaments reorganize through a process of severing and end-to-end reannealing (15, 16). In general, assembly and disassembly of IFs is believed to be regulated by phosphorylation or other posttranslational modifications (see ref. 17 for a review). Filament turnover involves an increase in the presence of short precursor IFs in the cell as well as the availability of a soluble pool of IF protein for the next round of IF polymerization. However, little is known about how the soluble pool of IF proteins is maintained. Similarly, subunit dynamics at the first step of IF assembly into ULFs remains to be understood.
In this study, we took advantage of the vimentin Y117L mutant (vimentinY117L) to study the dynamics of the first step of filament assembly in cells. VimentinY117L laterally associates into ULFs, but ULFs containing mutant vimentin do not longitudinally anneal and thus cannot form VIFs (18). We previously showed that this vimentin mutant forms particles in the cell that interact with the two other cytoskeletal polymers, microtubules, and actin microfilaments. We found that ULF particles are actively transported along microtubules and that sequestration of ULF particles by actin filaments prevents their transport (19). Here, we study the dynamics of the first step of VIF assembly by tagging the vimentinY117L with the photoconvertible protein mEos3.2. After photoconversion of mEos-vimentinY117L or mEos-vimentin in a restricted area of a cell, we could follow the turnover of vimentin subunits in ULF particles or filaments to compare the stability of ULFs versus mature IFs. Unexpectedly, we discovered that unlike mature intermediate filaments, ULFs are highly dynamic. Furthermore, we demonstrated that subunit exchange between ULFs and the soluble vimentin pool is ATP-dependent but independent of vimentin phosphorylation at serine 38, an important phospho-residue implicated in VIF disassembly (12, 20). We believe that elucidating the dynamic exchange machinery could pave the way to discovering new mechanisms of vimentin assembly regulation in cells.
Results
Rapid Exchange of Subunits in ULFs.
To study dynamic properties of vimentin precursors, we used vimentinY117L. In vitro assembly assays have shown that this mutant of vimentin laterally associates into ULF but fails to longitudinally anneal to form elongated VIFs (18). We tagged vimentinY117L with the photoconvertible protein mEos3.2 (mEos-vimentinY117L) and expressed it in the SW13 adenocarcinoma cell line clone that lacks cytoplasmic IFs (vim−). Previously, we demonstrated that expression of vimentinY117L in vim− cells leads to the formation of ULF particles, whereas coexpression of the mutant with wild-type vimentin results in incorporation of the mutant protein into a morphologically normal IF network (19). We observed that mEos-vimentinY117L formed mobile dot-shaped structures reminiscent of the ULF particles formed after the expression of GFP-vimentinY117L (19). Our strategy was to photoconvert mEos-vimentinY117L from green to red in a restricted area of the cytoplasm and monitor the incorporation of red vimentin that originated in the photoconverted area to nonconverted ULFs located outside it.
Immediately after photoconversion, most of the red fluorescence was contained within the original photoconverted area (Fig. 1A, first column). However, 1 min postconversion, photoconverted vimentin was clearly detectable in ULFs outside the initial photoconverted area (Fig. 1A, second column, and Movie S1). We demonstrated previously that a fraction of ULFs in a cell is highly motile and actively transported along microtubules (19). To make sure that the ULFs containing red vimentin observed outside the photoconverted area were not particles that had been photoconverted and then transported outside the photoconverted area, we tracked the location of all ULFs in the green channel. The ULFs outside the photoconversion area that initially had only green fluorescence gradually acquired red fluorescence (see white arrows in Fig. 1A, rows three and four). The same phenomenon was observed in mouse embryonic fibroblast (MEF) cells isolated from vimentin knockout mice and stably transfected with mEos-vimentinY117L (Fig. S1). Additionally, ULF in the photoconversion area lost fluorescence intensity in the red channel (Fig. 1B). These data suggest that photoconverted vimentin accumulated outside the photoconverted zone because of subunit exchange between ULF rather than ULF transport.
Fig. 1.

ULFs exchange vimentin subunits. (A) An area 10 µm in diameter of a mEos-vimentinY117L–expressing cell was photoconverted from green to red with 405-nm light (see circle), and images of the red channel (λ = 561 nm) were taken immediately and every 15 s after photoconversion for 5 min. Enlargements of the insets show that red fluorescence increase was faster in ULFs located close to the photoconverted area (blue Insets) than in ULFs far from to the photoconverted area (red Insets). Arrows point to the fluctuation of fluorescence in a single ULF over time. (Scale bar, 5 μm.) (B) Graph shows the decay of the average fluorescence intensity (AFI) in individual ULFs located inside the area of photoconversion. (C) Relative AFI per ULF was measured over time and the data points were grouped according to the radial circumference distance of the ULFs from the center of the photoconverted area. (D) The initial rate of exchange (relative AFI per second) was determined by calculating the slope over the first minute after photoconversion (C, gray box) for each radial distance zone. The result is the average ± SEM of 7,000 particles intensity measurements from 30 cells.
Fig. S1.

ULFs subunit exchange occurs in MEF cells. (A) mEos-vimentinY117L was stably expressed in embryonic fibroblasts isolated from a vimentin knockout mouse. Photoconversion of a 10-µm-diameter area of the cell from green to red was performed for 3 s (see circle), and images of the red channel (λ = 561 nm) were taken every minute after photoconversion for 5 min. (Scale bar, 10 μm.) (B) Three× zoom of the insets show the accumulation of red fluorescence in ULFs located outside of the photoconverted area. Five ULFs were manually tracked and labeled (see circles) to show how red fluorescence increased in single ULFs over time. The graph in C shows the intensity of individual ULF tracked in B as a function of time, and the graph in D shows the AFI per ULF as a function of time. Note that the high motility of MEF cells and high motility of ULF particles complicated large-scale quantification of individual ULF intensity.
Time-lapse imaging shows that red fluorescence quickly increased in ULFs that were close to the photoconverted area (Fig. 1A, blue insets), whereas ULFs located far from the area of photoconversion accumulated red protein much more slowly (Fig. 1A, red insets). To quantify the rate of red fluorescence increase, we selected ULFs that were stationary over the duration of the time-lapse (3 min) and located outside the converted area. Quantification of fluorescence in these ULF in the red channel confirmed that red increase was observed first in ULFs close to the converted area (5- to 10-µm zone) and later into the zones that were farther away (10- to 15- and 15- to 20-µm zones) (Fig. 1C). We measured the initial rate of accumulation of the photoconverted (red) vimentin as a function of distance between the ULFs and the center of the photoconversion zone (Fig. 1D). We defined initial rate as the rate of accumulation of red fluorescence during the first minute after photoconversion (the gray area in Fig. 1C). We found that the rate of red subunit accumulation in ULFs was much faster in ULFs that were located in the vicinity of the photoconverted area than in those farther away, indicating that the spreading of photoconverted subunits from the photoconverted area is a rate-limiting step in the exchange.
If accumulation of red fluorescence in ULF particles represents subunit exchange, the increase in red fluorescence should correlate with a decrease in green fluorescence of individual ULFs. This is difficult to observe using mEOS3.2-tagged vimentin, as green fluorescence of mEOS3.2 bleaches very quickly. Therefore, we created a fusion of vimentinY117L with mMaple3, a newly developed photoconvertible protein with improved photostability (21). Time-lapse imaging showed that, as with the mEos-ULF probe, red fluorescence accumulated in mMaple3-ULFs located outside the photoconverted area (Fig. S2). Measurements of the intensities of individual ULFs in both channels after photoconversion as a function of time demonstrated that accumulation of red fluorescence in the particles outside the converted zone is accompanied by the loss of green fluorescence and, as in the case of EOS3.2-tagged vimentin, the rate of green-red exchange is faster closer to the converted zone (Fig. 2). This result confirmed that the accumulation of red fluorescence in individual ULFs is a good read-out of subunit exchange.
Fig. S2.
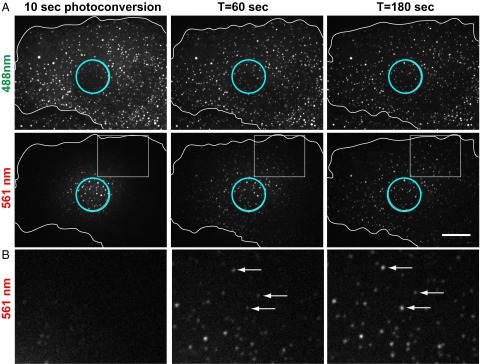
Use of mMaple3-vimentinY117L to probe ULF subunit exchange. mMaple3-vimentinY117L was stably expressed in SW13 vim− cells. (A) Photoconversion of a 10-µm-diameter area of the cell was performed for 10 s for maximal conversion from green to red of the ULF inside the conversion zone (see circle in the first column); 180 s after conversion, red fluorescence accumulated in ULFs all over the cell and green fluorescence came back in ULFs located inside the photoconverted area. (Scale bar, 10 µm.) (B) Three× zoom of the insets show the accumulation of red fluorescence in ULFs located outside of the photoconverted area. Arrows point to individual ULFs representing increase of red fluorescence in single ULFs over time. See Fig. 2 for red-green quantification.
Fig. 2.

Red fluorescence increase correlates with green fluorescence decrease in individual ULF. SW13 cell stably expressing mMaple3-vimentinY117L was photoconverted for 10 s from green to red with 405-nm light, and images of the red channel (λ = 561 nm) were taken immediately and every 15 s after photoconversion. The mean increase in intensity per individual ULF per second in both channels was determined by calculating the slope over the first minute after photoconversion for 11 cells (average ± SEM of 2,000 particles intensity measurements in both channels). The data points were grouped according to the radial circumference distance of the ULFs from the center of the photoconverted area to show that the green-to-red exchange occurred first in ULFs located close to the photoconverted area.
Microtubules and Actin Filaments Are Not Essential for ULF Subunit Exchange.
Because ULF farther from the area of photoconversion take longer to incorporate red subunits (Fig. 1), and ULFs are known to move along microtubules (19), it is possible that the movement of subunits or ULFs along microtubules could affect subunit exchange. To test this possibility, we determined the effect of microtubule depolymerization on the initial rate of subunit exchange. Microtubules were depolymerized by incubating cells at 4 °C for 1 h followed by transfer to 37 °C in medium containing 10 µM nocodazole. Immunostaining with a tubulin antibody previously showed that this treatment was sufficient to depolymerize most microtubules (19). As a control, cells were transferred from 4 °C to 37 °C in regular medium (in the absence of nocodazole) to allow microtubules to repolymerize. Direct observation and quantification of the initial rate of exchange for the ULFs located 5–10 µm from the center of the photoconversion zone demonstrated no significant difference between control and nocodazole-treated cells (Fig. 3), showing that microtubule-dependent transport is not essential for ULF subunit exchange. Moreover, because ULFs are static in nocodazole-treated cells (Fig. S3, Bottom), this result confirms that the increase in red intensity cannot be explained by fusion of ULF particles but truly represents exchange of green subunits for red.
Fig. 3.

Microtubules and actin filaments are not required for ULF subunit exchange. (A) Photoconversion experiments performed on control cells or cells treated with 10 µM nocodazole or with 5 µM LatB revealed that depolymerization of microtubules or actin filaments did not interfere with the accumulation of photoconverted molecules in ULF outside the photoconverted area (cyan circle). Images show ULFs in the red channel, located 5–10 µM form the center of the photoconverted zone, 2 min after photoconversion. (Scale bar, 5 µm.) (B) Graph shows the initial rate of exchange (average ± SEM of at least 625 measurements from 30 cells per condition) calculated in ULFs located in a radial circumference distance of 5–10 µM from the center of the photoconverted area.
Fig. S3.

LatB treatment increase ULF motility. mEos-vimentinY117L cells were treated with 5 µM LatB for 30 min to depolymerize actin filaments or with 10 µM nocodazole before photoconversion. Images of the red channel were taken every 2 s for 1 min to follow the motility of the ULFs. Every time frame was color-coded and the superimposition of all frames revealed ULF tracks. Rainbow tracks correspond to mobile ULFs and white dots represent stationary ULFs. Two examples of a cell for each treatment are presented to show that LatB increased the motility of ULF but nocodazole completely abolished any movement of ULF. (Scale bar, 10 µm.)
We previously reported that ULFs bind to actin filaments preventing their transport along microtubules (19). To test if binding to actin affects ULF subunit exchange, we tested the effect of actin filament depolymerization on the initial rate of ULF subunit exchange. Cells were treated with 5 µM latrunculin B (LatB) for 30 min to depolymerize actin filaments before photoconversion. The efficiency of actin filament depolymerization by LatB was confirmed using rhodamine-phalloidin staining (19). Comparison between the red channel 1 min after photoconversion in control versus LatB-treated cells shows that the absence of actin filaments did not slow down subunit exchange (Fig. 3). In fact, quantification of the initial rate of exchange showed a slight increase (P value of 0.1) after LatB treatment (Fig. 3B). One possible explanation is that even if soluble subunits of vimentin are not directly transported along microtubules, ULFs are. As we described previously, LatB treatment stimulates ULF transport (19). Time-lapse images of red ULF after photoconversion confirmed that in LatB-treated cells ULFs moved more rapidly than those in controls (see rainbow trajectories in Fig. S3, Middle). Furthermore, cases of ULFs traveling outside of the photoconverted area were clearly observed in LatB-treated cells (Fig. S3, Right, Middle). Accordingly, the increased transport of photoconverted ULFs outside the photoconverted area after LatB treatment likely increases the available pool of red subunits for the exchange into the 5- to 10-µm radial distance zone. Overall, depolymerization of the microtubule or actin cytoskeletons did not decrease the initial rate of exchange, showing that neither microtubules nor actin filaments are essential for ULF subunit exchange.
Y117L Vimentin Does Not Exchange in the Context of Mature Vimentin Filaments.
Previous studies have shown that VIFs are very stable because the exchange of fragments or smaller subunits along the length of mature filaments is very slow (16, 22). The present study revealed a very fast process of subunit exchange among ULFs assembled from vimentinY117L. There are two possibilities that could explain the difference between the highly dynamic properties of ULFs that we observed and the stability of mature filaments: either the mutation Y117L per se is responsible for faster exchange, or the ULF step of assembly allows the high rate of subunit exchange. To distinguish between these two possibilities, we expressed mEos-vimentinY117L in vimentin-containing (vim+) SW13 cells. In these cells, vimentinY117L copolymerized with endogenous wild-type vimentin and was incorporated into a morphologically normal vimentin filament network (Fig. 4C) (19). To better visualize individual filaments within the dense network, the imaging of photoconverted filaments was performed using total internal reflection fluorescence (TIRF) microscopy. As a control, we photoconverted a subset of filaments in SW13 vim+ cells expressing wild-type vimentin tagged with mEos3.2. In agreement with our previously published results, we observed the presence of some short and uniformly red segments of filaments that have been transported along microtubules outside the photoconverted zone (Fig. 4A) (16). However, no dual-color filaments were observed outside the photoconverted area in these cells, even 15 min after photoconversion (Fig. 4A). Within the same time period, the majority of ULFs in mEos- vimentinY117L SW13 vim− cells contained both red and green fluorescence (Fig. 4B). When mEos-vimentinY117L was expressed in SW13 vim+ cells it incorporated into filaments and, like in the wild-type vimentin case, dual-color filaments were not detected (Fig. 4C). This experiment demonstrates that the fast rate of subunit exchange in ULF cannot be explained by the Y117L mutation itself, but by the fact that filament assembly is halted at the ULF stage. Therefore, filament precursors appear to be extremely dynamic but mature filaments are much more stable.
Fig. 4.
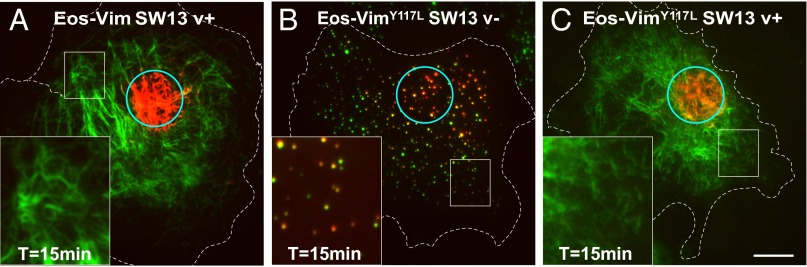
The Y117L mutation does not increase the rate of exchange in the context of mature vimentin filaments. As a control, mEos-vimentin (wild-type) was stably expressed in SW13 vim+ cells (A). mEos-vimentinY117L was stably expressed in SW13 vim− cells to form ULFs (B) or transfected in SW13 vim+ cells to copolymerize with endogenous vimentin and form mature filaments (C). Pictures of the green and red channel were taken immediately and 15 min after photoconversion of a small area of the cells (see cyan circle) using TIRF microscopy. Note that many yellow ULFs indicative of subunit exchange from green to red are present in SW13 vim− cells 15 min after photoconversion (B), but dual-colored filaments were not observed outside of the photoconverted area in SW13 vim+ cells (A–C). (Scale bar, 10 μm; Magnification: Insets, 2.5×.)
Vimentin Subunit Exchange in ULF Depends on ATP.
Our data suggest that the exchange rate in ULF is independent of active transport and is limited by the diffusion of subunits. To determine if another kind of energy-dependent mechanism is required for subunit exchange, we tested the effect of ATP depletion on exchange. Although a sustained lack of ATP is lethal for the cell, the effect of a transient ATP depletion can be fully reversible (23). Two different treatments were used to briefly deplete ATP. Cells were treated with either 20 mM sodium azide for 20 min or with 10 µM of the ionophore carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) for 30 min. Both treatments were performed in the absence of glucose to prevent ATP production by glycolysis (Methods). Surprisingly, even 2 min after photoconversion, no red fluorescence could be observed in ULFs outside the photoconverted area in ATP-depleted cells (Fig. 5A). Inhibition of subunit exchange by ATP depletion was fully reversible, because after a 1-h washout, the rate of exchange was indistinguishable from the control (Fig. 5), confirming that the inhibition of the exchange in ATP-depleted cells was not the result of nonspecific toxicity.
Fig. 5.

ATP depletion abolishes ULF subunit exchange. (A) Photoconversion experiments were performed on control cells, cells depleted of ATP by 20-min incubation with 1 µM FCCP, and FCCP-treated cells followed by a 1-h washout. In ATP-depleted cells, no accumulation of red fluorescence was detected outside of the photoconverted area (cyan circle) 2 min after photoconversion. However, exchange resumed 1 h after the washout of the drugs, showing that the cells survived the treatments. Images show ULFs in the red channel, located 5–10 µM form the center of the photoconverted zone, 2 min after photoconversion. (Scale bar, 5 μm.) (B) Graph shows the dramatic decrease of the initial rate of exchange in ATP-depleted cells after sodium azide or FCCP treatment. Measurements were taken in ULFs located in a radial circumference distance of 5–10 µM from the center of the photoconverted area (average ± SEM of at least 700 measurements from 30 cells per condition). ****P < 0.0001.
The exchange of vimentin between ULFs requires the presence of two pools of subunits: a pool of assembled ULF particles and a pool of a soluble form of vimentin that can freely diffuse in the cytoplasm. These two fractions should exist in a state of equilibrium. First, we compared pools of the soluble form of vimentin between cells expressing mEos-vimentin filaments and mEos-vimentinY117L ULFs. For these experiments, cells were lysed with 0.5% Triton X-100 and the insoluble fraction containing ULFs were pelleted by centrifugation, whereas the soluble material was collected in the supernatant. Western blot analysis demonstrated that mEos-vimentin was predominantly insoluble like endogenous wild-type vimentin (Fig. 6A). However, more than 50% of vimentinY117L was found in the soluble fraction (Fig. 6B, control). The amount of soluble vimentin available for exchange is defined by the rates of subunit dissociation and association with ULFs. To determine which one of these two steps requires ATP, we analyzed the pool of soluble mEos-vimentinY117L in cells after ATP depletion. The amount of soluble vimentin was dramatically reduced after ATP depletion compared with the control (Fig. 6B). Analysis of the total level of vimentin in the same condition showed that short-term ATP depletion does not affect the amount of vimentin, only its solubility. (Fig. 6B). Additionally, fluorescence microscopy revealed that cells expressing GFP-vimentinY117L contained a diffuse fluorescence between individual ULF particles, but after ATP depletion, the amount of diffuse fluorescence was decreased dramatically (Fig. 6C). In both cases, the effect of ATP-depletion was fully reversible because a 1-h washout was sufficient to fully restore the soluble pool of vimentin.
Fig. 6.

ATP depletion reduces the soluble pool of vimentin. Solubility tests were performed on mEos-vimentin (wild-type) cells which also coexpress endogenous vimentin (A) or on mEos-vimentinY117L cells (B). Cells were lysed in extraction buffer containing 0.5% Triton X-100. For ATP depletion, cells were treated with 20 mM sodium azide or 10 µM FCCP for 20 min prior to lysis. For washout experiments, cells were washed extensively after the azide or FCCP treatment and incubated for 1 h in fresh medium. Cell lysates were centrifuged at 265,000 × g to pellet insoluble proteins and collect soluble proteins in the supernatant. Equal amounts of each fraction were analyzed by Western blot using a chicken polyclonal vimentin antibody. ins, insoluble fraction; sol, soluble fraction; TL, total lysate. Quantification of the Western blots demonstrates that ATP depletion dramatically reduced the solubility of mEos-vimY117L (average ± SD from three experiments). (C) GFP-ULF expressing cells were treated with sodium azide or FCCP to deplete ATP. Pictures of control and ATP-depleted cells were taken and processed using the same parameters (exposure, contrast) to show that the diffuse fluorescent signal in between ULFs was decreased after ATP depletion, as confirmed by the quantification of the mean fluourescence intensity in regions between individual ULFs in unprocessed images (average ± SD of more than 300 regions from 35 cells). (Scale bar, 10 μm; Magnification: Insets, 3×.) ****P < 0.0001. (D) FCS measurements of GFP-vimentinY117L were performed on control versus cells treated with sodium azide. Typical correlation curves are shown (thin lines), where G is the amplitude and τ is the temporal correlation shift. Global fits (thick lines) were calculated across 20 measurements to determine the diffusion coefficient as described in material and method. Note that FCS curves taken in sodium azide-treated cells had a higher G0 than control cell indicating a relatively lower concentration of the soluble GFP-vimentinY117L in ATP-depleted cells. Nevertheless, the diffusion coefficient of both conditions is similar.
Further evidence of a soluble and highly diffusible pool of vimentin was obtained using fluorescence correlation spectroscopy (FCS) measurements. FCS measures fluctuations in fluorescence intensity as fluorescently labeled particles diffuse through the focal volume of the laser beam (24). The diffusion coefficient of the fluorescently labeled particles can be extracted from the fluctuations. Our FCS measurements on GFP-vimentinY117L indicated a diffusion coefficient of 22.9 µm2/s (Fig. 6D). FCS is an ultrasensitive single-molecule technique that can detect molecules at very low concentrations beyond what Western blot analysis can detect (25). For this reason, we could observe a freely diffusing pool of vimentin in ATP-depleted cells even if the vimentin-soluble pool was barely detected by Western blot. FCS measurements indicated a diffusion coefficient of 21.5 μm2/s in cells treated with sodium azide, which is similar to what was observed in the control (Fig. 6D). This result suggests that ATP was not required for the diffusion of the soluble vimentin subunits. Furthermore, FCS can give information about concentration. The system we used was not calibrated to give quantitative concentration measurements, but we could still obtain qualitative information on the relative concentration for measurements by comparing the y axis intercepts (sometimes called G0) of the correlation curves. FCS curves taken in sodium azide-treated cells had a higher G0 than in control cells, indicating a relatively lower concentration (Fig. 6D). In this way, we were able to confirm that sodium azide-treated cells had a lower concentration of diffusing vimentin than control cells.
Altogether, our data show that ATP is required for subunit exchange and to maintain a sizable pool of soluble vimentin. Vimentin solubility tests and FCS measurements strongly suggest that ATP depletion inhibits the dissociation of subunits from ULFs to the soluble pool rather than the spreading of soluble vimentin within the cytoplasm.
Soluble Vimentin Is Tetrameric.
To better understand the molecular regulation of the equilibrium between ULFs and soluble vimentin subunits, we analyzed the nature of the soluble subunit of mEos-vimentinY117L. Previous studies have suggested that the smallest vimentin oligomer present in the cell after vimentin filaments have been disassembled is a tetramer (10, 20). If the filament precursors assembled from mEos-vimentinY117L behave like normal filament precursors, the smallest oligomer should be a tetramer as well. We combined sedimentation velocity ultracentrifugation and size-exclusion chromatography to estimate the size of the soluble mEos-vimentinY117L oligomer compared with the size of soluble tetramers of endogenous vimentin released during filament disassembly (26). We used calyculin A to depolymerize vimentin filaments in SW13 vim+ cells, as described previously (20). Sucrose gradient ultracentrifugation experiments indicated Svedberg coefficient (S20,w) values of 5.6 and 6.8, whereas gel filtration on a Superose 6 column indicated Stokes radius (Rs) of 8.9 and 12.2 for soluble endogenous vimentin and soluble mEos-vimentinY117L, respectively (Table 1; see Fig. S4 for detailed analyses). These two parameters (S20,w and Rs) were used to estimate the size (Mr) of the soluble vimentin oligomers (Table 1; see Methods for calculation). For the endogenous soluble vimentin released in the presence of calyculin, the calculated Mr was 210 kDa, which is 3.7-fold the predicted mass of a monomer based on the amino acid sequence (57 kDa). This analysis demonstrates that the soluble oligomer of vimentin was a tetramer, which corroborates previous biochemical analyses of vimentin oligomers (10, 20). The calculated Mr of mEos-vimentinY117L was 349 kDa, which is 4.4-fold the predicted mass of a mEos-vimentin monomer (80 kDa). This result shows that the soluble mEOS-vimentinY117L was also tetrameric, suggesting that the filament precursors formed by vimentinY117L behave like endogenous vimentin filament precursors.
Table 1.
Hydrodynamic analysis of soluble vimentin
| Protein | Mr aa seq (kDa) | S20,w | Rs (nm) | Calculated Mr (kDa) | Oligo state |
| Vimentin | 57 | 5.6 | 8.9 | 210 | Tetrameric |
| mEos-vimY117L | 80 | 6.8 | 12.2 | 349 | Tetrameric |
The Svedberg coefficient (S20,w) and Stokes radius (Rs) of vimentin and mEos- vimentinY117L were calculated using the equation obtained from fitting curves to calibrations of the sucrose gradient or the gel filtration column using standard proteins. Mr aa seq is the predicted mass of the monomeric protein according to its amino acid sequence. The calculated Mr was obtained using the simplified Siegel–Monte calculation (M = 4,205 S Rs).
Fig. S4.

The soluble oligomer of vimentin is tetrameric. Soluble lysates from SW13 vim+ and SW13 mEos-vimentinY117L were separated by sedimentation velocity ultracentrifugation through a linear 5–20% sucrose gradient (A) or by size-exclusion chromatography using a Superose 6 column (B). The presence of endogenous vimentin (green) and mEos-vimentinY117L (red) were detected by Western blot using a chicken polyclonal vimentin antibody. A set of protein standards (blue) was used to calibrate the sucrose gradient and the Superose column. See Table 1 for Svedberg coefficients and Stokes radius values.
Phosphorylation of the Serine 38 Residue of Vimentin Is Not Required for ULF Subunit Exchange.
Typically, Ser/Thr phosphorylation promotes filament disassembly into ULFs and increases IF protein solubility (27). Because phosphorylation is a transient and ATP-dependent process, it could also play a role in the regulation of the fast subunit exchange that we observed here. To test this possibility, we first wanted to confirm that mEos-vimentinY117L was phosphorylatable by using Phos-tag technology. In SDS/PAGE using Phos-tag acrylamide, phosphorylated proteins bind to Phos-tag sites, retarding their migration and thus separating them from unphosphorylated proteins. As a control, we used SW13 vim+ cells untreated or treated with the phosphatase inhibitor calyculin A. Immunoblot analysis using a vimentin antibody showed the presence of a single band representing unphosphorylated protein without calyculin A in wild-type and ULF vimentin. Treatment of SW13 vim+ cells with the phosphatase inhibitor resulted in at least five bands (Fig. S5A), indicating that vimentin proteins were phosphorylated at different levels on multiple sites in these cells. The same phosphatase inhibition treatment to SW13 cells expressing mEos-vimentinY117L revealed the presence of only one band, which was shifted relative to the unphosphorylated control band, indicating that all of the mEos-vimentinY117L proteins were phosphorylated to the same extent. Next we tested whether the serine 38 phosphorylation site of vimentin regulates subunit exchange. This major phospho-residue of vimentin can be modified by at least six different kinases in vitro (27) and was shown to regulate vimentin filament disassembly in vivo (12, 20). Immunoblotting using phospho-specific serine 38 antibody after cells were treated with calyculin A showed that mEos-vimentinY117L became highly phosphorylated on serine 38 (Fig. S5B), to a greater extent than endogenous vimentin (Fig. S5C). However, photoconversion experiments comparing mEos-vimentinY117L and a nonphosphorylatable serine 38 version of the mEos-vimentinY117L probe (mEos-vimentinY117L/S38A) revealed no difference in the initial rate of subunit exchange. Therefore, the phosphorylation state of serine 38 residue is not involved in the regulation of the ULF subunit exchange (Fig. 7).
Fig. S5.
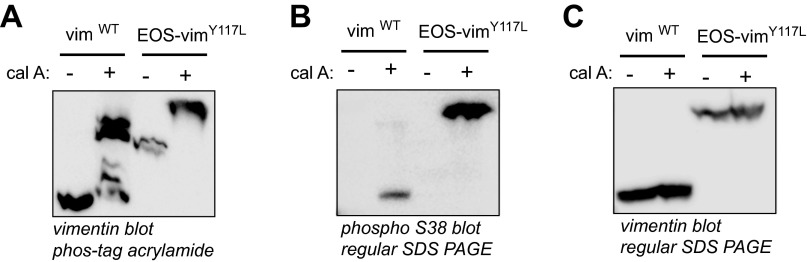
VimentinY117L can be phosphorylated on serine 38. SW13 vim+ and SW13 vim− stably expressing mEos-vimentinY117L were treated or not with the phosphatase inhibitor calyculin A (5 nM) for 30 min to stimulate vimentin phosphorylation. (A) Phosphorylated proteins were separated from unphosphorylated proteins using Phos-tag acrylamide SDS/PAGE. Vimentin immunoblot revealed the presence of at least five different bands of wild-type vimentin after treatment with calyculin A, indicating at least five different phosphorylation states. Only one band was shifted in vimentinY117L by the treatment with calyculin A. The same samples were separated by conventional SDS/PAGE and immunoblot with vimentin serine 38 phospho-specific antibody (B) or total vimentin antibody (C). The results show a robust phosphorylation of mEos-vimentinY117L on serine 38 after calyculin A treatment compared with wild-type vimentin after the same treatment. This result suggests that serine 38 is a major phosphorylation site of ULF.
Fig. 7.

Subunit exchange is independent of vimentin phosphorylation on serine 38. Photoconversion experiments were performed on mEos-vimentinY117L cell versus the nonphosphorylatable mutant mEos-vimentinY117L/S38A. Pictures show ULFs in the red channel, located 5–10 µM from the center of the photoconverted zone, 1 min after photoconversion. Quantification of the initial rate of exchange reveals that phosphorylation at serine 38 was not required for ULF subunit exchange. Measurements were taken in ULFs located in a radial circumference distance of 5–10 µM from the center of the photoconverted area (average ± SEM of at least 450 measurements from 18 cells per condition). (Scale bar, 10 μm.)
Discussion
Intermediate filament networks have always been considered the most stable components of the cytoskeleton. However, live-cell imaging demonstrates that IF networks are in fact quite dynamic and undergo rapid rearrangement in cells (12, 15, 16, 28–31). This reorganization consists of active transport of filaments and filament precursors and also the assembly and disassembly of filaments. Photoconversion approaches were used to demonstrate that although vimentin filaments are rapidly transported along microtubules, the most dominant form of mature filament dynamics occurs by slow severing and reannealing (15, 16). These experiments are consistent with in vitro data demonstrating that vimentin filaments elongate by end-to-end annealing of ULFs and short filaments (7). Because of technical limitations, very little attention has been given to the first steps of VIF polymer assembly in cells. The main reason is because under normal conditions, early filament precursors represent only a small part of the general vimentin pool, whereas most of the protein is incorporated into mature filaments, the most abundant form of vimentin in the cell. To make possible the analysis of the early stages of vimentin filament assembly, we took advantage of the vimentinY117L mutant. This mutant laterally associates into ULFs in vitro but cannot anneal longitudinally to form filaments (18). We showed previously that GFP-tagged vimentinY117L expressed in the absence of endogenous wild-type vimentin forms particles that interact with actin filaments and move along microtubules (19). These particles have uniform fluorescence and most likely represent individual ULFs, although at this point we cannot exclude that they actually represent clusters of several ULFs. In any case, we know that vimentin carrying the Y117L mutation is not denatured and is physiologically active because it can copolymerize into filaments in the presence of wild-type vimentin.
In the present study, we used vimentinY117L tagged with a photoconvertible probe, mEos3.2 or mMaple3. This process allowed us, to our knowledge for the first time, to study subunit exchange within ULF in live cells. Our surprising observation is that, unlike vimentin in mature IFs, vimentin in ULFs was very dynamic. Vimentin in ULFs exchanged with the soluble pool of vimentin tetramers within seconds; the rate of exchange was much faster than the diffusion of the soluble precursors in the cytoplasm. Interestingly, when mEos-vimentinY117L copolymerized with wild-type vimentin into the intermediate filament network, subunit exchange was not discernable even 15 min after photoconversion. This experiment strongly suggests that fast subunit exchange is not because of the Y117L mutation per se, but by the fact that filament assembly is halted at the ULF stage. Additionally, subunit exchange was not noticeable in VIFs containing wild-type mEos-vimentin (Fig. 4). This result is in agreement with the recent demonstration that subunit exchange in mature vimentin filaments is an extremely rare event both in vitro (8) and in vivo (16). Our results do not rule out the possibility that long filaments could incorporate short pieces of filaments or even filament precursors along their length, as suggested previously for vimentin and other IF polymers (22, 32–34), but the dynamics of subunit exchange in ULFs is at least several orders-of-magnitude faster than subunit exchange in mature vimentin filaments.
We previously showed that vimentinY117L forms ULF particles that move along microtubules (19). Therefore, movement of one ULF toward another and fusion of ULF particles can sometimes be observed during rigorous analyses of time-lapse sequences of GFP- or mEos-vimentinY117L. These events could bias the calculation of the initial rate of exchange because fusion of two ULFs would result in a large increase in red fluorescence. However, data presented in this report have shown that subunit exchange between ULFs is the major process responsible for the observed increase in red fluorescence in individual ULFs after photoconversion. First, photoconversion experiments done in the presence of nocodazole demonstrate that the red fluorescence increase in ULF still occurred in absence of microtubules (Fig. 3). In the absence of microtubule tracks, ULFs are stationary, preventing any events of ULF fusion to occur (Fig. S3). In this context, the most likely explanation for the red fluorescence increase is subunit exchange. Second, by using a fusion protein between vimentinY117L and the newly developed photoconvertible protein mMaple3, we were able to measure variation of intensity in both red and green channels. We observed that the increase in the red fluorescence intensity correlated to a decrease in the green fluorescence intensity in individual ULFs (Fig. 2 and Fig. S2), suggesting that the principal event resulting in the increase of red fluorescence intensity is the exchange of red subunits for green. Finally, we showed that the inhibition of subunit exchange caused by ATP depletion correlated with a dramatic reduction in the soluble pool of vimentin tetramers (Figs. 5 and 6). Altogether, these data strongly suggest that ULF particles rapidly exchange vimentin tetramers with the soluble pool.
One difference between ULFs and mature filaments is that ULFs are in equilibrium with the soluble pool of vimentin tetramers (Fig. 6 and Fig. S4). Surprisingly, the existence of this soluble pool (and therefore subunit exchange) requires the presence of ATP. ATP depletion completely blocked subunit exchange, locked vimentin in the ULF form, and depleted the soluble tetramer pool. Using FCS, we measured the diffusion of a residual soluble vimentin complex after ATP depletion and showed that the diffusion coefficient does not change. Therefore, our results cannot be explained by an ATP-dependent random intracellular motion, which is driven by active force fluctuations in the cytoplasm (35). Our results suggest that dissociation of vimentin tetramers from ULFs is an ATP-dependent process.
Reduced vimentin filament reorganization in the absence of ATP has been described previously (36, 37). These studies have shown that the collapse of vimentin filaments around the nucleus in response to microtubule depolymerization depends on ATP-dependent contraction of the acto-myosin cortex. In our case, the ATP-dependent mechanism responsible for subunit exchange is completely different because depolymerization of actin filaments with LatB had no effect on subunit exchange (Fig. 3). There are several potential explanations for the requirement of ATP in subunit exchange. An important aspect of the regulation of IF assembly and disassembly is IF phosphorylation. Because phosphorylation is a transient and ATP-dependent process, vimentin phosphorylation is an attractive candidate for the regulation of the fast subunit exchange. Typically, Ser/Thr phosphorylation of vimentin filaments promotes disassembly into ULFs and increases IF protein solubility (27). Specifically, phosphorylation of the serine 38 residue of vimentin triggers filament disassembly (12, 20). Because we found that this specific residue was highly phosphorylated on mEos-vimentinY117L, we tested the requirement of serine 38 phosphorylation for ULF dynamic exchange by expressing a nonphosphorylatable mutant mEos-vimentinY117L/S38A. Surprisingly, a robust subunit exchange was observed after the stable expression and photoconversion of SW13 cells expressing the phospho-mutant (Fig. 7). Because vimentin contains more than 35 phosphorylation sites in its head and tail domains, which are targeted by multiple kinases and phosphatases, we cannot exclude that phosphorylation at another site could be involved in the regulation of subunit exchange. However, our results indicate that the major phosphorylation site implicated in mature filament disassembly is not involved. Other posttranslational modifications should be considered in future study. Notably, sumoylation of keratin and vimentin regulates their solubility (38), but the effect of vimentin glycosylation remains elusive (39).
Another possible explanation for the ATP requirement in subunit exchange is that the vimentin tetramer forms a complex with an ATP-dependent chaperone to maintain solubility of the tetramer. The association of IF with small heat-shock proteins (sHSPs) and other chaperones was observed in a variety of cells expressing different types of IFs (40–44). A role for this interaction during IF assembly has been suggested because sHSPs were shown to influence IF solubility (42). However, the binding of vimentin and GFAP soluble subunits to sHSPs was demonstrated to be an ATP-independent process (40). Nevertheless, the implication of sHSPs in the maintenance of the soluble pool of vimentin should be considered because sHSPs cooperate with ATP-dependent chaperones and are induced by various stress conditions (see ref. 45 for review).
We propose that each step of VIF self-assembly is regulated in cells by different mechanisms (Fig. 8). We can hypothesize that in response to local signaling events in the cell, the breakdown of filaments by severing occurs through a mechanism still unknown but likely involves hyperphosphorylation of vimentin. After depolymerization to its building blocks, vimentin is maintained in equilibrium between ULFs and soluble tetramers by the local activation of an ATP-dependent cofactor (e.g., a kinase or an ATP-dependent chaperone). This energy-dependent cofactor stimulates the dissociation of the tetramer from ULF, preventing the longitudinal annealing of ULFs into filaments locally during vimentin network reorganization. Ultimately, the inactivation of this cofactor would allow the spontaneous self-assembly capacity of ULFs to anneal into short and longer filaments and reintegrate the filament network.
Fig. 8.

Model of VIF dynamics in cell. We propose that each step of VIF self-assembly is regulated in cell by different mechanisms. The breakdown of filaments by severing occurs through an unknown mechanism that likely involves hyperphosphorylation of vimentin. Vimentin at the ULF level is maintained in equilibrium with the tetrameric form by the local activation of an ATP-dependent cofactor (e.g., a kinase or an ATP-dependent chaperone) that stimulates the dissociation of the tetramer from ULF and locally prevents ULF from the longitudinal annealing that would form filaments during vimentin network reorganization. Ultimately, the inactivation of this cofactor would allow the spontaneous self-assembly capacity of ULFs to anneal into short and longer filaments.
Our study has provided, to our knowledge for the first time, a tool to study the dynamics of the initial step of VIF assembly, which is important during the establishment of a cell-specific filament network and also during filament reorganization in response to many cell-signaling events. The growing list of pathologies associated with IF aggregation reflects the importance of the cell’s ability to keep IF proteins in a soluble state during the proper reorganization of the IF network. We believe that the rapid subunit exchange within ULF that takes place during the first step of vimentin filament assembly is a critical process during physiological and pathological events that change the mechanical properties of the cell. Future work will be necessary to confirm that this mechanism is shared among other types of intermediate filaments.
Methods
Photoconvertible ULF Stable Cell Lines.
mEOS3.2-vimentinY117L or mMaple3-vimentinY117L were cloned into the pQCXIP retroviral vector and stably expressed in either SW13 vim− cells or embryonic fibroblasts derived from vimentin knockout mice (MEF vim KO). See SI Methods for details.
Live-Cell Imaging Microscopy.
For all live-cell experiments, cells were plated on glass coverslips ∼16 h before imaging. Cells were maintained at 37 °C + 5% CO2 during imaging using a Tokai-Hit stage-top incubator (Tokai-Hit) and Okolab gas mixer (Okolab). Live-cell confocal imaging was performed using a Nikon Eclipse U2000 inverted stand with a Yokogawa CSU10 spinning disk confocal head (Yokogawa Electric Corporation), and a 100× 1.40 NA lens. Images were acquired using an Evolve EMCCD (Photometrics) driven by Nikon Elements software. Photoconversion of mEos3.2-ULF or mMaple3-ULF from green to red was performed using illumination from a Heliophor LED light source in the epifluorescence pathway filtered with a 400-nm filter and confined by a diaphragm. Photoconversion time was 3 s and 10 s for mEos3.2 and mMaple3, respectively. The photoconversion zone was ∼10 μm in diameter and it was positioned to avoid the nuclear region, as ULFs are excluded from the nucleus and very few particles are present on the top or underneath the nucleus. Time-lapse sequences were acquired at 15-s intervals for 3 min using the 488- and 561-nm laser. Images were analyzed in Fiji, and assembled in Illustrator.
Live-cell TIRF images were collected on a Nikon Eclipse U2000 inverted microscope equipped with a Plan-Apo TIRF 100× 1.45 NA objective and a Hamamatsu CMOS Orca Flash 4.0 camera (Hamamatsu Photonics), controlled by MetaMorph 7.7.7.0 software (Molecular Devices). The angle of a 561-nm laser was manually adjusted until near total internal reflection was reached as judged by imaging of photoconverted mEos3.2-vimentin–expressing cells. To photoconvert, cells were exposed to UV light from an Hg+ light source for 10 s through a pinhole in the light path. Time-lapse sequences were acquired at 5-min intervals for 15 min using the 561-nm laser. Unless mentioned otherwise in the figure legend, the photoconversion experiments were performed using cell lines stably expressing the different photoconvertible probes (mEos3.2-vimentin, mEos3.2-vimentinY117L, or mMaple3-vimentinY117L).
Quantification of ULF Subunit Exchange.
Before quantifying individual ULF intensities, images in the red channel were bleach-corrected by scaling each image so that its mean was the same as the first image. This is known as ratio bleach correction. To measure relative intensities of individual ULFs, images from the green channel were used to track ULFs with Diatrack software (v3.01; Semasopht). This gave us the coordinates of the center of each ULF in each frame. We used these coordinates to measure the intensity of each ULF in each red frame by calculating the average fluorescence in a circle of fixed size centered at these coordinates. The measured intensities of the ULFs were normalized using an image taken in the red channel before photoconversion. The intensity of each ULF before conversion was subtracted from all its postconversion intensities; this accounted for any background fluorescence contributing to the intensity of the ULFs. To compare between movies, we also accounted for the degree of photoconversion in each cell. We estimated the amount of fluorescence caused by photoconverted proteins by measuring the intensity in the photoconverted region immediately after photoconversion. From this we subtracted the intensity in the same region before photoconversion. Each ULF intensity was then divided by this background corrected intensity of the photoconversion. Once these normalized intensities were computed, we plotted them versus time and calculated the slope over the linear range, corresponding to the first minute after photoconversion, to get the initial rate of exchange. To observe how the distance between a ULF and the converted region impacts the rate of fluorescence increase, ULFs were grouped as a function of their distance from the center of the converted region. Quantification was performed using custom software written in Python available at https://github.com/mollymolly/exchange. This software makes use of the Numpy and Matplotlib libraries (46, 47). All of the statistics were performed with Prism v6.0 (GraphPad software) using a two-tailed nonparametric test (Mann–Whitney test) with a confidence level of 95% (statistical difference P < 0.05).
FCS Measurement.
We performed FCS measurements with an ISS Alba system. The focal volume of the system was calibrated with rhodamine 110 using a 3D Gaussian point spread function model. Global fits were calculated across all measurements of the same type using VistaVision, ISS’s proprietary software (48). Data were fit with a 3D Gaussian excitation model with a single diffusion coefficient:
| [1] |
where G is the amplitude of the correlation, τ is the temporal correlation shift, N is the average number of particles in the effective measurement volume, D is the diffusion coefficient, ωxy is the radial beam waist, and z0 is the axial beam diameter.
For each measurement, a confocal image of the cell was acquired and a measurement location was selected away from ULFs. Each individual measurement was 5-s long and one or two measurements were taken in a cell before moving on to a new cell. Measurements were also taken in various locations within the cells—near the nucleus, near the periphery, and so forth—so the diffusion coefficient would not be influenced by location.
ATP Depletion and mEos-ULF Solubility.
Cells were incubated for 20 min in the presence of 20 mM sodium azide or 10 µM FCCP in PBS supplemented with 1 mM MgCl2 and 0.1 mM CaCl2 (PBS Mg2+Ca2+). As a control, cells were incubated for 20 min in PBS Mg2+Ca2+. For the washout conditions, sodium azide- or FCCP-treated cells were washed three times with PBS and incubated for 1 h at 37 °C in fresh medium. These cells were used for live-cell imaging or biochemistry analysis. For vimentin solubility analysis, the cells were lysed in a buffer containing 0.5% Triton X-100, 150 mM NaCl, 20 mM Tris⋅HCl pH 7.4, 2 mM EGTA, 2 mM EDTA, 1.5 mM sodium vanadate, 1 mM phenylmethyl sulfonylfluoride, and 10 µg/mL chymostatin, leupeptin, and pepstatin. Cell lysates were ultracentrifuged for 30 min at 265,000 × g and the pellet (insoluble fraction) and supernatant (soluble fraction) were denatured by boiling in Laemmli sample buffer.
Gel Filtration and Sucrose Gradient.
The soluble fraction from cells of two near-confluent 10-cm dishes was obtained as described in the previous section. For gel filtration, 500 µL of the soluble fraction was loaded on a Superose 6 column and 30 fractions were collected. For the sucrose density gradient centrifugations, the soluble fraction was loaded onto continuous sucrose gradients (5–20% in the corresponding extraction buffer), layered on top of a 70% sucrose cushion, and centrifuged for 20 h at 210,000 × g. Fractions were collected from the top of the gradient using the piston gradient fractionator (model 152, BioComp). Fractions collected from both techniques were analyzed by Western blot for the presence of vimentin using chicken polyclonal vimentin antibody (PCK-594P, BioLegend). The Stokes radius (Rs) of the soluble endogenous vimentin and mEOS-vimentinY117L were estimated based on the size-exclusion chromatography of standards with known Rs value (blue dextran, 27 nm; bovine thyroglobulin, 8.5 nm; horse spleen apo-ferritin, 6.1 nm; sweet potato β-amylase, 5.4 nm; and rabbit muscle aldolase, 4.8 nm). The sedimentation coefficient (S20,w) of the soluble endogenous vimentin and mEos-vimentinY117L were estimated based on the sedimentation through the sucrose gradient of standards with known S20,w value (Anhydrase, 2.8s; BSA, 4.6 s; and sweet potato β-amylase, 9.2 s). Next, 150–300 µg of proteins standards obtained from Sigma were diluted in extraction buffer before loading on the gel filtration column or sucrose gradient. The mass of the vimentin soluble subunit (calculated Mr) was obtained using the simplification of the Siegel–Monte calculation (M = 4,205 S Rs) as described previously (26).
Phosphorylation of mEos-Vimentin.
Cells untreated or treated with 5 nM calyculin A for 30 min were rinsed in ice-cold PBS supplemented with 10 mM β-glycerophosphate, 5 mM sodium fluoride, and 10 mM sodium pyrophosphate and lysed in laemmli buffer. Laemmli SDS/PAGE was carried out with 6% (wt/vol) acrylamide gels. For Phos-tag SDS/PAGE, 25 µM Phos-tag acrylamide and 50 µM MnCl2 was added to the 6% (wt/vol) resolving gel. After electrophoresis, Phos-tag gels were washed with transfer buffer supplemented with 1 mM EDTA for 10 min with gentle agitation according to the manufacturer’s protocol. Proteins were transfered to PVDF membranes and membranes were probed with chicken polyclonal antivimentin (PCK-594P, BioLegend) or rat monoclonal TM38 antivimentin pSer-38 (49).
SI Methods
DNA Constructs.
The original mEos3.2-vimentin plasmid was a generous gift from Michael Davidson (Florida State University, Tallahassee, FL). Using appropriate PCR-primers, a mEos3.2-vimentin cDNA was generated and cloned with AgeI and BamHI restriction sites into the pQCXIP retroviral vector as described previously (16). The mMaple3-vimentinY117L construct was created in two steps. First, a mMaple3-vimentin construct was created using the InFusion cloning kit (Clontech of Takara-Bio) to place two inserts (mMaple3 and vimentin) into the pQCXIP vector. The mMaple3 insert was created by PCR with the primers 5′-CCG CGG CCG CAC CGG TCG CCA CAA TGG TGA GC-3′ and 5′-AGA TCT GAG TCC GGA CTT ATA GAG TTC GTC CAT GCT GTC GGT GCT GTT CCT-3′ using vimentin-mMaple3 as a template (21). The vimentin insert was created by PCR using the primers 5′-TCC GGA CTC AGA TCT GGC AGC-3′ and 5′-GGG GGC GGA ATT CCG GATC-3′ using pQCXIP Eos3.2-vimentin as a template. The backbone vector, pQCXIP, was linearized with AgeI and BamHI restriction enzymes. The three pieces were fused together (such that mMaple3 was fused to the N terminus of vimentin) per the manufacturer’s instructions. Then, the Y117L-vimentin point mutation was introduced into both constructs using the following primers together with the PfuUltra High-Fidelity DNA Polymerase (Agilent Technologies, Stratagene) according to the protocol described by the manufacturer. Primer sense: 5′-GAC CGC TTC GCC AAC TTG ATC GAC AAG GTG CGC-3′; Primer antisense: 5′-GCG CAC CTT GTC GAT CAA GTT GGC GAA GCG GTC-3′. The S38A-vimentin point mutation was introduced into the mEos3.2-vimentinY117L construct using the following primers. Primer sense: 5′-TCC ACC CGC ACC TAC GCA CTG GGC AGC GCG CTG-3′; Primer antisense: 5′-CAG CGC GCT GCC CAG TGC GTA GGT GCG GGT GGA-3′.
Cell Culture, Transfection, and Stable Cell Lines.
SW13 vim+ and SW13 vim− cells were derived from human adenocarcinoma cells (50) and were maintained in Leibovitz’s L-15 medium supplemented with 10% FBS. The mEos-ULF and mMaple3-ULF SW13 stable cell lines and the mEOS-ULF MEF stable cell line were created by the infection of SW13 vim− cells with retrovirus carrying mEos 3.2-vimentinY117L. HEK 293 FT cells were used as packaging cells and were transfected with the pQCXIP vector containing mEos3.2-vimentinY117L or mMaple3-vimentinY117L and the helper plasmids pVSVG (Clontech) and pCL-Eco (Imgenex) encoding the gag/pol and env proteins required for virus production. Virus-containing medium was collected and filtered 48 and 72 h after the transfection of the packaging cells. Next, 8 µg/mL of polybrene was added to the freshly collected viruses and acceptor cells were incubated for 6 h with this virus/medium/polybrene mixture. Two days later, SW13 or MEF-infected cells were selected using 0.5 µg/mL or 2 µg/mL puromycin for 1 wk and survivor cells were tested for mEos-ULF expression. Transient transfection of SW13 vim+ cells with mEos3.2-vimentinY117L was performed using Xfect transfection reagent (Clontech Lab) according to the manufacturer’s recommendations.
Supplementary Material
Acknowledgments
We thank Dr. Michael Davidson (Florida State University) for the mEos3.2-vimentin cDNA, and Gina Daniel and Greg Smith (Northwestern University) for help with sucrose gradient experiments. This study was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Awards P01GM09697 and R01 GM52111, and American Heart Association Fellowship 13POST16210010 (to A.R.). Fluorescence correlation spectroscopy was performed at the Northwestern University Center for Advanced Microscopy supported by National Cancer Institute CCSG P30 CA060553.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. P.A.C. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1505303112/-/DCSupplemental.
References
- 1.Chung BM, Rotty JD, Coulombe PA. Networking galore: Intermediate filaments and cell migration. Curr Opin Cell Biol. 2013;25(5):600–612. doi: 10.1016/j.ceb.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrmann H, Strelkov SV, Burkhard P, Aebi U. Intermediate filaments: Primary determinants of cell architecture and plasticity. J Clin Invest. 2009;119(7):1772–1783. doi: 10.1172/JCI38214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mücke N, et al. Molecular and biophysical characterization of assembly-starter units of human vimentin. J Mol Biol. 2004;340(1):97–114. doi: 10.1016/j.jmb.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 4.Kirmse R, et al. A quantitative kinetic model for the in vitro assembly of intermediate filaments from tetrameric vimentin. J Biol Chem. 2007;282(25):18563–18572. doi: 10.1074/jbc.M701063200. [DOI] [PubMed] [Google Scholar]
- 5.Herrmann H, Häner M, Brettel M, Ku NO, Aebi U. Characterization of distinct early assembly units of different intermediate filament proteins. J Mol Biol. 1999;286(5):1403–1420. doi: 10.1006/jmbi.1999.2528. [DOI] [PubMed] [Google Scholar]
- 6.Herrmann H, et al. Structure and assembly properties of the intermediate filament protein vimentin: The role of its head, rod and tail domains. J Mol Biol. 1996;264(5):933–953. doi: 10.1006/jmbi.1996.0688. [DOI] [PubMed] [Google Scholar]
- 7.Winheim S, et al. Deconstructing the late phase of vimentin assembly by total internal reflection fluorescence microscopy (TIRFM) PLoS ONE. 2011;6(4):e19202. doi: 10.1371/journal.pone.0019202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nöding B, Herrmann H, Köster S. Direct observation of subunit exchange along mature vimentin intermediate filaments. Biophys J. 2014;107(12):2923–2931. doi: 10.1016/j.bpj.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prahlad V, Yoon M, Moir RD, Vale RD, Goldman RD. Rapid movements of vimentin on microtubule tracks: Kinesin-dependent assembly of intermediate filament networks. J Cell Biol. 1998;143(1):159–170. doi: 10.1083/jcb.143.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soellner P, Quinlan RA, Franke WW. Identification of a distinct soluble subunit of an intermediate filament protein: Tetrameric vimentin from living cells. Proc Natl Acad Sci USA. 1985;82(23):7929–7933. doi: 10.1073/pnas.82.23.7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Windoffer R, Wöll S, Strnad P, Leube RE. Identification of novel principles of keratin filament network turnover in living cells. Mol Biol Cell. 2004;15(5):2436–2448. doi: 10.1091/mbc.E03-09-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Helfand BT, et al. Vimentin organization modulates the formation of lamellipodia. Mol Biol Cell. 2011;22(8):1274–1289. doi: 10.1091/mbc.E10-08-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoon KH, et al. Insights into the dynamic properties of keratin intermediate filaments in living epithelial cells. J Cell Biol. 2001;153(3):503–516. doi: 10.1083/jcb.153.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kölsch A, Windoffer R, Würflinger T, Aach T, Leube RE. The keratin-filament cycle of assembly and disassembly. J Cell Sci. 2010;123(Pt 13):2266–2272. doi: 10.1242/jcs.068080. [DOI] [PubMed] [Google Scholar]
- 15.Uchida A, Çolakoğlu G, Wang L, Monsma PC, Brown A. Severing and end-to-end annealing of neurofilaments in neurons. Proc Natl Acad Sci USA. 2013;110(29):E2696–E2705. doi: 10.1073/pnas.1221835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hookway C, et al. Microtubule-dependent transport and dynamics of vimentin intermediate filaments. Mol Biol Cell. 2015;26(9):1675–1686. doi: 10.1091/mbc.E14-09-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snider NT, Omary MB. Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat Rev Mol Cell Biol. 2014;15(3):163–177. doi: 10.1038/nrm3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meier M, et al. Vimentin coil 1A-A molecular switch involved in the initiation of filament elongation. J Mol Biol. 2009;390(2):245–261. doi: 10.1016/j.jmb.2009.04.067. [DOI] [PubMed] [Google Scholar]
- 19.Robert A, Herrmann H, Davidson MW, Gelfand VI. Microtubule-dependent transport of vimentin filament precursors is regulated by actin and by the concerted action of Rho- and p21-activated kinases. FASEB J. 2014;28(7):2879–2890. doi: 10.1096/fj.14-250019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eriksson JE, et al. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J Cell Sci. 2004;117(Pt 6):919–932. doi: 10.1242/jcs.00906. [DOI] [PubMed] [Google Scholar]
- 21.Wang S, Moffitt JR, Dempsey GT, Xie XS, Zhuang X. Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging. Proc Natl Acad Sci USA. 2014;111(23):8452–8457. doi: 10.1073/pnas.1406593111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colakoğlu G, Brown A. Intermediate filaments exchange subunits along their length and elongate by end-to-end annealing. J Cell Biol. 2009;185(5):769–777. doi: 10.1083/jcb.200809166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bershadsky AD, Gelfand VI. ATP-dependent regulation of cytoplasmic microtubule disassembly. Proc Natl Acad Sci USA. 1981;78(6):3610–3613. doi: 10.1073/pnas.78.6.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elson EL, Magde D. Fluorescence xorrelation spectroscopy. 1. Conceptual basis and theory. Biopolymers. 1974;13(1):1–27. [Google Scholar]
- 25.Haustein E, Schwille P. Single-molecule spectroscopic methods. Curr Opin Struct Biol. 2004;14(5):531–540. doi: 10.1016/j.sbi.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Erickson HP. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol Proced Online. 2009;11:32–51. doi: 10.1007/s12575-009-9008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC. Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res. 2007;313(10):2098–2109. doi: 10.1016/j.yexcr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarke EJ, Allan VJ. Cytokeratin intermediate filament organisation and dynamics in the vegetal cortex of living Xenopus laevis oocytes and eggs. Cell Motil Cytoskeleton. 2003;56(1):13–26. doi: 10.1002/cm.10131. [DOI] [PubMed] [Google Scholar]
- 29.Helfand BT, Chang L, Goldman RD. Intermediate filaments are dynamic and motile elements of cellular architecture. J Cell Sci. 2004;117(Pt 2):133–141. doi: 10.1242/jcs.00936. [DOI] [PubMed] [Google Scholar]
- 30.Leube RE, Moch M, Kölsch A, Windoffer R. “Panta rhei”: Perpetual cycling of the keratin cytoskeleton. BioArchitecture. 2011;1(1):39–44. doi: 10.4161/bioa.1.1.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moch M, Herberich G, Aach T, Leube RE, Windoffer R. Measuring the regulation of keratin filament network dynamics. Proc Natl Acad Sci USA. 2013;110(26):10664–10669. doi: 10.1073/pnas.1306020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ngai J, Coleman TR, Lazarides E. Localization of newly synthesized vimentin subunits reveals a novel mechanism of intermediate filament assembly. Cell. 1990;60(3):415–427. doi: 10.1016/0092-8674(90)90593-4. [DOI] [PubMed] [Google Scholar]
- 33.Vikstrom KL, Lim SS, Goldman RD, Borisy GG. Steady state dynamics of intermediate filament networks. J Cell Biol. 1992;118(1):121–129. doi: 10.1083/jcb.118.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon M, Moir RD, Prahlad V, Goldman RD. Motile properties of vimentin intermediate filament networks in living cells. J Cell Biol. 1998;143(1):147–157. doi: 10.1083/jcb.143.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo M, et al. Probing the stochastic, motor-driven properties of the cytoplasm using force spectrum microscopy. Cell. 2014;158(4):822–832. doi: 10.1016/j.cell.2014.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hollenbeck PJ, Bershadsky AD, Pletjushkina OY, Tint IS, Vasiliev JM. Intermediate filament collapse is an ATP-dependent and actin-dependent process. J Cell Sci. 1989;92(Pt 4):621–631. doi: 10.1242/jcs.92.4.621. [DOI] [PubMed] [Google Scholar]
- 37.Tint IS, Hollenbeck PJ, Verkhovsky AB, Surgucheva IG, Bershadsky AD. Evidence that intermediate filament reorganization is induced by ATP-dependent contraction of the actomyosin cortex in permeabilized fibroblasts. J Cell Sci. 1991;98(Pt 3):375–384. doi: 10.1242/jcs.98.3.375. [DOI] [PubMed] [Google Scholar]
- 38.Snider NT, Weerasinghe SV, Iñiguez-Lluhí JA, Herrmann H, Omary MB. Keratin hypersumoylation alters filament dynamics and is a marker for human liver disease and keratin mutation. J Biol Chem. 2011;286(3):2273–2284. doi: 10.1074/jbc.M110.171314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slawson C, Lakshmanan T, Knapp S, Hart GW. A mitotic GlcNAcylation/phosphorylation signaling complex alters the posttranslational state of the cytoskeletal protein vimentin. Mol Biol Cell. 2008;19(10):4130–4140. doi: 10.1091/mbc.E07-11-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicholl ID, Quinlan RA. Chaperone activity of alpha-crystallins modulates intermediate filament assembly. EMBO J. 1994;13(4):945–953. doi: 10.1002/j.1460-2075.1994.tb06339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wisniewski T, Goldman JE. Alpha B-crystallin is associated with intermediate filaments in astrocytoma cells. Neurochem Res. 1998;23(3):385–392. doi: 10.1023/a:1022465702518. [DOI] [PubMed] [Google Scholar]
- 42.Perng MD, et al. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci. 1999;112(Pt 13):2099–2112. doi: 10.1242/jcs.112.13.2099. [DOI] [PubMed] [Google Scholar]
- 43.Planko L, et al. Identification of a keratin-associated protein with a putative role in vesicle transport. Eur J Cell Biol. 2007;86(11-12):827–839. doi: 10.1016/j.ejcb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 44.Kayser J, et al. The small heat shock protein Hsp27 affects assembly dynamics and structure of keratin intermediate filament networks. Biophys J. 2013;105(8):1778–1785. doi: 10.1016/j.bpj.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: The structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12(10):842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- 46.Hunter JD. Matplotlib: A 2D graphics environment. Comput Sci Eng. 2007;9(3):90–95. [Google Scholar]
- 47.Oliphant TE. Python for scientific computing. Comput Sci Eng. 2007;9(3):10–20. [Google Scholar]
- 48.Beechem JM. A second generation global analysis program for the recovery of complex inhomogeneous fluorescence decay kinetics. Chem Phys Lipids. 1989;50(3-4):237–251. doi: 10.1016/0009-3084(89)90052-2. [DOI] [PubMed] [Google Scholar]
- 49.Kosako H, et al. Specific accumulation of Rho-associated kinase at the cleavage furrow during cytokinesis: Cleavage furrow-specific phosphorylation of intermediate filaments. Oncogene. 1999;18(17):2783–2788. doi: 10.1038/sj.onc.1202633. [DOI] [PubMed] [Google Scholar]
- 50.Sarria AJ, Nordeen SK, Evans RM. Regulated expression of vimentin cDNA in cells in the presence and absence of a preexisting vimentin filament network. J Cell Biol. 1990;111(2):553–565. doi: 10.1083/jcb.111.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.