

Significance
Accumulation of lipofuscin in the retinal pigment epithelium precedes retinal degenerations and dystrophies responsible for blindness-causing retinal diseases. The mechanism behind lipofuscin formation in the retina or in any tissue is poorly understood. Here we show in mice that the dimerization of vitamin A is responsible for triggering the formation of more than 50% of ocular lipofuscin. Replacing three hydrogen atoms on vitamin A with deuterium inhibits vitamin A dimerization, resulting in reduced lipofuscin and transcriptional normalization of genes associated with inflammation without compromising retinal function. Thus, vitamin A deuterated at the carbon 20 position provides a clinically amiable tool to prevent vitamin A dimerization in humans to assess whether impeding such dimerization might prevent retinal degenerations such as Stargardt disease and age-related macular degeneration.
Keywords: C20-D3-vitamin A, age-related macular degeneration, bisretinoid, A2E, ALK-001
Abstract
Stargardt disease, an ATP-binding cassette, subfamily A, member 4 (ABCA4)-related retinopathy, is a genetic condition characterized by the accelerated accumulation of lipofuscin in the retinal pigment epithelium, degeneration of the neuroretina, and loss of vision. No approved treatment exists. Here, using a murine model of Stargardt disease, we show that the propensity of vitamin A to dimerize is responsible for triggering the formation of the majority of lipofuscin and transcriptional dysregulation of genes associated with inflammation. Data further demonstrate that replacing vitamin A with vitamin A deuterated at the carbon 20 position (C20-D3-vitamin A) impedes the dimerization rate of vitamin A—by approximately fivefold for the vitamin A dimer A2E—and subsequent lipofuscinogenesis and normalizes the aberrant transcription of complement genes without impairing retinal function. Phenotypic rescue by C20-D3-vitamin A was also observed noninvasively by quantitative autofluorescence, an imaging technique used clinically, in as little as 3 months after the initiation of treatment, whereas upon interruption of treatment, the age-related increase in autofluorescence resumed. Data suggest that C20-D3-vitamin A is a clinically amiable tool to inhibit vitamin A dimerization, which can be used to determine whether slowing the dimerization of vitamin A can prevent vision loss caused by Stargardt disease and other retinopathies associated with the accumulation of lipofuscin in the retina.
Stargardt disease, first described in 1909, is an autosomal recessive macular dystrophy affecting ∼1 in 10,000 people. The majority of people affected by the disease present with uncorrectable, decreased visual acuity during their teenage years, which most often progresses to legal blindness. To date, there is no approved intervention. Stargardt disease is marked by premature accumulation of lipofuscin in the retinal pigment epithelium (RPE), degeneration of the neuroretina, and subsequent loss of vision. The condition results from mutations in the ATP-binding cassette, subfamily A, member 4 (ABCA4) gene (1), which encodes a transmembrane flippase localized in photoreceptor outer segments. The flippase transports the phosphatidyl-ethanolamine-retinaldehyde Schiff base between the cytosol and the cytoplasmic disk surfaces (2). Mutations in ABCA4 also result in retinitis pigmentosa and cone-rod dystrophy and have been linked to age-related macular degeneration (AMD) (3, 4).
The accumulation of lipofuscin in the RPE is a common denominator in retinopathies associated with mutations in the ABCA4 gene. Also known as “wear and tear pigment,” lipofuscin accumulates as a byproduct of cumulative damage during aging. The brown-yellow, autofluorescent, electron-dense material is also found in cells of the liver, kidney, heart muscle, adrenals, nerve, and ganglion and is considered one of the most consistent morphologic features of aging with a rate of accumulation inversely related to longevity (5, 6). In Stargardt disease, changes in RPE lipofuscin-related autofluorescence (AF) signals are present in areas of imminent functional loss (7). The RPE serves as the outer blood–retinal barrier. The cell layer is responsible for the uptake of retinol from the circulatory system, converting retinol to 11-cis-retinal, and transporting 11-cis-retinal to the photoreceptors for photon recognition in a process called the vitamin A cycle; recycling of photoreceptor outer segments; and transporting ions, glucose, metabolites, and fluid to maintain the milieu of the neuronal retina. RPE senescence is thought to lead to neuroretina degeneration and vision loss (8).
How RPE lipofuscin forms is unknown. The retina ranks among the highest energy-consuming systems (9). Modulating energy use by depriving the eye of vitamin A (10), inhibiting enzymes involved in the vitamin A cycle (11, 12), or regulating photon catch (10) each result in a decrease in RPE lipofuscin. Modulating energy use leads to changes in the levels of intermediates of the vitamin A cycle (retinaldehyde, retinol, retinyl esters), changes in rhodopsin signaling and activation, changes in retinal energetics (glucose, high-energy phosphates, lipid profiles), and changes in cellular byproducts (reactive oxygen species, dimerized vitamin A, glycation endproducts, etc.). Many of these biochemical events have been proposed to contribute to RPE senescence and the formation of RPE lipofuscin (5, 6). As such, the contribution of any specific biochemical event to retinal lipofuscinogenesis remains elusive. Elucidating the minor and major drivers of lipofuscin generation is a main challenge toward better understanding the etiology of Stargardt disease and retinopathies associated with RPE lipofuscin accumulation.
Using an albino mouse model of Stargardt disease, it was shown that replacement of dietary vitamin A with vitamin A deuterated at the carbon 20 position (C20-D3-vitamin A) from birth to 1 y of age resulted in an 80% decrease in the vitamin A dimer, A2E, a significant decrease in RPE lipofuscin, and preservation of retinal function compared with control animals fed diets containing vitamin A (13). However, in humans, replacing vitamin A with C20-D3-vitamin A from birth is challenging because Stargardt disease is most often diagnosed after the onset of symptoms, usually during one’s teenage years. In addition, RPE lipofuscinogenesis is thought to involve melanin (14), which the albino mice lack. Albino Stargardt animals, and animals lacking melanin in general, also tend to be more susceptible to retinal degeneration compared with pigmented animals (15, 16). This suggests that melanin plays a role in retinal homeostasis, which was not modeled in albino mice.
Here, using pigmented mice, we used C20-D3-vitamin A as a tool to demonstrate that the dimerization of vitamin A contributes to the majority (∼50%) of lipofuscin in the RPE. We further show that the dimerization of vitamin A is a continuous pathologic event. Replacement of vitamin A with C20-D3-vitamin A at any point during the disease process prevents the dimerization of vitamin A, and thus the formation of lipofuscin without adverse effects on the retina electrophysiologic function. Data suggest that C20-D3-vitamin A might be used clinically to prevent vitamin A dimerization and subsequent lipofuscin accumulation in Stargardt disease and other retinopathies characterized by lipofuscin accumulation, and that disease progression can be monitored clinically by fundus AF.
Results
Fundus AF.
The mouse model for Abca4-related retinopathy has a neo cassette replacing the promoter and exon 1 of the Abca4 gene, thus abolishing gene expression (17). Similar to humans, Abca4 knockout (Abca4−/−) mice exhibit increased rates of vitamin A dimerization and lipofuscin accumulation compared with their wild-type counterparts (17, 18). Administration of C20-D3-vitamin A has been shown to inhibit vitamin A dimerization (19). Thus, to determine whether vitamin A dimerization was responsible for RPE lipofuscin formation, we administered a standard rodent diet to a cohort of Abca4−/− mice (control group) and compared signatures of retinal health with another cohort, in which we replaced vitamin A in the standard diet by the same amount of C20-D3-vitamin A (treatment group). Fundus AF intensity resulting from 488-nm excitation measured in vivo was used as a biomarker of lipofuscin content in the RPE. AF intensity with 790-nm excitation was also assessed, as it has been suggested to increase with melanolipofuscin formation (18). In vivo fundus AF resulting from 488-nm excitation was 89% greater in 9-mo-old Abca4−/− mice administered vitamin A at natural abundance compared with 9-mo-old Abca4−/− mice given C20-D3-vitamin A (AF intensity = 108 ± 14 control vs. 57 ± 5 treated; P < 0.05; Fig. 1 A and B). Likewise, AF from 790-nm excitation was 33% greater in the Abca4−/− control animals compared with the Abca4−/− C20-D3-vitamin A-treated animals (64 ± 9 control vs. 48 ± 2 treated; P < 0.05; Fig. 1 C and D).
Fig. 1.

C20-D3-vitamin administration reduces lipofuscin-related fundus AF. Representative fundus AF images taken with 488-nm (A) or 790-nm (C) excitation from 9-mo-old wild-type (WT) and Abca4−/− mice. Animals were fed with either a standard diet (control) or a diet in which vitamin A was replaced by C20-D3-vitamin A from weaning (treated). The bar charts illustrate average fundus AF intensities at 488 nm (B) and 790 nm (D), with SDs for each cohort of animals (n = 10 eyes of 10 animals). Unless noted with a bar, asterisks represent significant deviations (P < 0.05) from WT control animals fed a standard diet.
For additional comparison, we administered diets containing vitamin A or C20-D3-vitamin A to wild-type (Abca4+/+) animals. Abca4−/− animals administered vitamin A had 130% higher 488-nm AF and 88% higher 790-nm AF compared with wild-type animals given the same diet, confirming that the increase in 488-nm and 790-nm AF was caused by the Abca4 mutation. Treating Abca4−/− mice with C20-D3-vitamin A reduced both 488-nm and 790-nm AF levels to near those of the wild-type mice raised on normal vitamin A, demonstrating a correction of the AF-associated phenotype. In addition, treating wild-type mice with C20-D3-vitamin A reduced 488-AF by 28%, but had negligible effect on 790-nm AF.
Lipofuscin Granules.
To demonstrate that the reduction in AF levels correlated with a decrease in the RPE lipofuscin granule content, we used transmission electron microscopy to survey the RPE layer of the 9-mo-old Abca4−/− and wild-type animals administered the C20-D3-vitamin A-replaced or standard diet. Masked quantification of electron-dense lipofuscin granules was done using particle analysis software. For each eye, electron-dense, irregular, and/or granular bodies were quantified in 12 serial sections, sliced 100 nm apart (Fig. 2 A and B and SI Appendix, Fig. S1). Abca4−/− animals fed the standard diet had 3.6 times more lipofuscin granules compared with wild-type animals given the same diet (Fig. 2C and SI Appendix, Table S1), in accordance with the measured increase of 488-nm AF. When Abca4−/− animals were administered C20-D3-vitamin A, a 50% reduction in the lipofuscin granule content was observed compared with when the animals were administered vitamin A, again in accord with 488-nm AF measurements (Fig. 2C). C20-D3-vitamin A similarly reduced the amount of lipofuscin granules in wild-type animals by 22% compared with controls at 9 mo, although not with statistical significance (P > 0.05).
Fig. 2.

Inhibiting vitamin A dimerization prevents lipofuscin granule accumulation in the RPE. Representative transmission electron micrographs of a RPE cell taken from 9-mo-old Abca4−/− mutant mice administered either vitamin A (control, A) or C20-D3-vitamin A mice (treated, B). Lipofuscin granules were reconstructed from 20 serial sections taken at 100-nm intervals and overlaid in red on a representative section of the RPE shown in light gray. (C) Average lipofuscin volume per cytoplasmic volume, with 95% confidence intervals from animals described in A and B. −/−, Abca4−/− mutant mice; WT, wild-type, Abca4+/+, controls. Unless noted with a bar, asterisks represent significant deviations (P < 0.05, as determined by one-way analysis of variance) from wild-type animals given vitamin A (WT Control). (Scale bar, approximately 1 μm.)
Vitamin A Dimerization.
After determining that administering C20-D3-vitamin A could reduce RPE lipofuscin, we investigated whether reductions in lipofuscin correlated with amounts of vitamin A-derived ocular pigments. Organic extracts of eyes from 9-mo-old Abca4−/− mice administered a standard diet were dark orange in color (Fig. 3A). Ultra performance liquid chromatography (UPLC) analysis of the extracts with detection at long-wavelength absorption (445 nm) identified multiple orange pigments, two of which identified as the A2E and iso-A2E vitamin A dimers (Fig. 3B). In contrast, eye extracts from wild-type mice given the standard diet were a faint orange color, and although the number of pigments separated by UPLC were comparable (Fig. 3B), their magnitude was reduced by 89% compared with that of Abca4−/− mutant mice, indicating that the increased concentration of ocular pigments was a consequence of the Abca4 defect (Fig. 3C). Eye extracts from the treated Abca4−/− mice (Fig. 3A) were essentially similar in color to extracts of wild-type mice, and total ocular pigment content was decreased by 78% compared with the Abca4−/− animals fed vitamin A at natural abundance (Fig. 3C). C20-D3-vitamin A administered to affected animals normalized the concentration of A2E and other ocular pigments closer to wild-type levels, in accordance with 488-nm AF, suggesting that C20-D3-vitamin A normalized the rate of vitamin A dimerization in the genetically affected animals. Interestingly, the C20-D3-vitamin A treatment also reduced ocular pigments in wild-type mice by 67% compared with wild-type mice given a standard diet. The levels of A2E vitamin A dimer followed a similar trend as the levels of orange pigments (Fig. 3C).
Fig. 3.

C20-D3-vitamin A administration prevents vitamin A dimerization. (A) Alcohol extracts of retinas from 9-mo-old, Abca4−/− mutant mice administered vitamin A (control) or C20-D3-vitamin A from weaning (treated). Extracts from control eyes are less orange in color. (B) Representative UPLC chromatograms of eye extracts from A, along with that from wild-type (WT) mice administered vitamin A (control). Each peak represents an orange ocular pigment derived from vitamin A. (C) Average amounts of total orange ocular pigments and the A2E vitamin A dimer with SDs from the 9-mo-old animals described in the text, as determined by UPLC. Unless noted with a bar, asterisks represent significant deviations (P < 0.05) from wild-type animals given vitamin A (WT Control). (D) Average amounts of orange ocular pigment and SDs measured over time in the above groups of animals. (E) Average amounts of the A2E vitamin A dimer with SDs measured over time in the above groups of animals. Ten left eyes were averaged for each bar or point in the above panels (n = 10). −/−, Abca4−/− mutant mice; RAU, relative absorbance units; WT, wild-type.
To evaluate the rates of accumulation of ocular pigments, we quantified the total amounts of A2E and orange ocular pigments in 3-, 6-, and 9-mo-old mice. Data revealed that ocular pigments and A2E increased with age (Fig. 3 D and E). In Abca4−/− animals administered vitamin A, orange pigments accumulated ∼10 times faster than in wild-type animals receiving vitamin A at natural abundance. When administered C20-D3-vitamin A, accumulation rates were four times slower (five times for A2E) compared with accumulation rates in the same animals given vitamin A. A similar trend was observed for wild-type animals, in which the accumulation rate of orange pigments was attenuated by about six times on C20-D3-vitamin A supplementation.
Modulation of Complement.
After establishing that administration of C20-D3-vitamin A to Abca4−/− animals normalized vitamin A dimerization rates and the rate of lipofuscin accumulation to that seen in wild-type animals, we investigated whether transcription of genes encoding for components of the complement pathway, including complement substrates [complement component C3 (C3), complement factor B (Cfb)], soluble [complement factor h (Cfh), complement factor properdin (Cfp)], and cell surface [cluster of differentiation 59 (Cd59)] complement regulators, were altered. Transcription of C3 was up-regulated by 2.2-fold in Abca4−/− mice compared with wild-type controls (Fig. 4). In contrast, mRNA levels for Cfp and Cfb were both down-regulated by 2.3-fold and twofold, respectively, in the Abca4−/− mice compared with wild-type controls. Taken together, data suggest that the Abca4−/− mutation altered complement status. Administration of C20-D3-vitamin A to Abca4−/− mice reversed the above Abca4−/− mutation-related changes in DNA transcription such that mRNA levels of all three genes were now not statistically different between the Abca4−/−-treated mutants and the wild-type control animals. Administration of C20-D3-vitamin A to wild-type animals further shifted mRNA expression in the opposite direction of dysregulated mRNA levels of the Abca4−/− mice given a normal diet. There was no statistically significant difference in mRNA levels for Cfh (SI Appendix, Fig. S2). As an incidental finding, mRNA levels for the complement inhibitor Cd59 were up-regulated in both wild-type and Abca4−/− animals administered C20-D3-vitamin A compared with the same animals administered vitamin A (SI Appendix, Fig. S2).
Fig. 4.
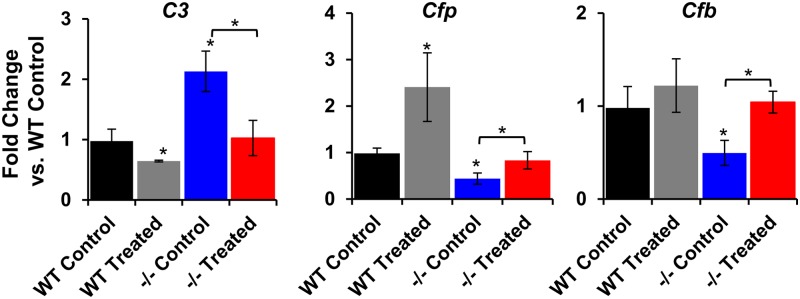
C20-D3-vitamin normalizes transcription of complement genes. Quantification of mRNA from 9-mo-old wild-type and Abca4−/− mice, administered either vitamin A (control) or C20-D3-vitamin A from weaning (treated). To generate each bar, total RNA from 5–10 eyes was extracted. −/−, Abca4−/− mutant mice; WT, wild-type. Unless noted with a bar, asterisks represent significant deviations (P < 0.05 by two-tailed, unpaired t test) from wild-type animals given vitamin A (black bars).
Electroretinography.
To assess any potentially adverse effects on retinal function resulting from long-term supplementation of C20-D3-vitamin A, we used electroretinography (ERG). Dark-adapted a- and b-waves (scotopic function; Fig. 5A) and light-adapted single flashes at 0.5 and 1 log cd·s/m2 and 20-Hz flicker responses (photopic function; Fig. 5B) were indistinguishable between 9-mo-old wild-type and Abca4−/− mice administered a normal diet, confirming that wild-type and Abca4−/− knockout mice are indistinguishable by electrophysiology at 9 mo (18). As expected, Abca4−/− mice administered C20-D3-vitamin A or vitamin A for 9 mo had identical dark-adapted a- and b-waves (Fig. 5A) and light-adapted single-flash ERGs at 0.5 and 1 log cd·s/m2, as well as 20-Hz flicker responses (Fig. 5B), demonstrating that no adverse events in electrophysiologic function resulted from long-term inhibition of dimerization with C20-D3-vitamin A. Likewise, wild-type mice administered C20-D3-vitamin A for 9 mo had identical ERG parameters (SI Appendix, Fig. S3).
Fig. 5.
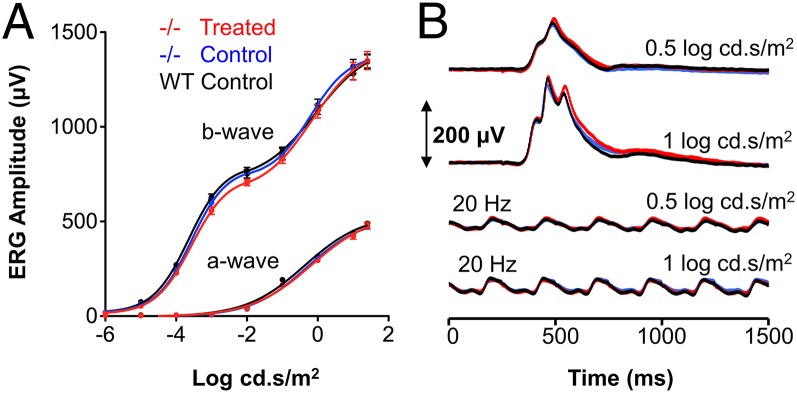
Long-term inhibition of vitamin A dimerization by C20-D3-vitamin A does not compromise retinal function. (A) Average dark-adapted a- and b-wave amplitudes in response to flashes of light of increasing intensity. (B) Average light-adapted, b-wave amplitudes with amplitudes elicited by light flickering at 20 Hz. Mice were given the control or C20-D3-vitamin A (treated) diet from weaning to 9 mo of age. All curves represent averages and SDs for 10 eyes (n = 10). −/−, Abca4−/− mutant mice; WT: wild-type. All ERG responses were identical between the three cohorts.
Variable Age at Treatment Initiation.
In a clinical setting, treatment is usually initiated on presentation of symptoms. To investigate the effects of treatment starting times, we administered C20-D3-vitamin A to Abca4−/− mice, starting from various ages [weaning (21 d) and 1.5, 3, and 6 mo of age], and measured AF signals at various points until the cohorts reached 9 mo of age (Fig. 6A). To one cohort, we administered the diet containing C20-D3-vitamin A from weaning to 3 mo of age and then switched back to normal vitamin A for the remaining 6 mo of treatment (Fig. 6A).
Fig. 6.

C20-D3-vitamin administration rapidly prevents vitamin A dimerization and lipofuscin accumulation. (A) Abca4−/− mutant mice were divided into six experimental cohorts, and each cohort was administered a diet containing C20-D3-vitamin A (treated) or vitamin A (control) over time, according to the panel. (B) Relative average 488-nm fundus AF intensities with SDs from animals described in A. Ten eyes were averaged for each data point (n = 10). The red lines correspond to the periods during which animals were administered C20-D3-vitamin A, as shown in A, whereas the blue lines correspond to the control diet. Treatment crossovers are highlighted by black arrows. Data were normalized so that untreated controls had a relative AF intensity of 1 at 3 mo. Data points at weaning and 1.5 mo are extrapolated from historical controls. (C) Relative average fundus fluorescence intensities with SDs, at 9 mo of age, at the conclusion of treatment regimens described in A and B. (D) Average amounts of total orange ocular pigments and of the A2E dimer, with SDs, in 9-mo-old animals, at the end of treatment, as determined by UPLC. *P < 0.05. P values compare significant deviations from wild-type animals fed vitamin A.
In all groups, 488-nm AF increased with age (Fig. 6B). However, there was a strong correlation between 488-nm AF intensity at 9 mo and duration of C20-D3-vitamin A administration: The longer the animals consumed C20-D3-vitamin A, the lower the 488-nm AF levels. Nine-month-old Abca4−/− mice administered C20-D3-vitamin A from weaning had the least intense AF, similar to age-matched wild-type mice reared on vitamin A throughout the 9-mo period, whereas 9-mo-old Abca4−/− mice given vitamin A had the most intense 488-nm AF, confirming data presented in Fig. 1 generated with different cohorts of animals. Nine-month-old mice administered C20-D3-vitamin A from 6, 3, and 1.5 mo of age showed progressively less intense 488-nm AF respectively compared with 9-mo-old mice administered vitamin A. Surprisingly, changes in AF from administering C20-D3-vitamin A were measurable in as little as 3 mo, irrespective of when the treatment was initiated. When mice were crossed over from the C20-D3-vitamin A diet back to normal vitamin A, 488-nm AF levels increased, which was also apparent within 3 mo. The above changes in 488-nm AF levels were mirrored by the concentrations of A2E and other orange ocular pigments, as presented in Fig. 6 C and D.
Discussion
Along with A2E (20–23), other dimers of vitamin A (24, 25), as well as higher-order vitamin A adducts (26) and other uncharacterized orange pigments, have been identified in lipofuscin granules and/or RPE extracts. Here, knocking out the Abca4 gene correlated with an increase in all orange pigments. Whether such pigments are primary insults that trigger lipofuscin formation or secondary symptoms of the aging retina is not known. Using C20-D3-vitamin A, we were able to impede the nonenzymatic Schiff base-mediated dimerization of vitamin A in pigmented mice, resulting in a parallel decrease in all orange pigments and in the amount of lipofuscin granules. Because only dimerization requires cleavage of carbon hydrogen bonds at carbon number 20 of vitamin A, the kinetic isotope effects resulting from the hydrogen/deuterium exchange are selective for the rate of vitamin A dimerization. With this tool, we showed that at least 50% of the amount of RPE lipofuscin at 9 mo (by counting lipofuscin granules and confirming by quantitative 488-nmAF) can be attributed to the propensity of vitamin A to dimerize. As lipofuscin accumulation is a hallmark of RPE senescence, this work suggests that vitamin A dimerization is a major chemical event responsible for RPE senescence and that inhibiting dimerization may impede RPE senescence and related retinopathies. C20-D3-vitamin A slows the rate of vitamin A dimerization by as much as sevenfold in test tubes (19), and the rate of accumulation of orange ocular pigments and A2E by four- to sixfold in animals, but does not entirely halt the dimerization. As such, the remaining lipofuscin may result from residual dimerization of vitamin A or from other, poorly understood events that may also be responsible for lipofuscinogenesis in other tissues.
Changes in the transcription of complement genes suggest that the Abca4−/− mutation provides an environment leading to persistent retinal inflammatory dysregulation. Along with lipofuscin accumulation, dysregulated inflammation is a hallmark of retinal senescence (27). The complement system comprises ∼20 proteins synthesized by the liver and the RPE (28). Chronic activation of complement is thought to play a role in the development and progression of retinopathies, such as AMD (29). For example, complement proteins such as C3 (30), CFB (31), and CFP (32), all dysregulated here in the mutant animals, are found in histologic specimens of AMD eyes, whereas variants of several genes encoding complement proteins are associated with a modulated risk to develop AMD. To date, the triggers or immunogens for complement activation are not known. However, using in vitro and animal models, several groups have shown that dimers of vitamin A, such as A2E, may activate complement (33–35). Our data demonstrate that inhibiting dimerization by C20-D3-vitamin A normalizes the transcriptional dysregulation of complement components. Interestingly, transcription of CD59, an inhibitor of the complement whose down-regulation may be involved in choroidal neovascularization (36), was up-regulated in both wild-type and mutant animals upon inhibiting the dimerization of vitamin A. Accordingly, the continuous formation of vitamin A dimers may provide immunogens resulting in chronic activation of complement. Taken together, the data suggest that the complement system may play a role in the severity of Stargardt disease.
The pigmented Abca4−/− mice used here start showing signs of electrophysiologic degeneration resulting from the Abca4 mutation after about 18 mo of age (18). Thus, interrogation by ERG, before any anticipated functional declines, provides a convenient means to evaluate long-term retinal safety of inhibiting vitamin A dimerization. Our data show that long-term inhibition of vitamin A dimerization does not result in retinal degeneration, and demonstrate that the nonenzymatic dimerization of the vitamin provides no measurable benefit, contrary to what has been suggested (37). The presented ERG data further reveal that vitamin A dimerization is an early event in disease etiology and triggers lipofuscin accumulation and transcriptional dysregulation of complement before electrophysiologic functional declines can be measured.
To date, other approaches to prevent the dimerization of vitamin A all rely on depriving the photoreceptors of vitamin A. These include inhibiting retinal pigment epithelium-specific 65-kDa protein (RPE65), retinol-binding-protein (RBP), or introducing molecules, such as foreign amines, into the eye to react with retinaldehyde. A necessary consequence of depriving the photoreceptors of vitamin A is rod-mediated or peripheral visual dysfunction (38, 39). However, patients with ABCA4-related retinopathies first lose central vision and rely on peripheral vision. Thus, worsening of the remaining peripheral vision in hopes of halting dwindling central vision is a challenging trade-off. Perhaps more important, depriving the photoreceptors of vitamin A may result in constitutive opsin signaling and retinal degeneration (40, 41). This is evidenced in chronic vitamin A deficiency in retinal dystrophies such as Leber congenital amaurosis, caused by mutations in genes involved in the vitamin A cycle and mutations in RBP, which result in diminished delivery of vitamin A to the eye and lead to retinal atrophy (42). In contrast, we could not identify any adverse effect on photopic and scotopic function resulting from long-term inhibition of vitamin A dimerization through prolonged administration of C20-D3-vitamin A.
Data also demonstrate that 488-nm AF can be used to evaluate interventions that attempt to modulate RPE lipofuscin. Quantitative AF is a noninvasive imaging modality that is becoming more accessible in clinical practice. Depending on the excitation wavelength, fundus AF derives from RPE lipofuscin or melanin/melanolipofuscin, both of which may be indicative of RPE health. In prior work, administration of C20-D3-vitamin A to albino Stargardt mice from birth resulted in reduced RPE AF, as measured ex vivo (13). Here, in pigmented mice, we detected in vivo, dynamic changes in AF in as little as 3 mo after the start or the stoppage of administration of C20-D3-vitamin A. The observed therapeutic rescue of Abca4-triggered increases in AF was verified through quantifying vitamin A dimers and lipofuscin granules. Of note was the increase in AF when treated mice were switched back to normal vitamin A, indicating that the disease process was not halted and that continuous treatment with C20-D3-vitamin A is required to prevent vitamin A dimerization in the eye. These relatively rapid phenotypic changes are in agreement with swine models, which showed that the vitamin A pool in the retina can be replaced with C20-D3-vitamin A within a couple of weeks (43). Taken together, the data provide a case for the use of quantitative AF as a biomarker to evaluate the disease progression and potential treatment effects in lipofuscin-associated retinopathies.
Preventative interventions should be safe, well-tolerated, and convenient for wide-scale implementation. As most lipofuscin-related retinopathies progress slowly, a modest inhibition of vitamin A dimerization might be sufficient to delay onset of vision loss by decades. C20-D3-vitamin A is a precise tool to prevent the dimerization of vitamin A without compromising retinal function. Such a tool may be used to evaluate whether impeding vitamin A dimerization benefits patients affected with Stargardt disease, age-related macular degeneration, or other retinopathies characterized by lipofuscin accumulation in the RPE.
Materials and Methods
Mice.
Pigmented, Abca4−/− mice (129S4/SvJae-ABCA4tm1Ght) were provided by Gabriel Travis (David Geffen School of Medicine, University of California, Los Angeles, CA) (14) and housed in the Biomedical Sciences Division, University of Oxford, United Kingdom. Pigmented wild-type control mice (129S2/SvHsd) were purchased from Harlan Laboratories. All experiments were conducted in female mice. Animals were kept in a 12-h light (∼100 lx)/dark cycle, with food and water available ad libitum. All procedures regarding the use of animals were approved by local and national ethical and legal authorities.
Treatment.
C20-D3-retinyl acetate (ALK-001) was provided by Alkeus Pharmaceuticals, blended with olive oil, and added to a grain-based diet at 15,000 IU/kg diet otherwise deficient in vitamin A. The control diet was the same grain-based diet but with the addition of nondeuterated retinyl acetate at 15,000 IU/kg diet in place of C20-D3-retinyl acetate. Both diets contained ca. 0.16 mg β-carotene/kg diet. Special Diets Services prepared the diets. Animals were switched to treatment or control diet at weaning (21 d of age) or when otherwise stated.
Fundus AF Imaging.
Standardized fundus AF imaging using a confocal scanning laser ophthalmoscope (SpectralisHRA; Heidelberg Engineering) was performed by the same examiner in all animals according to a previously published protocol (18, 44). The right eye of each animal was used for imaging. After alignment of the camera, using the near-infrared reflectance mode, the focus was set to the plane with the highest reflectivity. Subsequently, near-infrared and blue AF images were recorded using the automatic real time (ART) mode. Exposure time before recording blue AF images was 10–20 s to allow sufficient photopigment bleaching, but to avoid other processes leading to changes of the fluorescence signal (44). For quantitative analysis, mean gray levels (acquired with standardized signal detector sensitivity, unprocessed, 1,536 × 1,536 pixels) were measured by an unmasked reader as described (18) within a ring-shaped area between 250 and 450 pixel radii from the optic disk center, using ImageJ software (Version 1.43; National Institutes of Health, rsb.info.nih.gov/ij). The “electronic zero” was subtracted from each measured gray value to obtain the corrected gray level, which was used for all calculations.
A2E Quantification.
The cornea and lens were removed under PBS. Dissected eyecups were immediately snap frozen and stored at −80 °C until further processing. Frozen eyecups were placed into a 1.5-mL centrifuge tube containing 0.5 g of 1 mm zirconium oxide beads, 250 μL of 1-butanol containing 1 mg butylated hydroxytoluene per milliliter of butanol (butanolBHT), and homogenized with a bead mill homogenizer (BBY24M Bullet Blender STORM; Next Advance Inc.; setting seven for 5 min). We then added 400 μL saturated sodium chloride, rehomogenized for 10 min, and separated the resulting phases by centrifugation at 16,000 rpm for 5 min. We removed 200 μL of the butanol phase and added it to a 300-μL-capacity autosampler vial (Microsolv Technology Corp.), and the samples were held at room temperature in an autosampler until they were injected in the UPLC system. For UPLC analysis, we injected 100 μL of the above eye extracts into a Flexar FX-15 system with a PDA Detector (Perkin-Elmer). The system contained a 2.1 × 150 mm, 2.7 μm, C18 column (Brownlee SSP, Perkin-Elmer) with a 2.1 × 5 mm guard column with the same packing material. The column oven was set to 45 °C, and samples were eluted at 0.4 mL per minute with 50% by volume methanol containing 20% isopropanol and 0.01% trifluoroacetic acid (solvent A) and 50% (vol/vol) type 1 water containing 0.01% trifluoroacetic acid (solvent A) for 1 min. The gradient was changed to 100% solvent A over the course of 10 min and was further eluted for 15 min with 100% solvent A. We detected A2E and other vitamin A dimers at 445 nm. Under these conditions, the limit of detection for A2E was 3 pmol of A2E on column. To confirm retention times for A2E in eye extracts, we spiked eye extracts with 0.4 μL of an A2E stock solution (1 mg A2E/mL butanol-BHT). A2E standard was synthesized following published procedures (45). To confirm that all A2E was extracted, we re-extracted homogenates with another 250-μL portion of a butanol-BHT and analyzed the second extract by UPLC. No further A2E was detected. A pooled SD of 20% was used, and significance was calculated with a two-sided, two-tailed t test.
Electron Microscopy.
Eyecups (one per animal) were fixed at 4 °C in 2% (vol/vol) glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) containing 100 mM sucrose. Serial electron microscopy was performed by Renovo Neural Inc., and images were acquired using a Zeiss Sigma VP scanning electron microscope equipped with Gatan 3View in-chamber ultramicrotome. Masked quantification of electron-dense lipofuscin granules was done using Fiji software suite. For each eye, electron-dense, irregular, and/or granular bodies were quantified in 12 serial sections sliced 100 nm apart. Two eyes per treatment group were evaluated. Statistical analysis was performed using one-way analysis of variance (SI Appendix, Table S1).
Quantitative Reverse Transcription PCR.
After enucleation and removal of orbital tissue, cornea, and lens, the resulting eyecups were immediately snap frozen in liquid nitrogen and stored at −80 °C. RNA was extracted from the eyecups with RNeasy mini (RNeasy; Qiagen). Equal quantities of 300–400 ng RNA were used as a template for cDNA manufacture for each sample. cDNA was produced with a QuantiTect reverse transcriptase kit (Qiagen). Primers were designed for specific probe-binding regions using Roche Universal Probe Library (SI Appendix, Table S2). Quantitative reverse transcription PCR was performed to detect mRNA levels of mouse C3, Cfb, Cfp, Cd59, and Cfh. Reagents were obtained from Roche Diagnostics. Technique was based on FAM-labeled hydrolysis probes (Roche Diagnostics). Reactions were performed in triplicate in 20-µL aliquots. Fold-change was determined using the ΔΔCt method. Relative gene expression value ΔCt was calculated against β-actin for each cDNA sample. ΔΔCt values were calculated against 9-mo-old untreated wild-type samples.
ERG.
Animals were dark-adapted for at least 12 h before ERG responses were recorded from one eye, using a setup described previously (44). For dark-adapted (scotopic) testing, responses were elicited by flashes of white light on a dark background. Stimulus intensity was increased over ∼8 log units (SI Appendix, Table S3). For light-adapted testing, animals were pre-exposed to steady full-field white background illumination (30 cd/m2) for 10 min. Responses were then recorded to light flashes and to 20-Hz flicker of two intensities (0.5 and 1 log cd⋅s/m2) superimposed on the same background light. In single-flash ERGs, the b-wave amplitude (from a-wave trough to b-wave peak) was measured for all ERGs, whereas the a-wave amplitude (from baseline to a-wave trough) was measured only when recognizable as a distinct component (stimulus intensities ≥ −1 log cd⋅s/m2). Both amplitudes were measured in unfiltered recordings. Details of the ERG testing protocol and analysis are provided in SI Appendix, Table S3. For analysis of dose–response curves, data were fit to logistic functions (46).
Supplementary Material
Acknowledgments
Support was provided by the European Commission, FP7, Marie Curie Intra-European Fellowship 237238, ProRetina, Fight for Sight, Wellcome Trust Grant 086868/Z/08/Z, Health Foundation, Medical Research Council UK, Royal College of Surgeons of Edinburgh, National Institute for Health Research Oxford and Moorfields Biomedical Research Centers, and the National Institutes of Health, National Eye Institute (Grants 1R01EY021207 and 5P30EY019007).
Footnotes
Conflict of interest statement: I.W. is listed as an inventor on a patent related to treating macular degeneration.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1506960112/-/DCSupplemental.
References
- 1.Allikmets R, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 2.Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012;3:925. doi: 10.1038/ncomms1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allikmets R. Simple and complex ABCR: Genetic predisposition to retinal disease. Am J Hum Genet. 2000;67(4):793–799. doi: 10.1086/303100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cremers FP, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Human molecular genetics. 1998;7(3):355–362. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 5.Brunk UT, Terman A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med. 2002;33(5):611–619. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- 6.Gray DA, Woulfe J. 2005. Lipofuscin and aging: A matter of toxic waste. Sci Aging Knowledge Environ 2005(5):re1.
- 7.Duncker T, et al. Correlations among near-infrared and short-wavelength autofluorescence and spectral-domain optical coherence tomography in recessive Stargardt disease. Invest Ophthalmol Vis Sci. 2014;55(12):8134–8143. doi: 10.1167/iovs.14-14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonilha VL. Age and disease-related structural changes in the retinal pigment epithelium. Clin Ophthalmol. 2008;2(2):413–424. doi: 10.2147/opth.s2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niven JE, Laughlin SB. Energy limitation as a selective pressure on the evolution of sensory systems. J Exp Biol. 2008;211(Pt 11):1792–1804. doi: 10.1242/jeb.017574. [DOI] [PubMed] [Google Scholar]
- 10.Katz ML, Gao CL, Rice LM. Long-term variations in cyclic light intensity and dietary vitamin A intake modulate lipofuscin content of the retinal pigment epithelium. J Neurosci Res. 1999;57(1):106–116. doi: 10.1002/(SICI)1097-4547(19990701)57:1<106::AID-JNR11>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 11.Radu RA, et al. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci USA. 2003;100(8):4742–4747. doi: 10.1073/pnas.0737855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mata NL, et al. Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina. 2013;33(3):498–507. doi: 10.1097/IAE.0b013e318265801d. [DOI] [PubMed] [Google Scholar]
- 13.Ma L, Kaufman Y, Zhang J, Washington I. C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J Biol Chem. 2011;286(10):7966–7974. doi: 10.1074/jbc.M110.178657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feeney L. Lipofuscin and melanin of human retinal pigment epithelium. Fluorescence, enzyme cytochemical, and ultrastructural studies. Invest Ophthalmol Vis Sci. 1978;17(7):583–600. [PubMed] [Google Scholar]
- 15.Radu RA, et al. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci. 2008;49(9):3821–3829. doi: 10.1167/iovs.07-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peters S, Lamah T, Kokkinou D, Bartz-Schmidt KU, Schraermeyer U. Melanin protects choroidal blood vessels against light toxicity. Z Naturforsch C. 2006;61(5-6):427–433. doi: 10.1515/znc-2006-5-620. [DOI] [PubMed] [Google Scholar]
- 17.Weng J, et al. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell. 1999;98(1):13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 18.Charbel Issa P, et al. Fundus autofluorescence in the Abca4(-/-) mouse model of Stargardt disease—correlation with accumulation of A2E, retinal function, and histology. Invest Ophthalmol Vis Sci. 2013;54(8):5602–5612. doi: 10.1167/iovs.13-11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaufman Y, Ma L, Washington I. Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J Biol Chem. 2011;286(10):7958–7965. doi: 10.1074/jbc.M110.178640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eldred GE, Katz ML. Fluorophores of the human retinal pigment epithelium: Separation and spectral characterization. Exp Eye Res. 1988;47(1):71–86. doi: 10.1016/0014-4835(88)90025-5. [DOI] [PubMed] [Google Scholar]
- 21.Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature. 1993;361(6414):724–726. doi: 10.1038/361724a0. [DOI] [PubMed] [Google Scholar]
- 22.Sakai N, Decatur J, Nakanishi K, Eldred GE. Ocular age pigment “A2-E”: An unprecedented pyridinium bisretinoid. J Am Chem Soc. 1996;118(6):1559–1560. [Google Scholar]
- 23.Mihai DM, Washington I. Vitamin A dimers trigger the protracted death of retinal pigment epithelium cells. Cell Death Dis. 2014;5:e1348. doi: 10.1038/cddis.2014.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Y, et al. Retinal metabolism in humans induces the formation of an unprecedented lipofuscin fluorophore ‘pdA2E’. Biochem J. 2014;460(3):343–352. doi: 10.1042/BJ20140089. [DOI] [PubMed] [Google Scholar]
- 25.Li J, et al. Identification of a novel lipofuscin pigment (iisoA2E) in retina and its effects in the retinal pigment epithelial cells. J Biol Chem. 2013;288(50):35671–35682. doi: 10.1074/jbc.M113.511386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murdaugh LS, et al. Compositional studies of human RPE lipofuscin. J Mass Spectrom. 2010;45(10):1139–1147. doi: 10.1002/jms.1795. [DOI] [PubMed] [Google Scholar]
- 27.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509(7501):439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo C, Zhao J, Madden A, Chen M, Xu H. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp Eye Res. 2013;112:93–101. doi: 10.1016/j.exer.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 29.Schramm EC, et al. Genetic variants in the complement system predisposing to age-related macular degeneration: A review. Mol Immunol. 2014;61(2):118–125. doi: 10.1016/j.molimm.2014.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73(6):887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- 31.Anderson DH, et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog Retin Eye Res. 2010;29(2):95–112. doi: 10.1016/j.preteyeres.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolf-Schnurrbusch UE, Stuck AK, Hess R, Wolf S, Enzmann V. Complement Factor P in choroidal neovascular membranes of patients with age-related macular degeneration. Retina. 2009;29(7):966–973. doi: 10.1097/IAE.0b013e3181a2f40f. [DOI] [PubMed] [Google Scholar]
- 33.Radu RA, et al. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J Biol Chem. 2011;286(21):18593–18601. doi: 10.1074/jbc.M110.191866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Radu RA, Hu J, Jiang Z, Bok D. Bisretinoid-mediated complement activation on retinal pigment epithelial cells is dependent on complement factor H haplotype. J Biol Chem. 2014;289(13):9113–9120. doi: 10.1074/jbc.M114.548669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci USA. 2006;103(44):16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bora NS, et al. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. J Immunol. 2007;178(3):1783–1790. doi: 10.4049/jimmunol.178.3.1783. [DOI] [PubMed] [Google Scholar]
- 37.Maeda A, et al. Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J Biol Chem. 2009;284(22):15173–15183. doi: 10.1074/jbc.M900322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marmor MF, Jain A, Moshfeghi D. Total rod ERG suppression with high dose compassionate Fenretinide usage. Doc Ophthalmol. 2008;117(3):257–261. doi: 10.1007/s10633-008-9132-y. [DOI] [PubMed] [Google Scholar]
- 39.Kubota R, et al. Safety and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina. 2012;32(1):183–188. doi: 10.1097/IAE.0b013e318217369e. [DOI] [PubMed] [Google Scholar]
- 40.Lem J, Fain GL. Constitutive opsin signaling: Night blindness or retinal degeneration? Trends Mol Med. 2004;10(4):150–157. doi: 10.1016/j.molmed.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 41.Woodruff ML, et al. Spontaneous activity of opsin apoprotein is a cause of Leber congenital amaurosis. Nat Genet. 2003;35(2):158–164. doi: 10.1038/ng1246. [DOI] [PubMed] [Google Scholar]
- 42.Cukras C, et al. Exome analysis identified a novel mutation in the RBP4 gene in a consanguineous pedigree with retinal dystrophy and developmental abnormalities. PloS one. 2012;7(11):e50205. doi: 10.1371/journal.pone.0050205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mihai DM, Jiang H, Blaner WS, Romanov A, Washington I. The retina rapidly incorporates ingested C20-D₃-vitamin A in a swine model. Mol Vis. 2013;19:1677–1683. [PMC free article] [PubMed] [Google Scholar]
- 44.Charbel Issa P, et al. Optimization of in vivo confocal autofluorescence imaging of the ocular fundus in mice and its application to models of human retinal degeneration. Invest Ophthalmol Vis Sci. 2012;53(2):1066–1075. doi: 10.1167/iovs.11-8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Penn J, Mihai DM, Washington I. Morphological and physiological retinal degeneration induced by intravenous delivery of vitamin A dimers in rabbits. Dis Model Mech. 2015;8(2):131–138. doi: 10.1242/dmm.017194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.