

Significance
Nucleotide excision repair (NER) is a versatile repair machinery able to protect organisms from DNA damage. Defective NER leads to diseases like xeroderma pigmentosum (XP). XPA is a central NER protein that interacts with DNA in an unknown fashion. Here we present two crystal structures of the yeast homolog of XPA, Rad14, in complex with two NER substrate lesions. Rad14 binds to the damaged DNA from both sides of the lesion. Binding creates a sharp kink of the duplex by 70°. Each protein inserts a hairpin loop into the duplex to induce partial melting around the lesion. The structures provide insight into the mechanism of how XPA binds to kinked and lesion-containing DNA.
Keywords: XPA, Rad14, NER, AAF, cisplatin
Abstract
Nucleotide excision repair (NER) is responsible for the removal of a large variety of structurally diverse DNA lesions. Mutations of the involved proteins cause the xeroderma pigmentosum (XP) cancer predisposition syndrome. Although the general mechanism of the NER process is well studied, the function of the XPA protein, which is of central importance for successful NER, has remained enigmatic. It is known, that XPA binds kinked DNA structures and that it interacts also with DNA duplexes containing certain lesions, but the mechanism of interactions is unknown. Here we present two crystal structures of the DNA binding domain (DBD) of the yeast XPA homolog Rad14 bound to DNA with either a cisplatin lesion (1,2-GG) or an acetylaminofluorene adduct (AAF-dG). In the structures, we see that two Rad14 molecules bind to the duplex, which induces DNA melting of the duplex remote from the lesion. Each monomer interrogates the duplex with a β-hairpin, which creates a 13mer duplex recognition motif additionally characterized by a sharp 70° DNA kink at the position of the lesion. Although the 1,2-GG lesion stabilizes the kink due to the covalent fixation of the crosslinked dG bases at a 90° angle, the AAF-dG fully intercalates into the duplex to stabilize the kinked structure.
Defects of the nucleotide excision repair (NER) system cause the human disease xeroderma pigmentosum, which is characterized by hypersensitivity to sunlight, resulting from the inability of patients to repair UV-induced DNA lesions (1, 2). Eight XP complementation groups are known, of which seven are caused by mutations in genes involved in the NER process (3, 4). NER recognizes a large array of diverse lesions (5–9). It repairs UV- and cisplatin-induced intrastrand crosslinks (10, 11). As such, NER protects higher organisms from the harmful effects of sunlight but also establishes resistance against cisplatin therapeutics, which is a major problem associated with this type of chemotherapy (12). The NER system also repairs a wide variety of single base bulky DNA adducts formed by environmental carcinogens. NER is thus the most versatile known DNA repair system (5, 8, 9).
The structurally vastly different lesions are recognized either within a globally operating NER (global genome repair, GG-NER) (7, 13) or as part of a transcription coupled NER process (TC-NER), where the stalled RNA polymerase is the initial NER inducing signal (14–16). Several lines of evidence lead to the currently accepted hypothesis that the lesions are initially recognized by XPC with the help of UV-DDB (UV DNA damage binding protein) for cyclobutane pyrimidine dimer (CPD) lesions (17–20) followed by damage verification by XPD/TFIIH (21–24), XPA, and XPG (3) to finally assemble to the preincision complex (3, 25–27). This step seems to involve recruitment of the XPA protein, which is one of the proteins essential for both GG-NER and TC-NER (28). As such, mutations of the XPA proteins provide one of the strongest NER phenotypes (29). Although the function of most XP-proteins within the NER process is understood, the precise role of XPA has remained unclear XPA is known to interact with other NER proteins such as RPA, TFIIH, and ERCC1 (30–33) suggesting XPA to be a NER scaffold protein (3). Furthermore, detailed biochemical experiments showed that XPA binds to kinked DNA structures (34) and to DNA duplexes containing certain types of DNA lesions, such as cisplatin (10, 35, 36) adducts and bulky adducts (5, 37, 38). XPA was found to form a homodimer (38), and it was demonstrated that it forms a 2:1 complex with the respective DNA (37). Initial structural insights into the protein architecture were derived from an NMR structure of the DNA binding domain of XPA (39, 40). However, the precise structural basis of the interactions of XPA with its DNA substrates is not known (3, 28).
Here we present two structures of the Saccharomyces cerevisiae homolog of XPA, Rad14, in complex with DNA containing a cisplatin 1,2-GG intrastrand crosslink and a N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (AAF-dG) bulky adduct, which are representatives of typical NER substrates (6). The obtained structures shine light on how one of the major NER proteins interacts with DNA.
Overall Structure of Rad14 Bound to 1,2-GG Cisplatin and AAF-dG Containing DNA
We first investigated binding of Rad14 to double-stranded DNA containing different lesions in a central position (ODN1-5, see Table S1) using band shift assays (Fig. S1). We confirmed complex formation with DNA containing a cisplatin 1,2-GG (10, 36, 41) and an AAF-dG lesion (37, 38) (Fig. S1 D and E). We next studied the binding stoichiometry with AAF-dG containing DNA (Fig. S1E) and confirmed the expected formation of a 2:1 complex with Rad14 (37, 38).
Table S1.
Sequences of the oligonucleotides (ODN) used in the different experiments
| ODN | Oligonucleotide sequence | Application |
| 1 | 5′-TCCTCTCTTGGTTCTCTTCT-3′ | EMSA |
| 2 | 5′-TCTCTCG*CTCATCCAC-3′ | EMSA |
| 3 | 5′-TCTCTCFCTCATCCAC-3′ | EMSA |
| 4 | 5′-ACAGCGGYYGCAGGT-3′ | EMSA |
| 5 | 5′- GTCGTTCGGAAGACCCACTGCAAFGGGTTGATAATCGCCTCGCCATAG-3′ | EMSA |
| 6 | 5′-GCTCTACG*TCATCAC-3′ | Crystallization/Crosslinking experiment |
| 7 | 5′-TCTCTACGGTCATCAC-3′ | Crystallization |
| 8 | 5′-TCCTCTCTTG*TTCTCTTCT-3′ | Crosslinking experiment |
| 9 | 5′-CCGAGTCATTCCTG*CAGCGAGTCCATGGGAGTC AAAT-3′ | Crosslinking experiment |
| 10 | 5′-GCTGACAGATCAGAFCAATGACTAGCTGAG-3′ | Fluorescence polarization |
| 11 | 5′-GCTCTACFTCATCAC-3′ | Fluorescence polarization |
F, FITC-dU; G*, AAF-dG; GG, 1,2-GG cisplatin; YY, CPD/ (6-4)PP.
Fig. S1.

Binding assays of XPA and Rad14 with different DNA lesions. The indicated amounts of protein were incubated with DNA duplexes (33 fmol) containing the lesions. As control, undamaged dsDNA (UD) was used. Electrophoretic mobility shift assay (EMSA) of XPA with 1,2-GG (A, ODN1 see Table S1), AAF-dG (B, ODN2), and FITC-dU (C, ODN3). EMSA of Rad14 1,2-GG (D, ODN1), AAF-dG (E, ODN2) and UV-lesions [(6, 4)PP and CPD; F, ODN4]. (G) EMSA of Rad14188–302 (Rad14t) with AAF-dG containing DNA (ODN2). (H) Competitive EMSA for the determination of the binding constant of Rad14 to AAF-dG (ODN2). Indicated amounts of Rad14 were incubated with DNA containing AAF-dG (lanes 2–8) and unmodified DNA (lane 9). Competitive DNA was added to the mixture as indicated. The amount of the labeled DNA was counted and assigned as the percentage of bound DNA in the y axis. The x axis shows the logarithmic concentration of the added competition DNA. The curve was fitted as a one-site competition experiment. A binding constant of Kaff = 135 nM (±10 nM SEM) was determined for the affinity of Rad14 to DNA duplexes containing this bulky adduct.
For crystallization studies, we used two truncated versions of Rad14 (Rad14188–302/306), which represent the DNA binding domain. We confirmed that the truncated version of Rad14188–302 (Rad14t) retains the binding characteristics to DNA (Fig. S1G). Cocrystals were obtained with Rad14t in complex with a 15mer DNA duplex containing a single AAF-dG in the central position and with a 16mer duplex with a central cisplatin 1,2-GG lesion (Fig. 1 C and D). We first determined the structure of selenomethionine containing Rad14188–306 featuring a C-terminal strep-tag in complex with the AAF-dG containing DNA, which provided good structural data for the protein at 3.1-Å resolution. However, large parts of the DNA, including the area in which the damage was located, remained uninterpretable. We therefore substituted three thymidines with 5-iodo-uracils and replaced the tag. This new construct resulted in a different crystal form, of which the Rad14t moiety was solved by molecular replacement using the coordinates of the previously experimentally determined selenomethionine-labeled protein and the DNA was located by Fourier analysis. The structure was refined to a final resolution of 1.8 Å. In addition, the structure of the DNA-binding Rad14t fragment in complex with the 16mer DNA duplex containing a central cisplatin 1,2-GG lesion was determined to 2.8-Å resolution (Table S2).
Fig. 1.

Overall structures of the Rad14188–302-DNA complexes. (A) Schematic diagrams of the Rad14-AAF-dG and Rad14-1,2-GG complexes showing the different positions where the lesions were observed in orange and red, with the C1’–C1’ distance of the last base pair in the molten part of the structure. For the AAF-dG lesion (Left) two positions of the lesion and for the 1,2-GG lesion (Right) 4 positions of the lesion were observed (Fig. S3). For clarity, only one DNA orientation with the cisplatin lesion is shown here. (B) Schematic representation of the α/β-folding topology of Rad14188–302, with the central 3-stranded β-sheet. (C) Ribbon diagrams of the Rad14-cisplatin lesion DNA complex with the structure of the lesion and the DNA sequence. Rad14 is shown in green and gold (β-hairpin), the DNA backbone is shown in gray, and the cisplatin 1,2-GG lesion is shown in blue. Residues important for DNA kinking (Thr239, His258, Phe262, and Arg294) are shown as sticks. (D) Ribbon diagrams of the Rad14-AAF-dG lesion DNA complex with the structure of the lesion and the DNA sequence. Rad14 is shown in green and gold (β-hairpin), the DNA backbone is shown in gray, and the AAF-dG lesion is shown in blue. Residues important for DNA kinking (Thr239, His258, Phe262 and Arg294) are shown as sticks. In the DNA sequences, the unpaired, partially disordered DNA bases are depicted in light gray. Thymidines replaced by 5-iodo-uracils in the AAF-dG complex are shown in bold.
Table S2.
Data collection and refinement statistics
| SeMetRad14-AAF-dG | Rad14-AAF-dG | Rad14-1,2-GG | |
| Wavelength, Å | 0.979 | 0.976 | 0.873 |
| Resolution range, Å | 20–3.1 (3.2–3.1) | 65.3–1.8 (1.84–1.80) | 54.4–2.8 (2.9–2.8) |
| Space group | P 21 | P 41 | P 41 |
| Unit cell | a = 66.4, b = 51.7, c = 68.8 β = 111.98 | a = 53.8, b = 53.8 c = 130.4 | a = 54.4, b = 54.4 c = 130.8 |
| Total reflections | 59,565 (5,963) | 253,904 (14,638) | 39,291 (3,836) |
| Unique reflections | 7,284 (709) | 34,232 (1948) | 9,402 (933) |
| Multiplicity | 8.2 (8.4) | 7.4 (7.5) | 4.2 (4.1) |
| Completeness, % | 99.7 (99.2) | 99.8 (95.6) | 99.7 (98.1) |
| Mean I/sigma(I) | 21.3 (7.46) | 10.7 (2.6) | 9.1 (1.2) |
| Wilson B-factor | 88.69 | 31.3 | 58.5 |
| R-merge | 0.07 (0.27) | 0.08 (0.89) | 0.1495 (1.33) |
| Anomalous completeness | 0.995 (0.996) | 98.8 (96)0.4 | — |
| Anomalous multiplicity | 4.1 (4.1) | 3.4 (3.5) | — |
| CC1/2 | 0.998 (0.994) | 0.998 (0.593) | 0.992 (0.485) |
| CC* | 1 (0.999) | — | 0.998 (0.808) |
| Figure of merit | 0.33 | — | — |
| R-work | 0.21 (0.312) | 0.213 (0.363) | |
| R-free | 0.241 (0.347) | 0.256 (0.343) | |
| Number of nonhydrogen atoms | 3,172 | 4,088 | |
| Macromolecules | 3,049 | 4,074 | |
| Ligands | 80 | 14 | |
| Water | 43 | 0 | |
| Protein residues | 291 | 339 | |
| RMS (bonds) | 0.018 | 0.012 | |
| RMS (angles) | 2.15 | 1.50 | |
| Ramachandran favored, % | 96 | 97 | |
| Ramachandran outliers, % | 0 | 0 | |
| Average B-factor | 48.6 | 55.7 | |
| Macromolecules | 48.5 | 55.8 | |
| Ligands | 60.4 | 48.3 | |
| Solvent | 37 |
Statistics for the highest-resolution shell are shown in parentheses.
The Rad14 structures show that the protein assumes an overall α/β-fold (Fig. 1B) quite similar to the NMR structure (Fig. S2A) of the human XPA homolog, indicating high structural conservation (40, 42). In agreement with the 2:1 stoichiometry determined by gel shift experiments (Fig. 3 F and G and Fig. S1E), we see in both structures that two Rad14 molecules bind to the DNA duplex above and below the respective lesion. This binding is shown for the 1,2-GG cisplatin containing DNA in Fig. 1C and for the AAF-dG in Fig. 1D. Interestingly, the Zn-finger domains of both proteins are not in contact with the duplex DNA.
Fig. S2.

Comparison of the human XPA and its yeast homolog Rad14. (A) Structural superposition of the yeast Rad14 DNA-binding domain with the NMR structure (Protein Data Bank ID code 1XPA) of its human homolog (Sequence identity 26.5%, rmsd: 2.2 Å). (B) Sequence alignment of Rad14/XPA homologs. Sequences were aligned with Clustal Omega (76). The secondary structure prediction of the Saccharomyces enzyme was performed with psipred and is shown in the bottom line of the alignment. H represents α-helices, C coils, and E β-strands (77). The boxed area represents the structure shown in the manuscript. The color scheme follows that of Fig. 2. The cysteine residues that build the zinc finger are boxed in yellow.
Fig. 3.

Crosslinking experiment of the Rad14t-DNA complex and mass spectrometric analysis of the digested protein dimer. (A) Two Rad14t proteins (light and dark green) bind to one DNA strand containing the AAF-dG lesion (gray). The two lysines at a distance of 21.5 Å in the peptide sequence TECKEDY are highlighted in red. (B) SDS gel of the protein–DNA mixture incubated with increasing amounts of the Bis(NHS)PEG5 crosslinker. (Rad14t)2 bands are boxed in red. The bands were cut out and subjected to enzymatic digestion. (C) Crosslinked peptide sequences after enzymatic digest of the proteins. (D) The MS/MS-spectrum created in an attached HCD-cell of the mass spectrometer reveals the peptide sequence enabling peptide identification (the a- and b-ion series is shown in red, x- and y-ion series is shown in blue). (E) Mass spectrometric analysis revealing the PEG fragments from the crosslinker. The PEG chain fragments with a typical Δm = 44 Da. Electrophoretic mobility shift assay (EMSA) of Rad14fl (F) and XPAfl (G) proteins with a central FITC-dU lesion containing and undamaged DNA (UD; endstanding FITC labeled DNA ODN3 and ODN5, see Table S1). (H) SDS PAGE of the protein–DNA mixture incubated with increasing amounts of the Bis(NHS)PEG5 crosslinker. A 15mer (ODN6) and a 37mer (ODN9) DNA duplex containing an AAF-dG lesion were used. (Rad14t)2 bands are boxed in red. (I) Fluorescence depolarization data showing the DNA binding properties of Rad14t [blue dots: 30mer (ODN10) and purple dots: 15mer (ODN11)] and Rad14tF262A (cyan triangles) to a central FITC-dU lesion containing DNA duplexes.
In the structure with the 1,2-GG lesion (Fig. 1C), two Rad14 molecules bind to the undamaged segments of the DNA above and below the lesion. Each protein interrogates the duplex with a β-hairpin (residues 253–267), leading to stacking of Phe262 and His258 onto the last intact base pair on either DNA end and unpairing of the following bases (Fig. 1A). This intercalation on both ends creates a central 13-bp duplex recognition element with the lesion in the middle. The entire structure exhibits a pseudo C2 symmetry because the DNA is likewise bound in two different orientations, which differ by a 180° rotation around the central positioned lesion. Because the 1,2-GG lesion is a crosslink between two central dG bases, the lesion can be observed for each of the two possible DNA orientations in two different positions (in total four positions, see Fig. S3A). For clarity only one of the structures is shown in Fig. 1C. This observation already suggests that Rad14 binds lesion independently, mostly triggered by conformational effects. We used the anomalous signal of the Pt-atom to estimate the occupancies (DNA orientation 1: 21% and 27%; DNA orientation 2: 33% and 19%, see Fig. S3A). In the structure with the AAF-dG lesion (Fig. 1D), we observed that the DNA is also bound in two orientations with the AAF-dG lesion located in two different positions linked by the pseudo C2-symmetry axis. The two duplex orientations are occupied with an approximate 45–55% distribution as inferred from the anomalous signal of the iodine (Fig. S3B). Importantly, the 13mer DNA lesion recognition motif in both structures (AAF-dG and 1,2-GG) is fully base paired and the lesions are both not in a flipped out state. Instead, the structures exhibit a sharp kink of 70° at the position of the lesion showing that Rad14 does not recognize the lesion itself but likely senses the flexibility of the DNA created in both cases by the lesions (Fig. 2).
Fig. S3.

Alternative orientations of the DNA bound by two Rad14 proteins. (A) The dinucleotide 1,2-GG lesion occupies four alternative positions in the 13mer duplex wedged between the β-hairpins of the two Rad14 proteins. The occupancy was estimated from the anomalous signal of the platinum atoms. (B) The AAF-dG lesion takes up two opposite positions in the crystal structure.
Fig. 2.

Rad14t binds to lesion containing DNA by bending of the DNA. (A) Close up view of the β-hairpin structure with Phe262 and His258 stacking on top of the last base pair. The β-hairpin is shown in orange, Rad14t in green and the DNA as stick model. (B) Bending of the DNA duplex at the lesion position (red) by 70° reduces the P–P distance in the major groove to 11.7 Å. The dotted line represents the pseudo C2 symmetry axis. (C) Schematic representation of the bending process. The β-hairpin is shown in orange, the packing of α7 with Arg294 against the backbone is shown in green, and the stabilization of the bent by Thr239 is shown in blue. (D) Schematic representation of the interaction in the Rad14t-AAF-dG DNA (15mer duplex) complex showing the “fingers” domain mainly formed by the β-hairpin as well as residues Gln266, Lys229, and Thr230 and the “thumb” created by α7 with Arg294. Arg293 (light green) interacts with the backbone only in the AAF-dG structure. The dotted square indicates the dC base opposite the AAF-dG lesion, for which electron density is missing likely due to flexibility. (E) Schematic drawing of the interactions between Rad14t and cisplatin 1,2-GG DNA (16mer duplex) using the same color code as in D.
The DNA ends below and above the kinked 13mer recognition motif are unwound, they lack any H-bonding interactions and are partially disordered in the structure. The corresponding 5′ ends of the DNA duplex at both ends of the structure are bound in a cleft created by the β-hairpin, the end of helix α4, and the antiparallel β-sheet domain (Figs. 1B and 2A). The β-hairpin structure and this cleft are responsible for melting of the duplex above and below the 13mer recognition sequence. A recent DNA binding study by Sugitani et al. (43) suggests that the DNA binding properties of XPA are improved if the binding domain is extended at the C terminus by 20 amino acids (XPA98–239), of which 9 provide basic side chains. The improved binding can be explained with the presented structure. Addition of the amino acids extends helix α7 (Fig. 1D), which leads to improved packing against the DNA duplex backbone with additional charge interactions between the negatively charged backbone and positive amino acids.
DNA Bending and the Lesion Recognition Motif
The melting process, which is induced by the β-hairpin irrespective of the lesion (Figs. 1A and 2A), separates the bases at the duplex ends by about 18.5 Å (C1'–C1' distance base pair 1 and 15). The most prominent feature of the structures is the formation of a sharp 70° kink at the central position (Fig. 2 B and C) where the lesions are situated. Bending occurs into the major groove and is characterized by a reduction of the interstrand phosphodiester distance at the closest point of the concave side to 11.7 Å compared with 16.8 Å in B-DNA. The protein–DNA interfaces (Fig. 2 D and E) that enable these processes are composed of residues from the β-hairpin (Asn256, His258, Phe262, and Gln266), α4 (Lys229 and Thr230), the loop between α4 and α5 (Thr239), and α7 (Arg293 and Arg294). The most prominent feature of the aforementioned interfaces is the β-hairpin (Fig. 2A), which establishes the majority of protein–DNA interactions, representing the anchor point for the bending process. The DNA is held in place by intercalation of Phe262 and His258 and further interactions established by Lys229, Thr230, and Gln266, which hydrogen bond to the phosphate moieties of base 2 and 3 in the 5′ strand. His258 forms a hydrogen bond with the phosphate moiety of the base, which gives rise to partially distorted stacking interactions. Asn256 and Gln266 contribute hydrogen bonds to base 2 and the phosphodiester of base 4 (Fig. 2 D and E). These and all other interactions (with the exception of Asn256) observed in the complete protein DNA interface are exclusively achieved through phosphate backbone interactions, which ensure a sequence independent recognition mechanism. They can be described as interrogating “fingers” that tightly bind the duplex starting 4–5 base pairs away from the lesion. A second “thumb” like interaction with the DNA backbone is established by helix α7, which packs against the backbone close to the lesion. As observed by Sugitani et al., extending this helix would likely result in even tighter binding (43). The “thumb” holds the DNA with the help of Arg294, forming a charge interaction with the phosphodiester. The “thumb” is further supported by an interaction established by Thr239 located between α4 and α5. In the AAF-dG structures (see below), we observe in addition that Arg293 keeps the backbone in place. The combined action of the intercalated Phe262 and His258 residues and the arginines in the α7-“thumb” forces the DNA to bend into the major groove. Because the bent conformation is established by only two interactions, with Arg294 and Thr239, the structure supports the hypothesis that Rad14 binds to DNA structures that are easily bendable or which already adopt a bent conformation before Rad14 binding. In this respect, our structures also explain the observed binding of XPA/Rad14 to DNA with bulges (34) because such DNA structures are also kinked (44). The structures furthermore explain the observed cooperativity of the Rad14/XPA binding event that is also visible in our gel shift experiments (Fig. 3 F and G and Fig. S1E) (37). In our model, the first Rad14 might bend the duplex slightly, thus facilitating the second Rad14 binding step, which provides the ability to form the sharp kink.
Analysis of the (Rad14)2-DNA Complex in Solution
To prove that the crystallographically observed structures also exist in solution and to investigate the possibility that complex formation involves binding of Rad14 just to the DNA ends, we performed protein–protein crosslinking experiments. Analysis of the crystal structures shows that Lys233 of each Rad14 points toward each other at a distance of 21.5 Å (Fig. 3A). We therefore prepared a longer DNA 19mer duplex (ODN8, see Table S1) with a central AAF-dG lesion and incubated it with Rad14. To this solution we added the reactive Bis(NHS)PEG5 crosslinker (Fig. 3B), which places the reactive ester groups at a distance of ∼21.7 Å assuming an extended conformation. Analysis of the reaction by gel electrophoresis (Fig. 3B) confirmed the formation of a defined (Rad14)2-DNA species. Interestingly, formation of only one crosslinked species even in a concentration dependent fashion was observed arguing that the distance between the two Rad14 molecules is around 21.5 Å despite the increased length of the duplex. To prove that the crosslinker has bridged the Lys233 residues of the two monomers we next digested the crosslinked (Rad14)2 with trypsin and analyzed the fragments by HPLC-MS/MS. The data show a parent ion signal with the exact molecular weight (m = 2115.02 Da) of the expected crosslinked dipeptide species (Fig. 3C) that was further characterized as TECKEDY (marked in red in Fig. 3A) by MS sequencing (Fig. 3 D and E). We also detected the typical fragmentation pattern of the ethylene glycole (PEG) crosslinker with Δm = 44.03 Da confirming the presence of the crosslinker.
Despite the clear results, it cannot be fully excluded that the crosslinker reacts with endbound Rad14 with the DNA being in an unusual bent conformation. To exclude this possibility, we performed a crosslinking experiment side by side with AAF-dG containing duplexes of different lengths (15mer ODN6 and 37mer ODN9, Fig. 3H). Despite the length difference, similar crosslinking efficiencies were observed, which argues against this possibility. The crystallographically determined binding mode is furthermore supported by the observation that Rad14 also binds to longer duplexes only in the presence of a lesion (Fig. 3G). Undamaged duplexes are not recognized.
To further investigate the importance of the observed β-hairpin intercalation, we performed fluorescence depolarization studies with Rad14t and a Rad14t variant in which we replaced one of the intercalating residues of the β-hairpin, Phe262, by an alanine (Rad14tF262A). For this study FITC-dU containing duplexes of different lengths (15mer ODN11 and 30mer ODN10, see Table S1) were used, in which the fluorescein, needed for the detection, functions as the lesion. It is well known that Rad14 binds the fluorescein-containing base with significant affinity (see below) (45). Formation of the lesion recognition complex with FITC-dU and the full length proteins Rad14fl and XPAfl was demonstrated by a gel shift experiment (Fig. 3 F and G). As depicted in Fig. 3I, the F262A point mutation fully abolished the lesion recognition ability of Rad14t, thus solidifying the model that the intercalation of the β-hairpin into the duplex is crucial for lesion recognition.
Discussion
In contrast to other DNA repair mechanisms, NER successfully recognizes structurally vastly differing lesions. Although DNA binding of the NER proteins XPC (46–48) and XPE (49, 50) are structurally understood, for XPA, which exhibits the strongest NER phenotype, it is only known that it interacts with kinked DNA structures such as DNA containing bulges and also in special cases with damaged DNA (10, 51, 52). Structural information about the binding process is lacking. Our structure of the XPA homolog Rad14 in complex with lesion containing DNA now uncovers the mechanism of how XPA interacts with the DNA. The obtained crystallographic results are fully consistent with previous biochemical data that showed binding of XPA to kinked DNA structures (34). Importantly, Rad14/XPA, like Rad4/XPC (18), does not bind the lesion directly but recognizes weakened DNA duplex structures. We observe that Rad14/XPA binding goes in hand with formation of a sharp kink at the lesion site by 70°. DNA binding involves two Rad14 proteins. Each protein inserts a β-hairpin exactly 6 base pairs away from the lesion, which generates a 13mer recognition motif. The lesion is not in a flipped out state but stays inside the 13mer duplex motif to stabilize the sharply bent structure. Bending occurs into the major groove, which in the case of the cisplatin 1,2-GG lesion means toward the Pt-atom (Fig. 4A). The intercalation of the β-hairpin “fingers” separates the DNA strands to form single stranded regions above and below the 13mer Rad14 binding motif to which other NER proteins may bind. This DNA binding mode could be one element that allows XPA to interact with many other NER factors (31–33). The relatively few interactions between Rad14 and the duplex suggests that Rad14 binds best to already prekinked DNA structures (34).
Fig. 4.
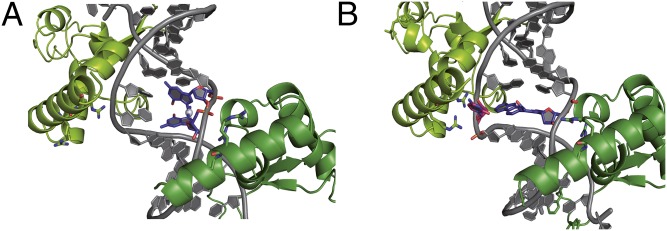
Rad14t recognizes lesions that can stabilize kinked structures. (A) Close-up view of the 1,2-GG lesion with the cisplatin unit (blue) intercalated between adjacent base pairs (gray) to stabilize the kinked structure formed by (Rad14t)2 (green). Arg294 is depicted in stick representation with the nitrogens in blue. (B) Close up view of the AAF-dG lesion in complex with Rad14t using the same color code as in A. The flipped out dC was modeled into the structure and is shown in red/blue.
This explains why Rad14 is able to bind to DNA containing bubbles, bulges and Y-junctions (34). All these substrates can form easily kinked DNA structures (52, 53). CPD lesions, on the other hand, are not recognized by Rad14, likely, because these lesions are less efficiently accommodated in a sharply kinked structure. Indeed, it is known that CPD lesions hardly influence the duplex structure and its stability (44). More importantly, however, is the fact that the stiff four-membered ring cyclobutane structure of the CPD lesion points into the major groove, which might block bending into this groove.
For single-base bulky adduct lesions such as AAF-dG, it is unclear to which extent they prekink the duplex. For AAF-dG and AF-dG lesions it was suggested that they exist in two different orientations with either the dG base or the AAF-unit inside the duplex (44). Melting point data of the AAF-dG–containing duplex used for crystallization and further thermodynamic studies (Fig. S4 and Table S3) show that the melting point is substantially reduced by ΔTm = 12 °C (ΔΔH = 3 kcal/mol) with no effects on ΔS (ΔΔS = 0 cal/molK). These data prove that the AAF-dG lesion creates a thermodynamically destabilized region in the duplex that can certainly be more easily kinked. More importantly, in the Rad14 structure with the AAF-dG lesion, we observe full intercalation of both the AAF and the dG unit into the duplex (Fig. 4B and Fig. S3B). The AAF-dG takes the place of a full base pair. The dC counterbase moves in response into a flipped out conformation (no electron density for the base) toward helix α7. Such a position of the AAF-dG fully inside the duplex was never observed, showing the unusual situation of the molecule in the kinked DNA duplex. It is most likely that this complete intercalation of the AAF-dG unit is needed to stabilizes the 70° kink using favorable π-stacking interactions. This model offers an explanation for binding of Rad14/XPA to FITC-dU containing DNA. This unit reduces the stability of the duplex by only a small amount. It may be that XPA impose some bending force to create a kink. Once the kink is temporarily formed, it may then be stabilized by intercalation of the flat and aromatic fluorescein unit. Further crystal structures are certainly needed to clarify this question.
Fig. S4.

Van't Hoff plots of Tm−1 vs. ln(Ct/4) derived from the melting curves for the control (undamaged DNA, green) and AAF-dG 15mer duplexes (blue) at concentrations between 2 μM and 7 μM. Ct represents the total oligonucleotide concentration.
Table S3.
Table of melting points and thermodynamic parameters of the AAF-dG lesion containing duplex used for crystallization
| Substrate | Tm, °C | ΔH, kcal/mol | ΔS, cal/mol K | ΔG298K, kcal/mol |
| dG:dC | 60 | −73 ± 0.5 | 274 | 8.7 |
| AAF-dG:dC | 48 | −70 ± 0.5 | 274 | 11.7 |
Our structure is also in agreement with previous detailed mutagenesis data (34, 54). The residues identified as critical contributors to the recognition of lesions by Rad14 are conserved in human XPA with the exception of Thr239, which is replaced by Lys151, Phe262, which is conservatively substituted by a tryptophane, and Gln266, which is replaced by a lysine. Although the side chain of Lys151 is somewhat longer, it would still be fully competent to fulfill the observed function of Thr239 in Rad14 (Fig. S5). The importance of the critical residues for DNA binding identified in our structure is supported by a lysine scanning mutagenesis study on human XPA where a K151E variant (corresponding to Thr239) was still able to interact with DNA but showed significantly decreased affinity to damaged DNA (54). In the same study, Lys141, Lys145, Lys179, Lys204, and Arg207 were analyzed as well. Intriguingly, the residues corresponding to Lys141 (Lys229), Lys179 (Gln266), and Arg207 (Arg294) all show impaired damaged DNA binding thus highlighting the importance of those residues for XPA function consequently validating our structural data.
Fig. S5.

Comparison of Rad14 bound to DNA and human XPA. (A) Superposition of a Rad14 monomer (green) bound to DNA (cyan) with XPA (light blue). Residues that have been implicated in DNA binding for XPA are shown in stick mode. The corresponding residues in Rad14 are also depicted. Human XPA residue numbers are shown in red; Rad14 residue numbers are shown in black. (B) Schematic representation of the Rad14 binding mode to DNA using the scheme introduced in Fig. 2D. Human XPA residues involved in DNA binding are depicted in red.
The structures reported here are unable to clarify the enigmatic role of XPA in the whole NER process. However, they provide mechanistic insight into how this important NER protein is able to interact with kinked DNA structures and lesion-containing duplexes that allow sharp bending. Binding requires interrogation of the duplex with a β-hairpin structure already observed in Rad4/XPC binding to DNA (18). Here we see similarities between both proteins. In summary our structures confirm that NER proteins and as such also XPA probe the structural integrity of the duplex, which is the secret behind the broad substrate promiscuity of NER (3).
Materials and Methods
For additional information on cloning of Rad14 and XPA, protein expression and purification, DNA synthesis, crystallization, data collection, structure determination and structure refinement, protein–DNA binding studies, protein-crosslinking experiments and LC-MS analysis, DNA melting temperature measurements, and fluorescence polarization measurements please see SI Materials and Methods.
SI Materials and Methods
DNA Preparation.
The AAF-dG phosphoramidite containing an isopropylphenoxyacetyl group at the N2-position for solid phase DNA synthesis was prepared as published (55, 56) and incorporated into DNA using ultra mild conditions for DNA synthesis (57). Oligonucleotide synthesis was performed on an ABI 394 Nucleic Acid Synthesis System (Life Technologies). Phosphoramidites for dA, dC, dG, dT, and CPG carriers were obtained from Glen Research or Link Technologies. The coupling time for the modified phosphoramidites was extended to 2 × 10 min. For the generation of the 1,2-GG cisplatin containing oligonucleotides, a modified protocol was used as published (58). Cisplatin was activated by treating the platinum salt with two equivalents of silver nitrate in an aqueous solution at a final concentration of 5 mM/10 mM. The mixture was protected from light and incubated at 18 °C for 15 h. Precipitated silver chloride was removed by centrifugation. The oligonucleotide 5′-TCTCTACGGTCATCAC-3′ was platinated with 3.0 equivalents of the activated platinum complex in a solution containing 100 mM NaClO4 at 37 °C for 4 h. The platinated oligonucleotide was purified by reverse phase HPL chromatography (Macherey-Nagel, Nucleosil 100–7 C18, 10 × 250 mm, 0.1 M triethylammonium acetate, linear gradient from 8% to 20% or 0–40% acetonitrile in 45 min). The purified strands were desalted using C18 cartridges (Waters, Sep-Pak C18 Classic Cartridge). The identity of the oligonucleotides were confirmed by matrix-assisted laser-desorption time-of-flight mass spectrometry and the purity determined to be >98% by capillary electrophoresis.
Melting Temperatures and Thermodynamic Parameters.
The duplexes of ODN6 (at concentrations between 1 and 7 µM; see Table S1) were dissolved in 200 µL of 10 mM Tris⋅HCl, pH 7.4, containing 150 mM NaCl. The samples were heated from 10 to 80 °C or cooled from 80 to 10 °C with a rate of 0.5 °C per min. Three melting profiles per sample concentration were performed with six different concentrations for each duplex. The enthalpy (ΔH°) and entropy (ΔS°) were calculated using a published protocol (59, 60). Van’t Hoff plot's of Tm−1 versus ln(Ct) to fit to Tm−1 = R/ΔH° ln Ct/4 + ΔS°/ΔH°, in which Tm is a melting transition point in K, Ct is the total duplex concentration, and R is the universal gas constant (1.978 cal/Kmol); ΔG° = ΔH° − TΔS°. Reported enthalpy and entropy values are the average of the determinations, and error estimates represent the SD of the data. The Gibbs free energy (ΔG°) of duplex formation was then calculated from these values with T = 298K.
Cloning of Rad14.
The sequence coding for the minimal DNA binding domain of the S. cerevisiae Rad14 protein (Rad14188–306) was amplified by PCR from genomic S. cerevisiae DNA using the AccuPrime Pfx DNA polymerase (Invitrogen) with the primers: forward: 5′-Pho-AATGGCGCCGAAATGTATTGAATGT-3′ and reverse: 5′-Pho-TCCC-GTATTTTTTCTCCCTTCTGTG-3′. The resulting PCR product was cloned via the StarGate method (IBA) into the pPSG-IBA3 with a C-terminal Strep-tagII. A truncated version of the minimal DNA binding domain (Rad14t188–302) lacking 4 amino acids on its C-terminal end was amplified by PCR using High Fidelity Phusion DNA polymerase (Finnzymes) and the following primers: forward: 5′-AGCGGCTCTTCAATGCTGGAAGTTCTGTTCCAGGGGCCAATGGCGCCGAAATGTATTGAATGTC-3′ and reverse: 5′-AGCGGCTCTTCTCCCCCTTCTGTGAGCCTTT-CCTTC-3′. The Rad14tF262A point mutation was generated through site-directed mutagenesis using the Q5 high fidelity polymerase (NEB) and the following primers: forward 5′-TTCGGGGACAGCGGCAAGAATGCAAC-3′ and reverse 5′-TGAGGGTTCGGCTTT-TCTAG-3′. Rad14t and Rad14tF262A were expressed with an N-terminal His-tag. A truncated version of the full length protein Rad14 (Rad14) lacking 9 amino acids on its N-terminal end was amplified by PCR using High Fidelity AccuPrime Pfx DNA polymerase (Invitrogen) and the following primers: forward: 5′-Pho-AATGGAGGCTAACAGGAAATTAGCAATAG-3′ and reverse: 5′-Pho-TCCCAATGT-CAATTTCTTCAGTTTCTAGCC-3′. The resulting PCR product was cloned via the StarGate method (IBA) into the pPSG-IBA3 with a C-terminal Strep-tagII.
Cloning of XPA.
The sequence coding for the minimal DNA binding domain of the human XPA protein (XPA98–219) was amplified by PCR from genomic DNA using the Phusion DNA Polymerase (NEB) with the following primers: forward: 5′-Pho-AATGGAATTTGATTATGTAATATGCG-3′ and reverse: 5′-Pho-TCCCA-AATTTCTTCTGTTTCATTTTTTCTC-3′. The resulting PCR product was cloned via the StarGate method (IBA) into the pPSG-IBA3 with a C-terminal Strep-tagII. The sequence coding for the full length human protein (XPAfl), was amplified by PCR from genomic DNA, using the Phusion DNA Polymerase (NEB) with the following primers: forward: 5′-Pho-AATGGCGGCGGCCGACGG-3′ and reverse: 5′-Pho-TCCCCATTTTTTCATATGTCAGTTCATGGCC-3′. The resulting PCR product was cloned via the StarGate method (IBA) into the pPSG-IBA35 with an N-terminal His-tag.
Protein Expression and Purification.
For protein expression, the constructs were transformed into Escherichia coli BL21 Star (DE3) (Invitrogen) cells and for the generation of selenomethionine derivatized protein E. coli B834(DE3)pLysS cells were used. Protein expression was induced by addition of isopropyl-β-d-galactoside (0.5 mM) at 25 °C (Rad14) or 28 °C (XPA) for 3 h, and the cells were grown either in Luria–Bertani or LeMaster (61) medium, respectively, supplemented with ZnCl2 (10 μM). All purification steps were carried out at 4 °C, and the purification was monitored by SDS/PAGE. For purification of Rad14-His, the cells were resuspended in 100 mM Tris⋅HCl pH 8.0, 100 mM NaCl, 1 mM DTT, 10% glycerol, and 10 µM ZnCl2. For the purification using the strep-tagged Rad14 and XPA, cells were resuspended in 100 mM Tris⋅HCl pH 8.0, 150 mM NaCl, and 1 mM EDTA. Buffers were supplemented with the Complete protease inhibitor mix (Roche). Cells were lysed in a French press, and the cell debris was removed by centrifugation before the supernatant was applied to the NiNTA column (GE Healthcare) or to the StrepTactin column (IBA), respectively. Protein elution was performed either with 500 mM imidazole (His-tag) or 2.5 mM desthiobiotin (Strep-tagII). Following tag-mediated affinity purification, the fractions were pooled and concentrated in MonoQ buffer A (100 mM Tris pH 7.5, 100 mM NaCl, 5% glycerol, 5 mM DTT, and 10 μM ZnCl2), and the protein was further purified by ion exchange (Q Sepharose from GE Healthcare) chromatography. The protein was eluted with a gradient of 5 column volumes with MonoQ buffer A containing 800 mM NaCl. Excess salt was removed by repeated concentration and dilution in 50 mM Hepes pH 8.0, 50 mM MgCl2, 5% glycerol, 5 mM DTT, and 10 μM ZnCl2 (crystallization buffer). In a final step, the protein was purified by size exclusion chromatography using a Superdex 75 column (GE Healthcare) in crystallization buffer. The correct mass of the protein was confirmed by SDS/PAGE and MALDI-TOF analysis. For purification of the selenomethionine derivatized protein, 10 mM DTT was added to each buffer.
Protein Expression and Purification for the Full-Length Proteins.
For Rad14fl protein expression, the constructs (pPSG-IBA3, C-terminal Strep-tagII) were transformed into E. coli BL21 Star (DE3) (Invitrogen) cells; for the generation of XPAfl protein (pPSG-IBA35, N-terminal His-tag), E. coli BL21(DE3)pLysS cells were used. Protein expression was induced by addition of isopropyl-β-d-galactoside (0.5 mM) at 25 °C (Rad14) or 37 °C (XPA) for 4 h and the cells were grown in Luria–Bertani (7) medium, respectively, supplemented with ZnCl2 (10 μM). All purification steps were carried out at 4 °C, and the purification was monitored by SDS/PAGE. For purification of XPA-His, the cells were resuspended in 25 mM Tris⋅HCl pH 7.5, 100 mM NaCl, 1 mM DTT, 10% glycerol, and 10 µM ZnCl2. For the purification of Rad14fl, cells were resuspended in 100 mM Tris⋅HCl pH 8.0, 800 mM NaCl and 1 mM EDTA, 5 mM DTT, 5% glycerol, and 10 µM ZnCl2. Buffers were supplemented with the Complete protease inhibitor mix (Roche). Cells were lysed by sonication and the cell debris was removed by centrifugation before the supernatant was applied to the NiNTA column (GE Healthcare) or to the StrepTactin column (GE Healthcare), respectively. Protein elution was performed either with 500 mM imidazole (His-tag) or 2.5 mM desthiobiotin (Strep-tagII). Following tag-mediated affinity purification, the fractions were pooled and concentrated in Heparin buffer A for Rad14 (100 mM Tris pH 7.5, 100 mM NaCl, 5% glycerol, 5 mM DTT (DTT) and 10 μM ZnCl2) and in Heparin buffer C for XPA (25 mM Tris pH 7.5, 50 mM NaCl, 10% glycerol, 1 mM DTT and 10 μM ZnCl2), and the protein was further purified by a heparin column (GE Healthcare) chromatography. Rad14fl was eluted with a gradient of 10 column volumes with Heparin buffer A containing 600 mM NaCl. Excess salt was removed by repeated concentration and dilution in Heparin buffer A. XPAfl was eluted with a gradient of 20 column volumes with Heparin buffer C containing 1 M NaCl. Excess salt was removed by repeated concentration and dilution in 50 mM Hepes pH 8.0, 50 mM MgCl2, 5% glycerol, 5 mM DTT, and 10 μM ZnCl2 (crystallization buffer). In a final step, XPAfl was purified by size exclusion chromatography using a Superdex 75 column (GE Healthcare) in crystallization buffer. The correct mass of both proteins were confirmed by SDS/PAGE and MALDI-TOF analysis.
Crystallization, Data Collection, Structure Determination, and Structure Refinement.
For cocrystallization experiments AAF-dG containing 15mer DNA (ODN6, see Table S1) was annealed in crystallization buffer to its counter strand without (5′-GTG ATG ACG TAG AGC-3′) or with 5-iododeoxyuridine (U*) (5′-GU*G AU*G ACG U*AG AGC-3′) (Metabion). For cocrystallization experiments with the cisplatin adduct a 16mer DNA strand (ODN7, see Table S1) was annealed to its counter strand in crystallization buffer. Before crystallization protein and DNA were mixed in a molar ratio of 1:1 protein:DNA (Strep-tagII) and 2:1 protein:DNA (His-tag) and incubated for 30 min at 4 °C. Initially, crystals for the selenomethionine labeled Strep-tagged protein in complex with AAF-containing DNA were obtained in 0.05 M Tris⋅HCl, pH 7, 0.1 M calcium acetate and 16–18% PEG8000 at 4 °C. His-tagged Rad14t protein in complex with AAF-dG and U*- or 1,2-GG cisplatin containing DNA was crystallized in 0.18 M ammonium nitrate and 40% 2-methyl-1,3,-propanediol (MPD) at 4 °C. Crystals were frozen in artificial mother liquor supplemented with 15% (vol/vol) ethylene glycol (SeMet-Rad14) or directly from the well (Rad14t) and stored in liquid nitrogen until data collection. Single anomalous dispersion (SAD) data on the SeMet-labeled Rad14-AAF complex crystals were collected at the microdiffractometer at the PXI beamline (Swiss Light source, Villigen, Switzerland). The crystals belonged to the space group P21, with unit cell dimensions of a = 66.4 Å, b = 51.7 Å, c = 68.8 Å and β = 109.6° and diffracted X-rays anisotropically to about 2.8 Å spacing. For structure determination, data collected on four different crystals were combined using AUTOPROC (62–64) and experimental phases where determined using PHENIX Autosol (65, 66). Initial model building was carried out with PHENIX Autobuild (67, 68). Iodine SAD data were collected at beamline ID29 of the European Synchrotron Radiation Facility (ESRF), diffracted X-rays to 1.8 Å and belonged to the tetragonal space group P41, with unit cell dimensions of a = 53.72 Å, b = 53.72 Å, and c = 130.55 Å. Crystals of Rad14t in complex with the 1,2-GG cisplatin DNA diffracted X-rays to 2.8 Å at the microfocus beamline ID23-2 (ESRF) and also belonged to the space group P41 with comparable unit cell constants as the AAF-dG complex. The structures were solved by molecular replacement (PHASER; refs. 69 and 70) using the coordinates of the Rad14-SeMet protein. Manual building of the model was performed with COOT (71) and structure refinement in Refmac5 (72) and Phoenix (68) using TLS (73, 74). Positions of the iodine and platinum atoms were determined and their occupancy estimated from the anomalous difference data (63). Structural figures were prepared with PyMOL (Delano Scientific).
Electromobility Shift Assays and Determination of the Binding Constant.
First, the counterstrand oligonucleotides were 5′-labeled with [γ-32P]ATP (3,000 Ci/mmol) using T4 polynucleotide kinase. Then, the damaged and undamaged oligonucleotides were mixed with their labeled complementary strands in equal molar amounts and annealed to form duplex DNA. The formation of the duplex DNA was checked by native TBE polyacrylamide gel electrophoresis. A standard DNA binding reaction mixture (10 μL) contained 33 fmol of the labeled duplex DNA and varying concentrations of purified Rad14 or XPA in binding buffer [25 mM Hepes-KOH pH 8.3, 100 mM KCl, 1 mM EDTA, 1 mM DTT, 45 μg/mL BSA and 10% (vol/vol) glycerol]. After incubation at 30 °C for 30 min, 2 μL of loading dye (250 mM Tris⋅HCl pH 7.5, 40% glycerol, 0.2% bromphenolblue) were added and electrophoresis was performed on a 4% nondenaturing polyacrylamide TBE gel at 4 °C, which was prerun for 30 min. Band shifts were visualized by autoradiography (Storm TM860, GE). Each gel-shift reaction and gel was performed three times. The binding constant was determined by a competitive binding experiment. The concentration of a radio labeled lesion-containing DNA was kept constant while increasing amounts of competing unlabeled DNA were added. The binding assay solutions contained 33 fmol 32P-labeled, lesion-containing DNA, 23 pmol purified Rad14 and varying amounts of lesion containing unlabeled DNA (Fig. S1H).
Fluorescence Polarization Measurements.
The DNA binding specificity and cooperativity of Rad14188–302 was determined in a fluorescence polarization experiment, using internal fluorescein-labeled DNA (30mer ODN10 and 15mer 11, see Table S1) substrate. The experiments were performed at 25 °C on a GeniusPro fluorescence photometer (Tecan). The excitation wavelength was set at 485 nm, and the emission wavelength was set at 535 nm. Measurements with Rad14t and Rad14tF262A were carried out in binding buffer (total volume of 100 µL) by incubating increasing amounts of protein with double stranded DNA containing an internal FITC-dU (5 nM) for 30 min at 25 °C. Measurements were carried out in 10 × 4-mm cuvettes at ambient temperature. Ten data points were taken per titration point. Each experiment was repeated three times.
Protein Crosslinking and LC-MS Analysis.
For the crosslinking experiments Rad14t was incubated for 30 min at 4 °C with a 19mer DNA duplex (ODN8, see Table S1) or a 37mer DNA duplex (ODN9) containing AAF-dG at its central position. Increasing amounts of Bis(NHS)PEG5 crosslinker were added to the protein-DNA complex (1:1–1:200) and the mixture was incubated for 2 h at 4 °C. After adding 2 µL of loading dye a SDS PAGE was performed. The protein band of interest was excised using a scalpel and subsequently in-gel digested as described by Shevchenko et al. (75). Trypsin and chymotrypsin were used for protein digestion. The samples were analyzed using an UltiMate 3000 nano liquid chromatography system (Dionex, Fisher Scientific) coupled to an LTQ-Orbitrap XL (Fisher Scientific). Ten microliters of the protein digest were injected, the samples desalted, concentrated on a µ-precolumn cartridge (PepMap100, C18, 5 µM, 100 Å, size 300 µm i.d. × 5 mm) and further processed on a custom made analytical column (ReproSil-Pur, C18, 3 µM, 120 Å, packed into a 75 µm i.d. × 150 mm and 8 µm picotip emitter). An 85-min multistep analytical separation was performed at a flow rate of 300 nL/min. In the first 50 min, a linear gradient was ramped up from 5% solvent B (acetonitrile containing 3% DMSO and 0.1% formic acid) and 95% solvent A (water containing 3% DMSO and 0.1% formic acid) to 95% solvent B. This level was held for 5 min and then ramped down again to 5% solvent B within 2 min. Mass spectrometric analyses were performed starting with a full mass scan in the mass range between m/z 300 and 1650. This survey scan was followed by three MS/MS scans using the FTMS mass analyzer and normalized collision energy of 70 in the HCD cell and three additional scans using the ion trap mass analyzer and normalized collision energy of 35. The Thermo Proteome Discoverer 1.1 software (Fisher Scientific) was used for protein identification. The Sequest search engine was used in combination with a uniprot database of the expression system (E .coli, complete proteome, download 12.2013) and the fasta file of the protein of interest “DNA repair protein RAD14” (S. cerevisiae, download 12.2013). As limit of detection a ratio of a threefold signal over the noise filter was applied. A maximum of three missed cleavage sites was allowed. The mass tolerances were 30 ppm for the precursor mass and 0.8 Da for the fragment ion mass. Dynamic modifications were cation: Na (Asp, Glu), oxidation (Met) as well as possible modifications of the crosslinker: Bis(NHS)PEG5 (Lys).
Supplementary Material
Acknowledgments
We thank Prof. Angelika Vollmar of the LMU Munich for access to radioactive laboratories and Jérôme Basquin and the staff of the MPI-Martinsried Crystallization Facility. Synchrotron access was supported by funds from the European Community's Seventh Framework Program (FP7/2007-2013) under grant agreement no. 226716. We also thank the beamline scientists at the SLS and ESRF for setting up the beamlines and support during data collection. We thank Dr. Markus Müller and Wolfgang Koelmel for helpful discussions, Johanna Bretzler for partly preparing and purifying XPA full length protein, and Karl-Peter Hopfner for initial crystal structure data interpretation and helpful discussions. This work was supported by Deutsche Forschungsgemeinschaft (SFB-646, SFB-749, the excellence cluster CIPSM and grant KI-562/2), Forschungszentrum FZ-82, the Fonds der Chemischen Industrie (FCI), and LMUexcellence. S.S. was supported by an FCI Liebig fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. S.B. is a guest editor invited by the Editorial Board.
Data deposition: Atomic coordinates have been deposited in the Protein Data Bank, www.ebi.ac.uk/pdbe (PDB ID codes 5a3d for AAF-dG and 5a39 for cisplatin).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1508509112/-/DCSupplemental.
References
- 1.Cleaver JE. Common pathways for ultraviolet skin carcinogenesis in the repair and replication defective groups of xeroderma pigmentosum. J Dermatol Sci. 2000;23(1):1–11. doi: 10.1016/s0923-1811(99)00088-2. [DOI] [PubMed] [Google Scholar]
- 2.Berneburg M, Lehmann AR. Xeroderma pigmentosum and related disorders: Defects in DNA repair and transcription. Adv Genet. 2001;43:71–102. doi: 10.1016/s0065-2660(01)43004-5. [DOI] [PubMed] [Google Scholar]
- 3.Naegeli H, Sugasawa K. The xeroderma pigmentosum pathway: Decision tree analysis of DNA quality. DNA Repair (Amst) 2011;10(7):673–683. doi: 10.1016/j.dnarep.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 4.Sancar A. Mechanisms of DNA excision repair. Science. 1994;266(5193):1954–1956. doi: 10.1126/science.7801120. [DOI] [PubMed] [Google Scholar]
- 5.Geacintov NE, et al. Thermodynamic and structural factors in the removal of bulky DNA adducts by the nucleotide excision repair machinery. Biopolymers. 2002;65(3):202–210. doi: 10.1002/bip.10239. [DOI] [PubMed] [Google Scholar]
- 6.Gillet LC, Schärer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106(2):253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 7.de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13(7):768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 8.Gunz D, Hess MT, Naegeli H. Recognition of DNA adducts by human nucleotide excision repair. Evidence for a thermodynamic probing mechanism. J Biol Chem. 1996;271(41):25089–25098. doi: 10.1074/jbc.271.41.25089. [DOI] [PubMed] [Google Scholar]
- 9.Huang JC, Hsu DS, Kazantsev A, Sancar A. Substrate spectrum of human excinuclease: Repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc Natl Acad Sci USA. 1994;91(25):12213–12217. doi: 10.1073/pnas.91.25.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones CJ, Wood RD. Preferential binding of the xeroderma pigmentosum group A complementing protein to damaged DNA. Biochemistry. 1993;32(45):12096–12104. doi: 10.1021/bi00096a021. [DOI] [PubMed] [Google Scholar]
- 11.Zamble DB, Mu D, Reardon JT, Sancar A, Lippard SJ. Repair of cisplatin—DNA adducts by the mammalian excision nuclease. Biochemistry. 1996;35(31):10004–10013. doi: 10.1021/bi960453+. [DOI] [PubMed] [Google Scholar]
- 12.Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478(1-2):23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 13.Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer. 2001;1(1):22–33. doi: 10.1038/35094000. [DOI] [PubMed] [Google Scholar]
- 14.Mellon I, Spivak G, Hanawalt PC. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 1987;51(2):241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 15.Sweder KS, Hanawalt PC. Preferential repair of cyclobutane pyrimidine dimers in the transcribed strand of a gene in yeast chromosomes and plasmids is dependent on transcription. Proc Natl Acad Sci USA. 1992;89(22):10696–10700. doi: 10.1073/pnas.89.22.10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tornaletti S, Hanawalt PC. Effect of DNA lesions on transcription elongation. Biochimie. 1999;81(1-2):139–146. doi: 10.1016/s0300-9084(99)80046-7. [DOI] [PubMed] [Google Scholar]
- 17.Volker M, et al. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8(1):213–224. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 18.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449(7162):570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 19.Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 2003;22(19):5293–5303. doi: 10.1093/emboj/cdg489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luijsterburg MS, et al. Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J Cell Biol. 2010;189(3):445–463. doi: 10.1083/jcb.200909175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathieu N, Kaczmarek N, Rüthemann P, Luch A, Naegeli H. DNA quality control by a lesion sensor pocket of the xeroderma pigmentosum group D helicase subunit of TFIIH. Curr Biol. 2013;23(3):204–212. doi: 10.1016/j.cub.2012.12.032. [DOI] [PubMed] [Google Scholar]
- 22.Kuper J, Wolski SC, Michels G, Kisker C. Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 2012;31(2):494–502. doi: 10.1038/emboj.2011.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathieu N, Kaczmarek N, Naegeli H. Strand- and site-specific DNA lesion demarcation by the xeroderma pigmentosum group D helicase. Proc Natl Acad Sci USA. 2010;107(41):17545–17550. doi: 10.1073/pnas.1004339107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pugh RA, Wu CG, Spies M. Regulation of translocation polarity by helicase domain 1 in SF2B helicases. EMBO J. 2012;31(2):503–514. doi: 10.1038/emboj.2011.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugasawa K, Akagi J, Nishi R, Iwai S, Hanaoka F. Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Mol Cell. 2009;36(4):642–653. doi: 10.1016/j.molcel.2009.09.035. [DOI] [PubMed] [Google Scholar]
- 26.Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15(7):465–481. doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- 27.Tapias A, et al. Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J Biol Chem. 2004;279(18):19074–19083. doi: 10.1074/jbc.M312611200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi T, et al. Mutational analysis of a function of xeroderma pigmentosum group A (XPA) protein in strand-specific DNA repair. Nucleic Acids Res. 1998;26(20):4662–4668. doi: 10.1093/nar/26.20.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satokata I, Tanaka K, Yuba S, Okada Y. Identification of splicing mutations of the last nucleotides of exons, a nonsense mutation, and a missense mutation of the XPAC gene as causes of group A xeroderma pigmentosum. Mutat Res. 1992;273(2):203–212. doi: 10.1016/0921-8777(92)90081-d. [DOI] [PubMed] [Google Scholar]
- 30.He Z, Henricksen LA, Wold MS, Ingles CJ. RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature. 1995;374(6522):566–569. doi: 10.1038/374566a0. [DOI] [PubMed] [Google Scholar]
- 31.Li L, Lu X, Peterson CA, Legerski RJ. An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol Cell Biol. 1995;15(10):5396–5402. doi: 10.1128/mcb.15.10.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park CH, Mu D, Reardon JT, Sancar A. The general transcription-repair factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J Biol Chem. 1995;270(9):4896–4902. doi: 10.1074/jbc.270.9.4896. [DOI] [PubMed] [Google Scholar]
- 33.Tripsianes K, et al. Analysis of the XPA and ssDNA-binding surfaces on the central domain of human ERCC1 reveals evidence for subfunctionalization. Nucleic Acids Res. 2007;35(17):5789–5798. doi: 10.1093/nar/gkm503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Camenisch U, Dip R, Schumacher SB, Schuler B, Naegeli H. Recognition of helical kinks by xeroderma pigmentosum group A protein triggers DNA excision repair. Nat Struct Mol Biol. 2006;13(3):278–284. doi: 10.1038/nsmb1061. [DOI] [PubMed] [Google Scholar]
- 35.Missura M, et al. Double-check probing of DNA bending and unwinding by XPA-RPA: An architectural function in DNA repair. EMBO J. 2001;20(13):3554–3564. doi: 10.1093/emboj/20.13.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997;16(21):6559–6573. doi: 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, et al. Cooperative interaction of human XPA stabilizes and enhances specific binding of XPA to DNA damage. Biochemistry. 2005;44(19):7361–7368. doi: 10.1021/bi047598y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang ZG, Liu Y, Mao LY, Zhang JT, Zou Y. Dimerization of human XPA and formation of XPA2-RPA protein complex. Biochemistry. 2002;41(43):13012–13020. doi: 10.1021/bi026064z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuraoka I, et al. Identification of a damaged-DNA binding domain of the XPA protein. Mutat Res. 1996;362(1):87–95. doi: 10.1016/0921-8777(95)00038-0. [DOI] [PubMed] [Google Scholar]
- 40.Ikegami T, et al. Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nat Struct Biol. 1998;5(8):701–706. doi: 10.1038/1400. [DOI] [PubMed] [Google Scholar]
- 41.Schweizer U, Hey T, Lipps G, Krauss G. Photocrosslinking locates a binding site for the large subunit of human replication protein A to the damaged strand of cisplatin-modified DNA. Nucleic Acids Res. 1999;27(15):3183–3189. doi: 10.1093/nar/27.15.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchko GW, Ni S, Thrall BD, Kennedy MA. Structural features of the minimal DNA binding domain (M98-F219) of human nucleotide excision repair protein XPA. Nucleic Acids Res. 1998;26(11):2779–2788. doi: 10.1093/nar/26.11.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sugitani N, Shell SM, Soss SE, Chazin WJ. Redefining the DNA-binding domain of human XPA. J Am Chem Soc. 2014;136(31):10830–10833. doi: 10.1021/ja503020f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukin M, de Los Santos C. NMR structures of damaged DNA. Chem Rev. 2006;106(2):607–686. doi: 10.1021/cr0404646. [DOI] [PubMed] [Google Scholar]
- 45.Krasikova YS, et al. Comparative analysis of interaction of human and yeast DNA damage recognition complexes with damaged DNA in nucleotide excision repair. J Biol Chem. 2013;288(15):10936–10947. doi: 10.1074/jbc.M112.444026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sugasawa K, et al. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2(2):223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 47.Sugasawa K, et al. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15(5):507–521. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hey T, et al. The XPC-HR23B complex displays high affinity and specificity for damaged DNA in a true-equilibrium fluorescence assay. Biochemistry. 2002;41(21):6583–6587. doi: 10.1021/bi012202t. [DOI] [PubMed] [Google Scholar]
- 49.Fujiwara Y, et al. Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J Biol Chem. 1999;274(28):20027–20033. doi: 10.1074/jbc.274.28.20027. [DOI] [PubMed] [Google Scholar]
- 50.Scrima A, et al. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135(7):1213–1223. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li L, Peterson CA, Lu X, Legerski RJ. Mutations in XPA that prevent association with ERCC1 are defective in nucleotide excision repair. Mol Cell Biol. 1995;15(4):1993–1998. doi: 10.1128/mcb.15.4.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buschta-Hedayat N, Buterin T, Hess MT, Missura M, Naegeli H. Recognition of nonhybridizing base pairs during nucleotide excision repair of DNA. Proc Natl Acad Sci USA. 1999;96(11):6090–6095. doi: 10.1073/pnas.96.11.6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buterin T, Meyer C, Giese B, Naegeli H. DNA quality control by conformational readout on the undamaged strand of the double helix. Chem Biol. 2005;12(8):913–922. doi: 10.1016/j.chembiol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 54.Camenisch U, Dip R, Vitanescu M, Naegeli H. Xeroderma pigmentosum complementation group A protein is driven to nucleotide excision repair sites by the electrostatic potential of distorted DNA. DNA Repair (Amst) 2007;6(12):1819–1828. doi: 10.1016/j.dnarep.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z, Rizzo CJ. Synthesis of the C8-deoxyguanosine adduct of the food mutagen IQ. Org Lett. 2001;3(4):565–568. doi: 10.1021/ol006968h. [DOI] [PubMed] [Google Scholar]
- 56.Gillet LC, Schärer OD. Preparation of C8-amine and acetylamine adducts of 2′-deoxyguanosine suitably protected for DNA synthesis. Org Lett. 2002;4(24):4205–4208. doi: 10.1021/ol026474f. [DOI] [PubMed] [Google Scholar]
- 57.Gillet LC, Alzeer J, Schärer OD. Site-specific incorporation of N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (dG-AAF) into oligonucleotides using modified ‘ultra-mild’ DNA synthesis. Nucleic Acids Res. 2005;33(6):1961–1969. doi: 10.1093/nar/gki335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei M, Cohen SM, Silverman AP, Lippard SJ. Effects of spectator ligands on the specific recognition of intrastrand platinum-DNA cross-links by high mobility group box and TATA-binding proteins. J Biol Chem. 2001;276(42):38774–38780. doi: 10.1074/jbc.M106374200. [DOI] [PubMed] [Google Scholar]
- 59.Allawi HT, SantaLucia J., Jr Thermodynamics and NMR of internal G.T mismatches in DNA. Biochemistry. 1997;36(34):10581–10594. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- 60.Zaliznyak T, Bonala R, Johnson F, de Los Santos C. Structure and stability of duplex DNA containing the 3-(deoxyguanosin-N2-yl)-2-acetylaminofluorene (dG(N2)-AAF) lesion: A bulky adduct that persists in cellular DNA. Chem Res Toxicol. 2006;19(6):745–752. doi: 10.1021/tx060002i. [DOI] [PubMed] [Google Scholar]
- 61.LeMaster DM, Richards FM. 1H-15N heteronuclear NMR studies of Escherichia coli thioredoxin in samples isotopically labeled by residue type. Biochemistry. 1985;24(25):7263–7268. doi: 10.1021/bi00346a036. [DOI] [PubMed] [Google Scholar]
- 62.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 63.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vonrhein C, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Terwilliger TC, et al. Decision-making in structure solution using Bayesian estimates of map quality: The PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 6):582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zwart PH, et al. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 67.Terwilliger TC, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64(Pt 1):61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 4):458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 70.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 74.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39(1):109–111. [Google Scholar]
- 75.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 76.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292(2):195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]