

Significance
Neurons use RNA modification to control specific functions by generating increased complexity and dynamism relative to the static genome. Known mechanisms of neuronal RNA modification include neuron-specific regulation of alternative splicing and mRNA editing by adenosine deaminases. Here, we identify a previously unidentified mechanism for RNA modification in neurons that is mediated by the RNA ligase RtcB. RtcB activity acts in neurons to regulate axon regeneration, and this neuronal function is independent of RtcB’s previously identified roles in tRNA and XBP1 mRNA ligation. RtcB’s function in axon regeneration is also independent of its cofactor archease, and RtcB translocates from the cell body of neurons to the stumps of injured axons. Taken together, these data establish a new function and mechanism for RtcB in inhibiting axon regeneration.
Keywords: axon regeneration, RNA ligation, RtcB
Abstract
Activity of the RNA ligase RtcB has only two known functions: tRNA ligation after intron removal and XBP1 mRNA ligation during activation of the unfolded protein response. Here, we show that RtcB acts in neurons to inhibit axon regeneration after nerve injury. This function of RtcB is independent of its basal activities in tRNA ligation and the unfolded protein response. Furthermore, inhibition of axon regeneration is independent of the RtcB cofactor archease. Finally, RtcB is enriched at axon termini after nerve injury. Our data indicate that neurons have co-opted an ancient RNA modification mechanism to regulate specific and dynamic functions and identify neuronal RtcB activity as a critical regulator of neuronal growth potential.
The RNA ligase RtcB is the only known RNA ligase in metazoans. RNA ligation by RtcB is required for the maturation of intron-containing tRNAs (1–3), and also, it is required to process the transcription factor xbp-1 mRNA and activate the unfolded protein response (UPR) (4–6). Other than these two basic cellular processes, which are likely common to all metazoan cells, no functions for RNA ligation or RtcB are known. The nervous system is a site of expanded RNA processing after transcription. For example, neurons regulate alternative premRNA splicing in response to activity (7–10) and are highly enriched for mRNA editing (11–13). Here, we define a neuron-specific function for RtcB activity in regulating axon regeneration and show that this neuronal function is independent of RtcB’s activities in tRNA and xbp-1 ligation.
RtcB activity in neurons inhibits axon regeneration. We assayed axon regeneration in the GABA motor neurons of Caenorhabditis elegans using single-neuron laser axotomy (14). Mutants in the single C. elegans ortholog of RtcB, rtcb-1(gk451) (5), exhibited increased regeneration to the dorsal nerve cord (DNC) at 24 h after injury, consistent with previous data (Fig. 1 A and B) (15). Increased DNC regeneration was reduced to WT levels by introduction of a single-copy WT RtcB transgene (Fig. 1A). The increase in DNC regeneration is not caused by the trivial explanation that RtcB animals are narrower than the WT. At 6 h after injury, a time point at which neurons in WT animals are just initiating regeneration (15–17), a substantial fraction of axons in RtcB mutants had already regenerated to the DNC (Fig. 1C). Furthermore, WT animals did not regenerate as well as RtcB mutants, even when given additional time to regenerate (Fig. 1C). Finally, rescuing the overall growth defects of RtcB mutants did not alter the effect of loss of RtcB on axon regeneration (Fig. 2). Thus, loss of RtcB results in faster and more successful axon regeneration. Increased regeneration depends on loss of RtcB in neurons, because expressing WT RtcB under a GABA-specific promoter restored DNC regeneration to WT levels (Fig. 1A). DNC regeneration levels were not restored when the rescue construct contained a point mutation that eliminates ligase activity (H428A) (18–21), showing that RtcB inhibits axon regeneration cell autonomously through its ligase activity.
Fig. 1.

Neuronal activity of RtcB inhibits axon regeneration. (A) Percentage of axons that regenerated back to the DNC after 24 h. Number of axons cut per genotype from left to right: 62, 99, 18, 46, 51, and 24. (B) Representative micrographs of GABA motor neurons 24 h after axotomy in control animals and RtcB mutants. VNC indicates ventral nerve cord. DNC indicates dorsal nerve cord. (Scale bar: 10 μm.) (C) DNC regeneration of control and RtcB mutant animals 6 h after axotomy. Number of axons cut per genotype from left to right: 113 and 35. (D) DNC regeneration of control and RtcB mutant animals 48 h after axotomy. Number of axons cut per genotype from left to right: 105 and 47. Error bars indicate 95% confidence interval. N.S., not significant; **P < 0.005; ***P < 0.0005.
Fig. 2.

RtcB inhibits axon regeneration independently of its role in tRNA ligation. (A) tRNAs containing introns are cleaved by the splicing endonuclease complex containing TSEN2. The resulting tRNA halves are ligated by RtcB and its cofactor archease. (B) Percentage of axons that regenerated back to the DNC after 24 h. Error bars indicate 95% confidence interval. Number of axons cut per genotype from left to right: 47, 26, 30, 18, 15, 38, and 24. (C) Representative micrographs 96 h after egg collection of WT, rtcb-1(gk451), tsen-2(wp15), and arch-1(wp16) with or without prespliced tRNAs. (Scale bar: 500 μm.) (D) Quantification of body length measurements at 96 h after egg collection (20 animals per genotype). Error bars indicate SEM. NS, not significant; ***P < 0.0005.
RtcB’s neuronal regeneration function is independent of its function in tRNA maturation. Intron-containing tRNAs are processed in a two-step mechanism that requires cleavage and release of the intron by the splicing endonuclease complex and subsequent ligation of the 5′ and 3′ exons by a ligation complex containing RtcB (Fig. 2A) (22). Consequently, RtcB mutants have tRNA processing deficits that cause growth defects and shortened lifespan, and these deficits can be rescued by providing prespliced tRNAs that do not contain introns and therefore, do not require RtcB for processing (5). However, prespliced tRNAs had no effect on the axon regeneration phenotype of RtcB mutants (Fig. 2D). To confirm that RtcB inhibits axon regeneration independently of tRNA ligation, we generated mutants for two additional components of the tRNA splicing pathway—TSEN2 and archease—using the CRISPR/cas9 system (23). TSEN2 is a catalytic component of the splicing endonuclease complex, and it is required for intron cleavage and release (24–26). Archease is an RtcB cofactor that promotes tRNA ligation by enabling the formation of an RtcB–guanylate intermediate (27, 28). Mutation of the C. elegans TSEN2 and archease orthologs encoded by tsen-2/Y56A3A.11 and arch-1/C43H8.1, respectively, resulted in growth defects that were rescued by prespliced tRNAs, similar to RtcB mutants (Fig. 2 C and D). However, unlike RtcB, neither TSEN2 nor archease mutants had any phenotype in axon regeneration (Fig. 2B). Together, these data indicate that, although RtcB mutants have defects in tRNA processing (5), loss of this activity does not account for the effect of RtcB activity in neurons on axon regeneration.
RtcB’s neuronal regeneration function is independent of its function in the UPR. Other than tRNA ligation, the sole known function of RtcB is in ligating the xbp-1 mRNA during the IRE1/XBP1 branch of the UPR (4–6). Because RtcB mutants cannot ligate the xbp-1 mRNA after it is cleaved by IRE-1 under conditions of protein stress, xbp-1 mRNA fragments accumulate, and XBP-1 transcriptional targets fail to express (5). However, xbp-1 mutants, which also fail to activate the IRE1/XBP1 branch of the UPR (29, 30), do not have an increase in axon regeneration (Fig. 3A), consistent with previous results (15). Rather, unlike RtcB mutants, xbp-1 mutants displayed a defect in axon regeneration—although they initiated growth in response to injury at approximately normal levels, this regenerative growth was often dysmorphic (Fig. 3B). Dysmorphic growth included axons that formed growth cones that grew toward the ventral nerve cord and axons that sprouted processes from the base of the axon near the ventral nerve cord instead of forming a growth cone near the injury site. To determine whether lack of the UPR contributes to the regeneration phenotype of RtcB mutants, we expressed preligated xbp-1S in the RtcB mutant background. When expressed in the intestine of either WT animals or RtcB mutants, xbp-1S expression resulted in activation of the UPR (as indicated by activation of the Phsp-4::GFP UPR reporter) independently of protein stress, consistent with the idea that loss of the UPR in RtcB mutants is specifically caused by lack of xbp-1 ligation (Fig. 3C). However, expression of xbp-1S in GABA neurons or panneuronally had no effect on axon regeneration (Fig. 3A). We also tested UPR activity in archease mutants, because archease is required in cultured cells along with RtcB to efficiently ligate the xbp-1 mRNA (4). We found that archease mutants fail to activate the UPR reporter (Fig. 3D), similar to RtcB mutants (5). However, archease mutants do not have a phenotype in axon regeneration (Fig. 2B). Taken together, these data indicate that RtcB regulates axon regeneration independently of its function in ligating the xbp-1 mRNA and activating the UPR.
Fig. 3.

RtcB inhibits axon regeneration independently of its role in the UPR. (A) Percentage of axons that regenerated back to the DNC after 24 h. Number of axons cut per genotype from left to right: 47, 59, 44, 21, 57, and 24. (B) Percentage of axons showing dysmorphic growth in WT, rtcb-1(gk451) mutants, and xbp-1(zc12) mutants. Number of axons cut per genotype from left to right: 47, 59, and 44. (C, Top) Differential interference contrast (DIC) and (C, Middle and Bottom) fluorescence micrographs of mutant animals either without or with prespliced xbp-1 (xbp-1s) expression in gut under the regulation of spl-1 promoter. Dashed circles indicate GFP expression from injection markers in the pharynx. Dashed squares indicate corresponding magnified regions in Bottom. (D, Top) DIC and (D, Middle and Bottom) fluorescence micrographs of arch-1(wp16) mutants expressing the Phsp-4::GFP UPR reporter; arch-1 heterozygous control animals express the Phsp-4::GFP UPR reporter on treatment with tunicamycin, but arch-1(wp16) homozygote animals fail to activate the reporter. Dashed circles indicate the Pmyo-2::GFP balancer used to identify heterozygous arch-1(wp16) mutants. Error bars indicate 95% confidence interval. NS, not significant; *P < 0.05; **P < 0.005; ***P < 0.0005.
RtcB has an injury-dependent localization in neurons. tRNA ligation occurs in the nucleus, whereas ligation of xbp-1 takes place in the cytoplasm (31–35). We determined RtcB localization in intestinal cells and GABA neurons with a functional red fluorescent protein (RFP)-tagged protein that rescues the UPR (5) and axon regeneration (Fig. 1A). In intestinal cells, RtcB was found in both the nucleus and cytoplasm (Fig. 4A) (5). Consistent with its role as an RtcB cofactor for both tRNA ligation and xbp-1 ligation (Figs. 2 and 3) (27, 28), archease was also found in the nucleus and cytoplasm of intestinal cells and colocalized with RtcB (Fig. 4A). By contrast, neuronal RtcB was tightly associated with a perinuclear compartment and only weakly present in the remainder of the cell body; little or no RtcB was detectable in neuronal processes (Fig. 4B). A similar localization pattern was also observed with GFP-tagged RtcB, with the GFP tag located at either the N terminus or at an internal site (Fig. S1). Archease localization in neurons was nuclear–cytoplasmic, similar to its localization in intestine and different from neuronal RtcB. Catalytically inactive TagRFP-RtcB(H428A) displayed similar expression and localization as the WT protein (Fig. 4C), indicating that, although RtcB’s catalytic activity is required for its function in regeneration (Fig. 1A), this activity does not mediate its localization in neurons. Because RtcB functions to regulate axon regeneration, we determined localization after injury and found that, although RtcB is excluded from the commissures of uninjured neurons, laser injury resulted in the accumulation of RtcB at the tips of injured axons (Fig. 4D).
Fig. 4.

RtcB exhibits injury-dependent localization in the GABA motor neurons. (A) Expression of RFP-tagged WT RTCB-1 and GFP-tagged ARCH-1 in the intestine. Both RTCB-1 and ARCH-1 are diffusely localized in the cytoplasm and nucleus. The dashed squares in Left are magnified in Right. Arrowheads indicate the expression of RTCB-1 and ARCH-1 in the nucleus of the gut. Similar localization was observed in all animals in multiple transgenic lines (two of two lines; >10 animals for each). (B) Expression of RFP-tagged WT RTCB-1 and GFP-tagged ARCH-1 in the GABAergic neurons. ARCH-1 is robustly expressed throughout the cell bodies, including the nucleus. In contrast, RTCB-1 shows punctate localization at the perinuclear area. Row 4 shows the magnified images of rows 1–3. Arrowheads indicate RFP-tagged RTCB-1. Similar localization was observed in all animals in multiple transgenic lines (two of two lines; >10 animals for each). *Cell bodies. (C) Localization of catalytically inactive RTCB-1 (H428A) in the GABAergic neurons. Arrowheads indicate RFP-tagged RtcB. Similar localization was observed in all animals in multiple transgenic lines (two of two lines; >10 animals for each). *Cell bodies. (D) Expression of WT RTCB-1 in the GABAergic neurons at 20 h after injury. The dashed squares indicate the tips of injured axons that are magnified in Bottom. The arrow indicates the intact axon. Arrowheads indicate accumulated RFP-tagged RTCB-1 near the injured axon tips. RFP was detected in 21 of 27 severed axons (78%; 95% confidence interval is 59–90%). *Distal stump as evidence of axotomy.
Fig. S1.
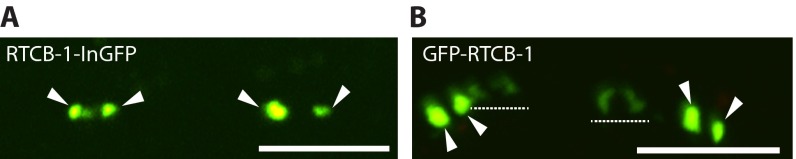
Expression of GFP-tagged RtcB shows similar localization with expression of RFP-tagged RtcB in the GABA motor neurons. (A) Expression of internal GFP-tagged WT RTCB-1 in the GABAergic neurons. RTCB-1::InGFP shows punctate localization at the perinuclear area. Arrowheads indicate RTCB-1::InGFP. Similar localization was observed in multiple animals (one line; >10 animals). (Scale bar: 10 μm.) (B) Expression of N-terminal GFP-tagged WT RTCB-1 in the GABAergic neurons. GFP::RTCB-1 shows punctate localization at the perinuclear area. Arrowheads indicate GFP-tagged RTCB-1. Some neurons show diffused expression of GFP::RTCB-1 in the cell body (indicated by dashed lines). Similar localization was observed in multiple animals (one line; >10 animals). (Scale bar: 10 μm.)
Our results identify a novel, neuron-specific role for the RNA ligase RtcB in inhibiting axon regeneration. The rapid initiation of axon regeneration after injury in RtcB mutants and the robust growth of mutant axons back to targets in the DNC indicate that RtcB activity normally functions to suppress an intrinsic growth capacity of neurons. Suppressing regeneration may allow for proper control of neuronal wiring or may occur to conserve resources needed to address trauma normally associated with axonal injury. In metazoan cells, the known functions of RtcB in tRNA and xbp-1 mRNA ligation require its cofactor archease (4, 28). Our in vivo data show that, like RtcB mutants (5), archease mutants have defects in tRNA maturation and the UPR (Figs. 2 and 3). However, loss of RtcB but not loss of archease results in increased axon regeneration. RtcB mutants have reduced tRNA levels and also, accumulate tRNA fragments (5), and neurological phenotypes are linked to accumulation of tRNA fragments and disruptions of tRNA processing (36–39). Because archease mutants also fail to ligate tRNAs (Fig. 2 C and D) (28) but do not have altered regeneration (Fig. 2B), neither lack of mature tRNAs nor accumulation of unligated tRNA fragments mediate RtcB’s regeneration function. Similarly, neither failure to activate the UPR nor accumulation of unligated xbp-1 mRNA fragments account for the increased regeneration in RtcB mutants (Fig. 3). Rather, our data indicate that RtcB activity in neurons acts independently of known targets to inhibit axon regeneration. Furthermore, this role in regeneration is independent of archease at the level of both RtcB function and RtcB’s subcellular localization. RtcB is a component of a neuronal RNA trafficking granule (40), and our finding that RtcB traffics to the tip of injured axons (Fig. 4) suggests that RNA ligation may inhibit regeneration at the site of injury. Together, our data suggest that neurons use RtcB in a cell- and context-dependent way to modify RNA and regulate cellular functions.
Methods
C. elegans.
C. elegans were fed Escherichia coli OP50 and maintained on nematode growth media (NGM) plates at 20 °C. The following strains were used:
SJ17 xbp-1(zc12);zcIs4[hsp-4p::GFP];
XE1728 rtcb-1(gk451) I/hT2[bli-4(e937) let-?(q782) qIs48]; wpIs63[prespliced tRNA rescue];oxIs12[Punc-47:GFP, lin-15+];
XE1743 rtcb-1(gk451)/hT2[bli-4(e937) let-?(q782) qIs48];wpIs63[prespliced tRNA rescue];zcIs4[Phsp-4::GFP];
XE1744 rtcb-1(gk451)/hT2[bli-4(e937) let-?(q782) qIs48];wpIs63[prespliced tRNA rescue];zcIs4[Phsp-4::GFP];wpEx251[Pspl-1::xbp-1s];
XE1745 oxIs12[Punc-47:GFP, lin-15+];wpEx252[Punc-47::TagRFP::rtcb-1 + Pmyo-2::mCherry];
XE1746 oxIs12[Punc-47:GFP, lin-15+];wpEx253[Punc-47::TagRFP::rtcb-1 + pRF4(rol-6)];
XE1747 rtcb-1(gk451)/hT2[bli-4(e937) let-?(q782) qIs48];wpIs63[prespliced tRNA rescue];oxIs12[Punc-47:GFP, lin-15+];wpEx254[Punc-47::TagRFP::rtcb-1 (H428A) + Pmyo-2::mCherry];
XE1735 uthIs270[rab-3p::xbp-1s (constitutively active) + myo-2p::tdTomato];oxIs12[Punc-47:GFP, lin-15+];
XE1736 rtcb-1(gk451)/hT2[bli-4(e937) let-?(q782) qIs48];wpIs63[prespliced tRNA rescue];uthIs270[Prab-3::xbp-1s (constitutively active) + Pmyo-2::tdTomato];oxIs12[Punc-47:GFP, lin-15+];
XE1737 rtcb-1(gk451)/hT2[bli-4(e937) let-?(q782) qIs48];wpIs63[prespliced tRNA rescue];oxIs12[Punc-47:GFP, lin-15+];wp245[Punc-47::xbp-1s + Pmyo-2::mCherry];
XE1743 arch-1(wp16)/hT2[bli-4(e937) let-?(q782) qIs48];zcIs4[Phsp-4::GFP];
XE1741 wpEx249[Pspl-TagRFP::rtcb-1 + Pspl-1::arch-1::GFP + Pmyo-2::mCherry];
XE1742 wpEx250[Punc-47::TagRFP::rtcb-1 + Punc-47::arch-1::GFP + pRF4(rol-6)];
XE1760 wpEx258[Punc-47::GFP::rtcb + Pmyo-2::mCherry]; and
XE1761 wpEx259[Punc-47:: rtcb::InGFP + Pmyo-2::mCherry].
To make transgenic animals with extrachromosomal arrays, the relevant constructs were microinjected (41, 42). The concentration of each injected plasmid was Punc-47::TagRFP-rtcb-1 (2 ng/μL), Punc-47::TagRFP-rtcb-1(H428A) (2 ng/μL), Punc-47::GFP-rtcb-1 (2 ng/μL), Punc-47::rtcb-1_InGFP (2 ng/μL), Punc-47::arch-1-GFP (3 ng/μL), and Pspl-1::xbp-1s (20 ng/μL). pRF4(rol-6) (40 ng/μL) or Pmyo-2::mCherry marker (2 ng/μL) was used as an injection marker. Transgenic worms were selected based on Pmyo-2::mCherry expression or roller phenotype generated by pRF4(rol-6) dominant overexpression.
CRISPR Mutations.
tsen-2 And arch-1 mutants were generated using CRISPR as described previously (23). The mutation in arch-1(wp16) resulted in a 7-bp deletion, which led to the first 13 amino acids remaining the same followed by mismatched amino acids until an early stop at 48 amino acids. The mutation in tsen-2(wp15) was a 2-bp deletion that resulted in the first 16 amino acids remaining the same followed by mismatched until an early stop at 18 amino acids.
UPR Fluorescent Reporter Assay.
UPR assay was performed as described previously (5, 43). Animals at the L4 stage were placed on NGM plates with 5 μg/μL tunicamycin for ∼24 h. RtcB homozygotes [rtcb-1(gk451);zcIs4[hsp-4::GFP]] or archease homozygotes [arch-1(wp16);zcIs4[Phsp-4::GFP]] were isolated from balanced heterozygotes. Heterozygous rtcb-1(gk451)/hT2;zcIs4[Phsp-4::GFP] or arch-1(wp16)/hT2;xcIs4[Phsp-4::GFP] served as a control. To rescue the UPR defects in RtcB mutants, the transgene encoding Pspl-1::xbp-1s (20 ng/μL) was injected into the balanced RtcB heterozygotes. Animals were mounted on 3% (wt/vol) agarose pads, imaged with UltraVIEW VoX (PerkinElmer) Spinning Disk Confocal Microscopy, and subsequently processed with Volocity (Improvision).
Cell Biology.
Transgenic animals expressing relevant transgenes were examined for localization at 4th larval stage (L4) or young adult stage. pFR4(rol-6) was used as an injection marker to efficiently observe the colocalization of RTCB-1 with ARCH-1 in GABAergic neuronal cell bodies on the ventral nerve cord. To test the RTCB-1 localization in GABAergic neurons after axotomy, selected axons in transgenic animals expressing TagRFP-RTCB-1 were cut using a Micropoint Laser from Photonic Instruments (10 pulses at 20 Hz). The axotomized animals were recovered to NGM plates; 24 h later, the animals were mounted on 3% agarose pads and imaged. Postaxotomy images were acquired with an Olympus DSU mounted on an Olympus BX61 Microscope, an Andor Neo sCMOS Camera, and a Lumen Light Source and subsequently, processed with ImageJ.
Molecular Biology.
Gateway recombination (Invitrogen) was used to generate Punc-47::arch-1::GFP, Pspl-1::TagRFP::rtcb-1, and Punc-47::TagRFP::rtcb-1(H428A). Entry clones were generated using Phusion High-Fidelity DNA Polymerase (NEB) or Q5 High-Fidelity DNA Polymerase (NEB). To generate the entry clone encoding TagRFP::rtcb-1(H428A), site-directed mutagenesis was used to introduce the point mutation (H428A) in WT TagRFP::rtcb-1 in the entry vector, which was previously generated (5).
To create the entry clone encoding GFP::rtcb-1, the rtcb-1 coding regions were amplified from the TagRFP::rtcb-1 entry clone, and GFP was amplified with primers with the flexible linker sequences. To generate the rtcb-1::internal GFP entry clone, the rtcb-1 cDNA coding region was amplified from the pTE049 plasmid encoding rtcb-1 cDNA in entry vector, and the GFP coding region was amplified with primers with the flexible linker sequences. The final plasmids were assembled in a two-fragment Gibson reaction. To prepare the xbp-1s entry clone, the xbp-1 coding sequence was amplified from genomic DNA extract and subsequently, processed by the site-directed mutagenesis to remove the intron that is edited by RtcB. The arch-1 cDNA entry clone was prepared by PCR amplification using the cDNA library as a template.
Primers are as follows: RtcB H428A Forward (Fw): ggagctggacgagcactttc, RtcB H428A Reverse (rev): tgcacatgttgttccaaaagtttccac, xbp1 sp Rev: ctgattcaaaggcacggcgat, xbp1 sp Fw: ctgttggtactggtcctatccacctccatcaacaac, xbp1 ATG Fw: ggggacaagtttgtacaaaaaagcaggctttatgagcaactatccaaaacgt, xbp1B Rev: ggggaccactttgtacaagaaagctgggtaggttttggaagaaatttaggtcgattg, Arc Fw 221: ggggacaagtttgtacaaaaaagcaggcttcatgccgagcacatcaatgat, Arc Rev 221: ggggaccactttgtacaagaaagctgggtaatgtcaacaatgacatagatatcg, RtcB 4 InGFP Rev: ctttactcatagatgctgaacctgccgatgaaccaactccaactgga, Rtcb 4 InGFP Fw: actatacaaatccgccggatccgcctcccgtggtgcaattccaatgctt, InGFP 4 RtcB Fw: gagttggttcatcggcaggttcagcatctatgagtaaaggagaagaacttttc, InGFP 4 RtcB Rev: attgcaccacgggaggcggatccggcggatttgtatagttcatccatgcc, nGFP 4 RtcB Fw: aagagctgattaaggagaacatgagtaaaggagaagaacttttc, nGFP 4 RtcB Rev: tgtgcgaggcatacttcctcctcctccacttttgtatagttcatccatgcc, RtcB 4 nGFP Fw: actatacaaaagtggaggaggaggaagtatgcctcgcacatttgaagaag, and RtcB 4 nGFP Rev: agttcttctcctttactcatgttctccttaatcagctcttcg.
Laser Axotomy.
Laser axotomy was performed as described previously (44). Briefly, L4 stage worms were mounted on microscope slides visualized with a Nikon Eclipse 80i Microscope using a 100× Plan ApoVC Lens (1.4 N.A.). The two or three of seven most posterior ventral and dorsal D-type (VD/DD) GABA motor neurons were cut using a Photonic Instruments Micropoint Laser at 10 pulses and 20 Hz. Worms were recovered to plates and scored for regeneration 6, 24, or 48 h after axotomy. Axons were only scored if the severed distal stump was present to confirm successful axotomy. Axons were scored for regeneration back to the DNC. All axotomies were carried out in strains containing the oxIs12[Punc-47::GFP] genetic background; 95% confidence intervals were determined using the Wald method, and two-tailed P values were determined using Fisher’s exact test.
Body Length Measurements and Analysis.
To examine the body lengths of tRNA mutant animals during development, 50 gravid adults were placed on a seeded NGM plate to lay eggs for 2 h. Eggs laid during this period were incubated at 20 °C for 48, 72, or 96 h. Mutant animals were identified under a Zeiss M2BIO Fluorescence Dissecting Microscope for lack of green fluorescence. Before each experiment, two to three animals were picked onto an unseeded NGM plate and allowed to move freely for 10 min. A picture of each animal was taken with a Point Gray Camera (FL3-FW-03S3M-C) in WormLab (MBF Bioscience). A photo of a micrometer was taken under the same condition during each experiment for length calibration. ImageJ software (NIH) was used to measure body length by drawing a freehand midline from the tip of the nose to the tip of the tail of each animal. The resulting body lengths were averaged for a total of 20 animals per genotype per time point.
Supplementary Material
Acknowledgments
We thank Markus Englert and Dieter Söll for discussion. We also thank WormBase and the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs Grant P40 OD010440. S.G.K. was funded by NIH Training Grant T32 GM 7223-37. S.M.H. was funded by the James Hudson Brown–Alexander Brown Coxe Postdoctoral Fellowship and the Department of Genetics Research Fellowship at Yale University. B.H. and M.R.K. were funded by NIH Grant R01 NS036918. W.A. was funded by NIH Medical Scientist Training Program Training Grant T32GM007205. Work in the laboratory of M.H. is supported by the NIH.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1502948112/-/DCSupplemental.
References
- 1.Englert M, Sheppard K, Aslanian A, Yates JR, 3rd, Söll D. Archaeal 3′-phosphate RNA splicing ligase characterization identifies the missing component in tRNA maturation. Proc Natl Acad Sci USA. 2011;108(4):1290–1295. doi: 10.1073/pnas.1018307108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Popow J, et al. HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science. 2011;331(6018):760–764. doi: 10.1126/science.1197847. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka N, Shuman S. RtcB is the RNA ligase component of an Escherichia coli RNA repair operon. J Biol Chem. 2011;286(10):7727–7731. doi: 10.1074/jbc.C111.219022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jurkin J, et al. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 2014;33(24):2922–2936. doi: 10.15252/embj.201490332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosmaczewski SG, et al. The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep. 2014;15(12):1278–1285. doi: 10.15252/embr.201439531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu Y, Liang F-X, Wang X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol Cell. 2014;55(5):758–770. doi: 10.1016/j.molcel.2014.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie J, Black DL. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature. 2001;410(6831):936–939. doi: 10.1038/35073593. [DOI] [PubMed] [Google Scholar]
- 8.An P, Grabowski PJ. Exon silencing by UAGG motifs in response to neuronal excitation. PLoS Biol. 2007;5(2):e36. doi: 10.1371/journal.pbio.0050036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee J-A, et al. Depolarization and CaM kinase IV modulate NMDA receptor splicing through two essential RNA elements. PLoS Biol. 2007;5(2):e40. doi: 10.1371/journal.pbio.0050040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iijima T, et al. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell. 2011;147(7):1601–1614. doi: 10.1016/j.cell.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67(1):11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 12.Rosenthal JJC, Seeburg PH. A-to-I RNA editing: Effects on proteins key to neural excitability. Neuron. 2012;74(3):432–439. doi: 10.1016/j.neuron.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li JB, Church GM. Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nat Neurosci. 2013;16(11):1518–1522. doi: 10.1038/nn.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrne AB, Edwards TJ, Hammarlund M. In vivo laser axotomy in C. elegans. J Vis Exp. 2011;51(2011):e2707. doi: 10.3791/2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nix P, et al. Axon regeneration genes identified by RNAi screening in C. elegans. J Neurosci. 2014;34(2):629–645. doi: 10.1523/JNEUROSCI.3859-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323(5915):802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edwards TJ, Hammarlund M. Syndecan promotes axon regeneration by stabilizing growth cone migration. Cell Rep. 2014;8(1):272–283. doi: 10.1016/j.celrep.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Englert M, et al. Structural and mechanistic insights into guanylylation of RNA-splicing ligase RtcB joining RNA between 3′-terminal phosphate and 5′-OH. Proc Natl Acad Sci USA. 2012;109(38):15235–15240. doi: 10.1073/pnas.1213795109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desai KK, Bingman CA, Phillips GN, Jr, Raines RT. Structures of the noncanonical RNA ligase RtcB reveal the mechanism of histidine guanylylation. Biochemistry. 2013;52(15):2518–2525. doi: 10.1021/bi4002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okada C, Maegawa Y, Yao M, Tanaka I. Crystal structure of an RtcB homolog protein (PH1602-extein protein) from Pyrococcus horikoshii reveals a novel fold. Proteins. 2006;63(4):1119–1122. doi: 10.1002/prot.20912. [DOI] [PubMed] [Google Scholar]
- 21.Genschik P, Drabikowski K, Filipowicz W. Characterization of the Escherichia coli RNA 3′-terminal phosphate cyclase and its ς54-regulated operon. J Biol Chem. 1998;273(39):25516–25526. doi: 10.1074/jbc.273.39.25516. [DOI] [PubMed] [Google Scholar]
- 22.Popow J, Schleiffer A, Martinez J. Diversity and roles of (t)RNA ligases. Cell Mol Life Sci. 2012;69(16):2657–2670. doi: 10.1007/s00018-012-0944-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedland AE, et al. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013;10(8):741–743. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winey M, Culbertson MR. Mutations affecting the tRNA-splicing endonuclease activity of Saccharomyces cerevisiae. Genetics. 1988;118(4):609–617. doi: 10.1093/genetics/118.4.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trotta CR, et al. The yeast tRNA splicing endonuclease: A tetrameric enzyme with two active site subunits homologous to the archaeal tRNA endonucleases. Cell. 1997;89(6):849–858. doi: 10.1016/s0092-8674(00)80270-6. [DOI] [PubMed] [Google Scholar]
- 26.Paushkin SV, Patel M, Furia BS, Peltz SW, Trotta CR. Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3′ end formation. Cell. 2004;117(3):311–321. doi: 10.1016/s0092-8674(04)00342-3. [DOI] [PubMed] [Google Scholar]
- 27.Desai KK, Cheng CL, Bingman CA, Phillips GN, Jr, Raines RT. A tRNA splicing operon: Archease endows RtcB with dual GTP/ATP cofactor specificity and accelerates RNA ligation. Nucleic Acids Res. 2014;42(6):3931–3942. doi: 10.1093/nar/gkt1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Popow J, Jurkin J, Schleiffer A, Martinez J. Analysis of orthologous groups reveals archease and DDX1 as tRNA splicing factors. Nature. 2014;511(7507):104–107. doi: 10.1038/nature13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calfon M, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 30.Urano F, et al. A survival pathway for Caenorhabditis elegans with a blocked unfolded protein response. J Cell Biol. 2002;158(4):639–646. doi: 10.1083/jcb.200203086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Robertis EM, Black P, Nishikura K. Intranuclear location of the tRNA splicing enzymes. Cell. 1981;23(1):89–93. doi: 10.1016/0092-8674(81)90273-7. [DOI] [PubMed] [Google Scholar]
- 32.Nishikura K, De Robertis EM. RNA processing in microinjected Xenopus oocytes. Sequential addition of base modifications in the spliced transfer RNA. J Mol Biol. 1981;145(2):405–420. doi: 10.1016/0022-2836(81)90212-6. [DOI] [PubMed] [Google Scholar]
- 33.Lund E, Dahlberg JE. Proofreading and aminoacylation of tRNAs before export from the nucleus. Science. 1998;282(5396):2082–2085. doi: 10.1126/science.282.5396.2082. [DOI] [PubMed] [Google Scholar]
- 34.Sidrauski C, Cox JS, Walter P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell. 1996;87(3):405–413. doi: 10.1016/s0092-8674(00)81361-6. [DOI] [PubMed] [Google Scholar]
- 35.Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73(6):1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- 36.Kasher PR, et al. Impairment of the tRNA-splicing endonuclease subunit 54 (tsen54) gene causes neurological abnormalities and larval death in zebrafish models of pontocerebellar hypoplasia. Hum Mol Genet. 2011;20(8):1574–1584. doi: 10.1093/hmg/ddr034. [DOI] [PubMed] [Google Scholar]
- 37.Namavar Y, et al. PCH Consortium Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain. 2011;134(Pt 1):143–156. doi: 10.1093/brain/awq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanada T, et al. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature. 2013;495(7442):474–480. doi: 10.1038/nature11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weitzer S, Hanada T, Penninger JM, Martinez J. CLP1 as a novel player in linking tRNA splicing to neurodegenerative disorders. Wiley Interdiscip Rev RNA. 2015;6(1):47–63. doi: 10.1002/wrna.1255. [DOI] [PubMed] [Google Scholar]
- 40.Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Neuron. 2004;43(4):513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 41.Mello C, Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- 42.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10(12):3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor RC, Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013;153(7):1435–1447. doi: 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byrne AB, Edwards TJ, Hammarlund M. In vivo laser axotomy in C. elegans. J Vis Exp. 2011;51(2011):pii:2707. doi: 10.3791/2707. [DOI] [PMC free article] [PubMed] [Google Scholar]