

Abstract
Ion–dipole interactions in biological macromolecules are formed between atomic or molecular ions and neutral protein dipolar groups through either hydrogen bond or coordination. Since their discovery 30 years ago, these interactions have proven to be a frequent occurrence in protein structures, appearing in everything from transporters and ion channels to enzyme active sites to protein–protein interfaces. However, their significance and roles in protein functions are largely underappreciated. We performed PDB data mining to identify a sampling of proteins that possess these interactions. In this review, we will define the ion–dipole interaction and discuss several prominent examples of their functional roles in nature.
Keywords: Ion–dipole interaction, ABC transport receptors, ion channel, serine protease, IMP dehydrogenase, SNARE, AP180, phage tail proteins
Introduction
Ion (charge)–dipole interactions are fundamental attractive forces, akin to hydrogen bonds and Van der Waals interactions, that are formed between ions or charged molecules and dipolar groups with partial charges. They encompass a broad number of more specifically defined interactions, because they include charge–neutral hydrogen bonds and coordinations. In biological macromolecules, these ubiquitous interactions and their roles in protein function are largely underappreciated. Originally, conventional wisdom stated that ions interacted solely through charge coupling with counterions or salt links with countercharged residues in proteins. This changed significantly with the X-ray structure of the sulfate-binding protein (SBP), the primary high-specificity, high-affinity sulfate ABC transport receptor, which revealed that the sulfate was completely buried, desolvated, and bound solely by neutral dipolar groups (described subsequently).1 In this instance, ion–dipole interactions govern charged ligand specificity, desolvation, and charge compensation.
In this brief review, we will define ion–dipole interactions and discuss several diverse functional roles they play in biological and biochemical processes.
Ion–Dipole Definition
There are several notable features that define an ion–dipole interaction in proteins. First, it is an ion or charged molecule that interacts exclusively through neutral polar dipoles (Fig. 1). The dipolar groups include main chain NH and CO groups as well as the polar groups of Ser, Thr, Asn, Gln, Tyr, and Trp side chains. They completely lack formal charges; therefore, salt links and cation/anion coordination to charged atoms are excluded. As expected, the dipolar NH groups from both main chains and side chains bind anions and negatively charged molecules, whereas the CO groups from both sources bind metallic cations and positively charged molecules. Hydroxyl side chains act in a bifunctional manner, because the proton and oxygen can interact with negative and positive charges, respectively.
Figure 1.
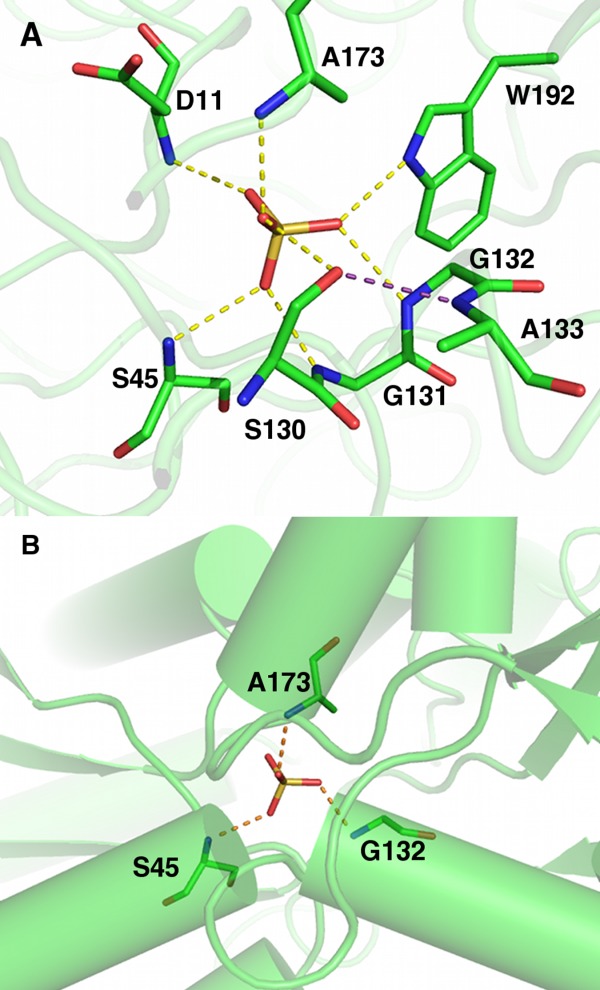
Salmonella typhimurium SBP-sulfate complex X-ray structure (PDB ID 1sbp). (A) SBP sulfate-binding site. Dashed lines represent ion–dipole interactions via hydrogen bonds, and cooperative hydrogen bonds are colored purple. Carbons are colored green; nitrogen, blue; oxygen, red; sulfur, gold. (B) Helix N-termini in the sulfate-binding site, each providing a main chain NH dipole.
Second, the charged atoms or ligands are completely desolvated and buried within the protein. The ion–dipole interactions are sufficiently favorable to overcome the desolvation energies of these ions, which are frequently very high. For example, the solvation energies of sulfate and potassium are estimated to be approximately −300 and −80 kcal/mol, respectively.2–4 These solvation energies arise from the classic ion–dipole interaction between ions and water dipoles. In some cases the unbound protein residues are solvated prior to binding as well, which only contributes to the energetic barrier. In addition, during the binding process, the ion must be transferred from a high dielectric constant medium (water = ∼80) to a relatively low dielectric constant environment (protein = ∼4).5 Altogether, this poses what should be a significant barrier to binding. The strength of a single hydrogen bond in protein–ligand interactions has been estimated from protein and substrate engineering experiments to be −1 to −3 kcal/mol for average hydrogen bond lengths,6–8 meaning that the energy provided by primary hydrogen bonds alone are insufficient to surmount the energetic barrier. Nevertheless, the protein binding sites appear to be more hospitable to charged ligands than aqueous solvent (further discussed in the section entitled “Ion channel selectivity filter”).
Third, dipolar groups involved in ion–dipole interactions are often engaged in secondary interactions that maintain rigid, optimized binding geometry. The SBP-sulfate structure is a hallmark of the two types of secondary interactions that are also frequently observed in other structures. Type 1 is interactions with dipolar backbone amides located in the first turns or N-termini of α helices [Fig. 1(B)]. The obvious assumption was that helix macrodipoles, macroscopic dipoles theorized to originate from the vector sum of the partially aligned peptide dipoles along the helical axis,9,10 play a significant role in ion charge compensation. However, computational analysis of the SBP-sulfate complex indicated that the first turn of the helix accounted for 80% of the dipole effect and the first two turns provided 95%.11 Moreover experimental studies indirectly probing the contribution of α helices and the peptide group to charge compensation showed little or no contribution.12 Further studies into helix macrodipoles in other proteins showed that ions are rarely located along helical axes, where the macrodipole effect would be greatest, and ion binding seems to be related to stereoelectronic requirements rather than long distance electric fields.13 Moreover computational studies of the chloride channel suggest that the contribution of helix macrodipoles to anion binding is only marginal.14
Type 2 is the “cooperative hydrogen bond” where a bifunctional side chain OH donor also accepts a hydrogen bond via its oxygen atom, demonstrated by the Ser residue in the SBP binding site [Fig. 1(A)].
Ion–dipole interactions lack distinct subgroups. There seems to be no preferential binding of backbones or side chains based on the nature of the charge or the specific type of charged ligands (metal, organic, atomic, and molecular). There are numerous examples of each type of ligand binding primarily backbone or side chain atoms and those that are fairly evenly mixed. However, the aim of this article is to present some of the functional aspects of ion–dipole interactions; thus, not all binding combinations are represented.
Ion–Dipoles in Nature
While a few examples had been identified previously,16 we recently performed a rough data mining of the PDB looking for ion–dipole interactions in proteins to determine their functional roles in nature. Using the program Pymol,17 we evaluated the biological assemblies of all the PDBs with ligands that were formally defined as “ions.” From there we excluded those ligands that were within 3.5 Å (hydrogen bond and coordination distance) of a water molecule or within 2.39 Å (covalent bond distance) of a side chain and had greater than 10% solvent accessible surface area. Manual curation of the resulting list identified a number of new unknown functional associations. While this is by no means a comprehensive list, it does highlight the versatility and crucial roles of ion–dipole interactions.
Substrate/solute binding
The prototypical ion–dipole interaction was revealed by the X-ray structure of the ligand-bound Salmonella typhimurium SBP.1,18 Overall, SBP adopts the canonical “Venus fly trap” fold with two non-contiguous globular domains separated by a cleft region.19,20 The sulfate binds in the cleft and is completely engulfed by domain closure through a bending motion of the hinge between the two domains (Fig. 1). The sulfate molecule is completely dehydrated. It is held in place through seven dipolar/hydrogen bonds distributed between the two domains, one Ser side chain OH and six NH groups, five from backbone amides and one from a Trp side chain [Fig. 1(A)]. In the case of oxyanion binding, the ion-dipole interactions serve three major purposes, providing exquisite specificity through the precise geometry of the interacting hydrogen bonds, stripping waters from the anion and dipolar groups, and compensating for the ion charge in preparation for transport across the membrane via membrane-bound components. The ensemble of dipolar groups is responsible for sulfate charge compensation. As mentioned previously, SBP has both Type I and II secondary interactions. SBP has three helix termini NH donors [Fig. 1(B)] and one cooperative interaction between the Ser side chain hydroxyl and an adjacent backbone amide [Fig. 1(A)]. Combined together, these features provide rigidity and precise geometry to the ion-binding site.
Ion channel selectivity filter
Ion channels, including potassium, sodium, and chloride channels, exemplify coordination-based ion–dipole interactions. Ion–dipole interactions contribute to the selectivity filter of these ion channels. In chloride channels the ion–dipole configuration is similar to SBP.21 The chloride is bound by two backbone NH groups and two OH groups from serine and tyrosine residues. Furthermore, the serine OH is involved in cooperativity identical to that in SBP.
An excellent example of ion–dipole via coordination combined with secondary “cooperative” interactions is seen in the S4 potassium site in the homotetrameric potassium channel KcsA, which is the final selectivity filter site located adjacent to the internal cavity (Fig. 2).22 In KcsA the K+ ion is coordinated by oxygens from the backbone carbonyl and side chain hydroxyls of Thr 75 from each monomer of the homotetramer. The hydrogen from the side chain hydroxyl is in turn expected to orient toward the adjacent Thr carbonyl oxygen, forming a pinwheel cooperative hydrogen bond configuration (Fig. 2).
Figure 2.
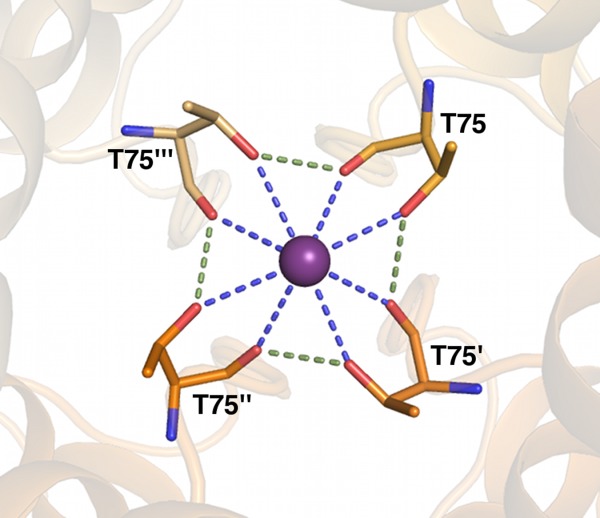
Streptomyces lividans KcsA potassium channel X-ray structure (PDB ID 1bl8) potassium site S4. Dashed lines represent ion–dipole interactions via coordination, and cooperative hydrogen bonds are colored green. Oxygens are colored red; nitrogens, blue; and potassium, purple. Different chain carbons are colored as different shades of orange and labeled with primes.
The actual role of the ion–dipole is a heavily debated topic, deeply embroiled in the various proposed ion-selectivity mechanisms. For simplicity sake, we will focus on those mechanisms described for K+ channels (reviewed in Ref.23). The “field strength” mechanism relies on the principle that for a specific ion, the energy of the ion–dipole will be favorable over the energy of the hydrated ion. Based on the geometry of the cavity, the coordinating carbonyl dipoles will have sufficient strength to overcome the energy of hydration. This field strength will be diminished in the presence of larger atoms, whereas smaller solvated ions will be too bulky to form proper dipolar interactions. The “snug fit” mechanism centers on the idea that the position of the carbonyls lining the cavity is optimized for the solvation of K+ ions only. Smaller ions such as Na+ would need higher energy to enter the carbonyl cage of the selectivity filter. The “ligand-ion attraction versus ligand-ligand repulsion” mechanism is primarily enthalpy driven. This mechanism purports that while positive ions will be attracted to the channel carbonyls, the carbonyls are going to be repulsed by one another. In this mechanism, only a specific ion (K+) will be able to achieve a balance between the attractive and repulsive forces, which would allow it to pass through the channel. These are highly simplified descriptions of a rich and extensive body of literature, and we cannot comment on the accuracy of one mechanism over the other. However, no matter which mechanism is favored, it is clear that the ion–dipole and cooperativity play critical roles in the action of the selectivity filter.
Catalytic intermediate stabilization
Ion–dipoles can be used directly as a part of catalysis. This is most directly observed with the oxyanion hole in serine proteases, exemplified by elastase. Elastase is in the chymotrypsin protein family and is capable of breaking down most connective tissue components in humans.24 In brief, the catalytic mechanism of elastase is as follows. Three amino acids, a His, Asp, and Ser compose the “catalytic triad” that engages in a nucleophilic attack upon a substrate carbonyl group. The serine then covalently bonds the substrate, forming a negatively charged tetrahedral intermediate that is stabilized through ion–dipole interactions with the “oxyanion hole,” the backbone amides of Gly 193 and Ser 195 (Fig. 3). The tetrahedral intermediate then becomes an acyl-enzyme intermediate by proton-assisted leaving group expulsion, and the protein substrate is cleaved. The tetrahedral oxyanion and its ion dipole interactions have been directly observed in a neutron crystal structure, which allows for the determination of hydrogen positions.24 This structure verified that the two oxyanion lone pairs directly form ion–dipole interactions with the Gly 193 and Ser 195 backbone NH groups. In this case, the ion–dipole geometry acts to stabilize the charged tetrahedral intermediate, and this reduces the activation energy for proton transfer and subsequent substrate cleavage.
Figure 3.
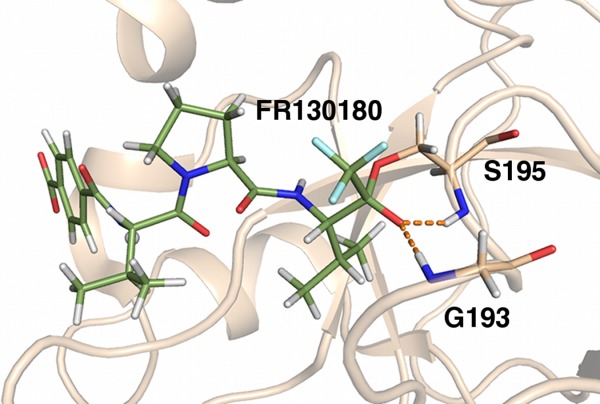
Porcine pancreatic elastase complexed with a peptidyl inhibitor FR130180 neutron structure (PDB ID 3hgn) oxyanion hole. Hydrogenated protein was crystallized in a precipitant solution prepared in D2O, which caused the exchangeable hydrogens to be replaced with deuteriums prior to neutron diffraction. The Ser and Gly stabilize the charged catalytic intermediate. Dashed lines represent ion–dipole interactions. Protein carbons are colored tan; FR130180 carbons, green; nitrogen, dark blue; oxygen, red; hydrogens, white; and deuteriums, light cyan.
Allosteric catalysis regulation
In addition to directly affecting catalysis, ion–dipoles can also regulate catalysis allosterically. This is seen with K+ in inosine-5′-monophosphate (IMP) dehydrogenases.25 IMP dehydrogenase catalyzes the conversion of IMP to xanthosine-5′-monophosphate. The enzyme has a basal catalysis rate in the absence of K+; however, in the presence of K+ the rate increases approximately 100-fold. The conserved ion–dipole K+ binding site is relatively distant from the active site and composed of six backbone carbonyls, three from a loop containing the catalytic Cys and three from an α helix C-terminus in the adjacent subunit (Fig. 4). K+ activation is fairly specific. While K+ activates the reaction, Na+ has little effect. Li+, Mg2+, and Ca2+ were shown to inhibit K+ activation without blocking the basal reaction. Studies showed that K+ increases the closure of a critical flap necessary for catalysis, and it adjusts the closed conformation to a more open-like state, which makes the catalytic conformational changes more facile. In this way the K+ acts as a “ball and socket joint” accelerating the critical catalytic conformational changes.25
Figure 4.
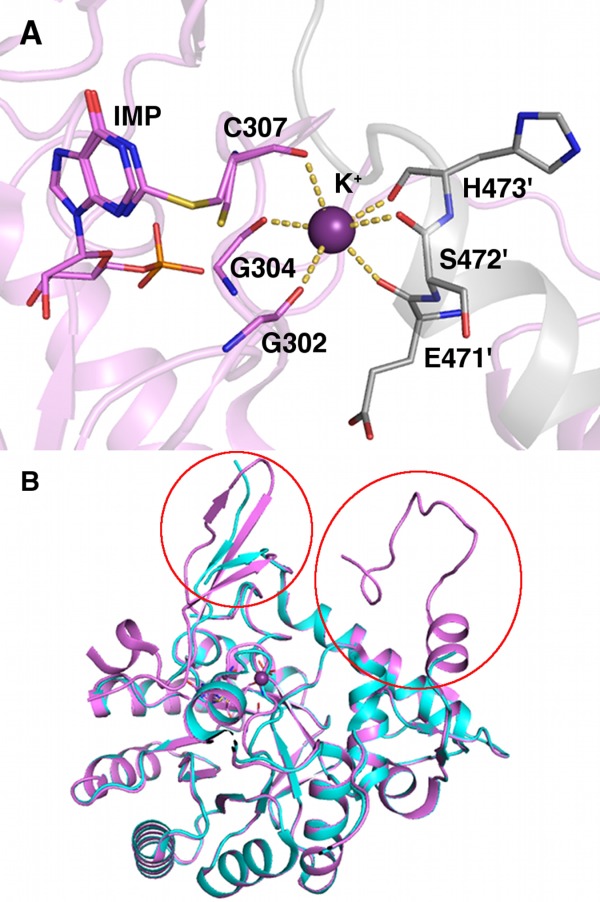
Vibrio cholerae inosine 5′-monophosphate (IMP) dehydrogenase X-ray structure. (A) IMP dehydrogenase potassium-binding site (PDB ID 4qne). Dashed lines represent ion dipole via coordination. Protein carbons are colored pink; symmetry-related carbons, gray; nitrogen, blue; oxygen, red; sulfur, yellow; phosphorus, orange; and potassium, purple. Symmetry-related residues are labeled with primes. (B) Potassium-bound superposed on apo-IMP dehydrogenase (colored cyan, PDB ID 4qq3). Areas structured upon K+ binding noted with red circles.
Inter- and intraprotein interactions: structural stabilization
In some cases the “ion” in the ion–dipole refers to charged residues. These can facilitate a number of inter- and intraprotein interactions and largely act as structural stabilizers. An example of the interprotein ion–dipole interaction is seen in the crystal structure of the core synaptic fusion SNARE complex composed of syntaxin, synaptobrevin, and synaptosome-associated protein of relative molecular mass 25K (SNAP 25).26 This complex forms a twisted, four α helical bundle and is involved in the exocytosis of synaptic vesicles that are critical for neurotransmission. The complex is formed during the fusion of neurotransmitter-bearing vesicles. Following fusion, the complex is bound by α-SNAP, which recruits the ATPase, N-ethylmaleimide-sensitive factor (NSF). Subsequent ATP hydrolysis disassembles the complex, resetting it for further interaction. While the bulk of the protein complex is composed of leucine zipper-like interactions, embedded in the complex core is a conserved three hydrogen bond ion–dipole interaction, referred to as the “ionic” or “0” layer. This interaction occurs between an Arg guanidinium side chain from synaptobrevin-II and the side chain carbonyl groups of three Gln from syntaxin-1A and the N- and C-terminal portions of SNAP25 [Fig. 5(A)]. Unlike many of the other interactions, this ion–dipole has no second shell interactions, such as cooperativity or hydrogen bond arrays. In fact, it is entirely shielded by hydrophobic interactions, which factors into its function. The ionic layer was initially hypothesized to be related to dissociation.26 Further studies confirmed that this layer, in the 3Q:1R ratio, was critical for NSF-mediated dissociation of the complex, and that its role was unrelated to α-SNAP binding or complex reassembly.27 Given that these residues are highly conserved between SNAREs, it seems likely that this is the preferred release mechanism for the fusion protein complex.
Figure 5.
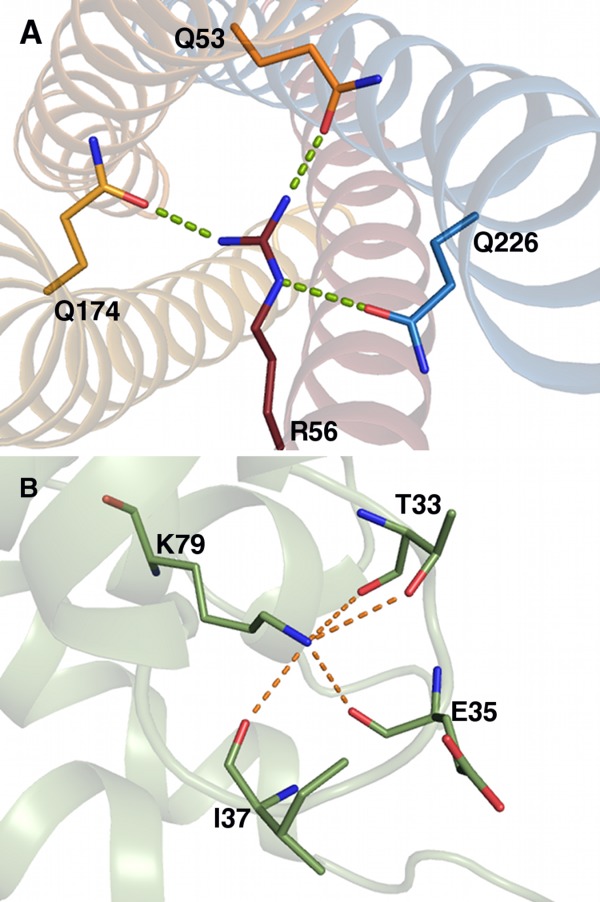
(A) Rattus norvegicus SNARE complex X-ray structure (PDB ID 1sfc). The synaptobrevin Arg interacts with Glns from syntaxin and the N- and C-terminal segments of synaptosome-associated protein of relative molecular mass 25 kDa (SNAP 25). Dashed lines represent ion–dipoles. Syntaxin carbons are colored dark blue; synaptobrevin carbons, red; SNAP 25 carbons, orange; nitrogen, blue; and oxygen, red. (B) Drosophila melanogaster AP180 X-ray structure (PDB ID 1sfc). The Lys stabilizes the active site loop, maintaining an active conformation. Carbons are colored green; nitrogen, blue; and oxygen, red.
In addition to interprotein interactions, charged residue ion–dipoles can exist as part of intraprotein interactions. The AP180 protein family exemplifies this type of interaction. This protein family is made up of assembly proteins for clathrin-mediated endocytosis. The AP180 protein family contains a highly conserved Lys 79, located adjacent to a phosphoinositide-binding site.28 The phosphoinositide-binding site is composed of two positively charged residue segments. The geometry of these segments is maintained by a near circular binding site loop between two α helices (α2-α3). Lys 79 stabilizes this loop through dipolar interactions with four backbone carbonyl oxygens and a hydroxyl side chain of an invariant Thr [Fig. 5(B)]. Undoubtedly, ion–dipole interacting charged residues play important roles in protein function, likely many more than the structural stabilization described here; however, given the sheer number of unique X-ray structures, the task of identifying them is daunting.
Coiled-coil stabilization
One of the more surprising results of the data mining was the identification of ions sequestered in coiled coils and engaged in ion–dipole interactions. This motif is fairly common among pathogenic factors including the tail needle of bacteriophages and trimeric autotransporter adhesins (TAA).29 In the gp26 tail needle protein of P22 bacteriophage, there are two different ion-binding sites located in Domain II, an approximately 100-residue-long trimeric coiled coil.30 The first site binds Ca2+ in an octahedral cage comprised of the side chain carbonyl oxygens from symmetry-related Asn 63s and Gln 66s [Fig. 6(A)]. The second binding site harbors Cl−, which is bound by the side chain NHs of symmetry-related Asn 94s [Fig. 6(B)]. Attempts to exchange the chlorine with bromine failed to displace the chlorine in the site, indicating high specificity. The authors hypothesized that these sites acted as binding guides to ensure the correct specificity and register of the complex, which would offset the reduced stability contributed by polar residues in a hydrophobic binding site.
Figure 6.
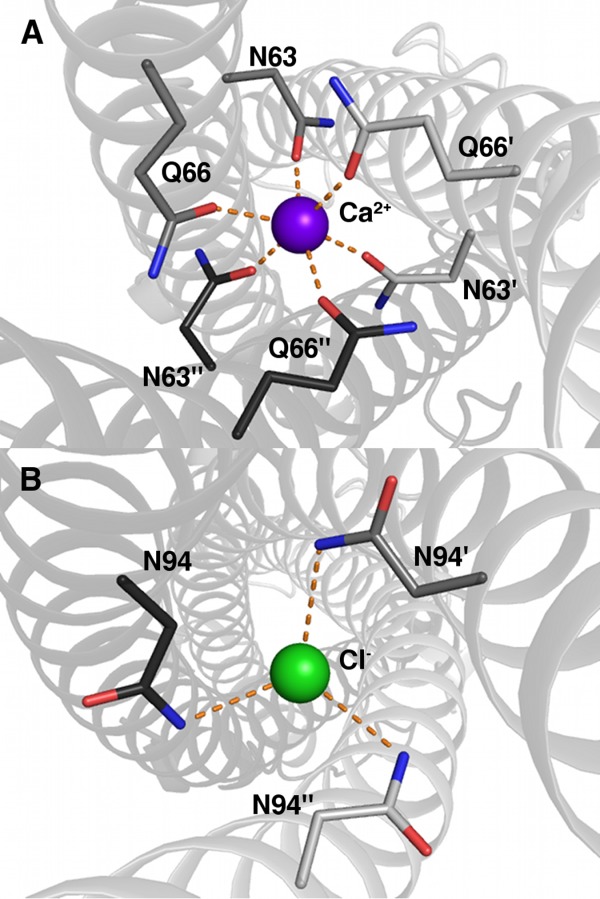
Enterobacteria phage P22 tail needle protein gp26 X-ray structure (PDB ID 3c9i) showing ion–dipole interactions by way of calcium (A) and chloride (B) coordination. Dashed lines represent ion–dipole interactions. (A) Calcium-binding site. Oxygens are colored red; nitrogens, blue; and calcium, purple. Different chain carbons are colored as different shades of gray and labeled with primes. (B) Chlorine-binding site. The protein is labeled and colored as in (A), and chlorine is colored green.
In the TAAs, we see slightly different functional attributes. TAAs have more repetitive motifs with the polar residue, primarily Asn, in the d position of the leucine zipper heptad (referred to as the N@d motif).29 These ion–dipoles are frequently bound to anions such as chlorine but tend to be less specific about their ligand. Experiments showed that the sites could frequently be exchanged with larger atomic ions and molecular ions such as nitrate, although the binding site geometry stayed relatively fixed. Unlike gp26, these binding sites had extensive secondary interactions. The Asn side chain carbonyl frequently formed water bridges to adjacent side chains, specifically the Thr frequently found at position e of the heptad. In this case, the authors theorized that the ion–dipoles served two functions.29 The first was structural specificity. Relocation of the Asn to a different residue of the heptad induced significant stammer and stutter to the coiled coil. The second was facilitating the transport of these highly hydrophobic proteins to the outer membrane. They believe that the improved solvation in the unfolded state would allow the protein to easily traverse the pore and fold correctly on the other side.
Summary
Overall, ion–dipole interactions occur frequently in macromolecules and serve a broad range of critical functions. When evaluating the various functions some patterns begin to emerge. Ion–dipoles are frequently utilized when high specificity or fixed geometry is needed. In the case of transporters or ion channels, they act as gatekeepers, ensuring that only the appropriate atomic or molecular ion can traverse the membrane. In the case of enzymes such as serine proteases, the ion–dipole interaction holds the enzymatic intermediate in a fixed orientation allowing the reaction to proceed. Ion-dipoles are common in situations where fast transitions need to occur, such as transport or quick pKa cycling.8 They also commonly act as a source of structural stability. This can occur by inducing allosterically activating conformational shifts, holding loops in catalytically active conformations, maintaining unfolded protein solubility during cross-membrane transit, and mediating protein–protein interfaces, both directly and by providing scaffolding for correctly orienting monomers to one another. We are only beginning to scratch the surface on the full extent of ion–dipole functionality. Because of the vast number of available structures, we did not focus the abundant ion–dipole interactions found between DNA/RNA phosphate backbones and proteins, especially in ribosomes, and the full extent of their functionality. Currently, our understanding of ion–dipole interactions, combined with secondary or cooperative interactions, and their binding process energetics is limited, and this represents an open field that could significantly improve how we model ligand binding and understand residue interactions.
Acknowledgments
The authors would like to thank Dr. Phil Jeffrey for his thoughtful conversations and generous help data mining the PDB. We would also like to thank Dr. Steven Ludtke and Dwight Noel for providing time and assisting with the Prism computer cluster, which made the data mining possible.
References
- Pflugrath JW, Quiocho FA. Sulphate sequestered in the sulphate-binding protein of Salmonella typhimurium is bound solely by hydrogen bonds. Nature. 1985;314:21–27. doi: 10.1038/314257a0. [DOI] [PubMed] [Google Scholar]
- Cannon R, Pettitt BM, McCammon JA. Sulfate anion in water: model structural, thermodynamic, and dynamic properties. J Phys Chem. 1994;98:6225–6230. [Google Scholar]
- Kelly CP, Cramer CJ, Truhlar DG. Aqueous solvation free energies of ions and ion-water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton. J Phys Chem B. 2006;110:16066–16081. doi: 10.1021/jp063552y. [DOI] [PubMed] [Google Scholar]
- Marcus Y. Ion solvation. Chichester, UK: Wiley; 1985. [Google Scholar]
- Honig B, Yang AS. Free energy balance in protein folding. Adv Prot Chem. 1995;46:27–58. doi: 10.1016/s0065-3233(08)60331-9. [DOI] [PubMed] [Google Scholar]
- Vermersch P, Tesmer J, Quiocho F. Protein–ligand energetics assessed using deoxy and fluorodeoxy sugars in equilibrium binding and high resolution crystallographic studies. J Mol Biol. 1992;226:923–929. doi: 10.1016/0022-2836(92)91041-m. [DOI] [PubMed] [Google Scholar]
- Fersht A, Shi J, Knill-Jones J, Lowe D, Wilkinson A, Blow D, Brick P, Carter P, Waye M, Winter G. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314:235–238. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- Harris TK, Turner GJ. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- Wada A. The alpha-helix as an electric macro-dipole. Adv Biophys [VOL] 1976:1–63. [PubMed] [Google Scholar]
- Hol WG, van Duijnen PT, Berendsen HJ. The alpha-helix dipole and the properties of proteins. Nature. 1978;273:443–446. doi: 10.1038/273443a0. [DOI] [PubMed] [Google Scholar]
- Aqvist J, Luecke H, Quiocho FA, Warshel A. Dipoles localized at helix termini of proteins stabilize charges. Proc Natl Acad Sci USA. 1991;88:2026–2030. doi: 10.1073/pnas.88.5.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He JJ, Quiocho FA. Dominant role of local dipoles in stabilizing uncompensated charges on a sulfate sequestered in a periplasmic active transport protein. Protein Sci. 1993;2:1643–1647. doi: 10.1002/pro.5560021010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti P. An assessment of the effect of the helix dipole in protein structures. Protein Eng. 1994;7:471–474. doi: 10.1093/protein/7.4.471. [DOI] [PubMed] [Google Scholar]
- Faraldo-Gómez J, Roux B. Electrostatics of ion stabilization in a ClC chloride channel homologue from Escherichia coli. J Mol Biol. 2004;339:981–1000. doi: 10.1016/j.jmb.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Vyas NK, Vyas MN, Quiocho FA. Crystal structure of M tuberculosis ABC phosphate transport receptor: specificity and charge compensation dominated by ion–dipole interactions. Structure. 2003;11:765–774. doi: 10.1016/s0969-2126(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Schrodinger LLC. 2010. ). The PyMOL molecular graphics system, Version 1.3r1.
- Pflugrath JW, Quioccho FA. The 2 A resolution structure of the sulfate-binding protein involved in active transport. J Mol Biol. 1988;2000:163–180. doi: 10.1016/0022-2836(88)90341-5. [DOI] [PubMed] [Google Scholar]
- Mao B, Pear MR, McCammon JA, Quiocho FA. Hinge-bending in L-arabinose-binding protein. The "Venus's-flytrap" model. J Biol Chem. 1982;257:1131–1133. [PubMed] [Google Scholar]
- Quiocho FA, Ledvina PS. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol Microbiol. 1996;20:17–25. doi: 10.1111/j.1365-2958.1996.tb02484.x. [DOI] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Noskov SY, Roux B. Ion selectivity in potassium channels. Biophys Chem. 2006;124:279–291. doi: 10.1016/j.bpc.2006.05.033. [DOI] [PubMed] [Google Scholar]
- Tamada T, Kinoshita T, Kurihara K, Adachi M, Ohhara T, Imai K, Kuroki R, Tada T. Combined high-resolution neutron and X-ray analysis of inhibited elastase confirms the active-site oxyanion hole but rules against a low-barrier hydrogen bond. J Am Chem Soc. 2009;131:11033–11040. doi: 10.1021/ja9028846. [DOI] [PubMed] [Google Scholar]
- Riera TV, Zheng L, Josephine HR, Min D, Yang W, Hedstrom L. Allosteric activation via kinetic control: potassium accelerates a conformational change in IMP dehydrogenase. Biochemistry. 2011;50:8508–8518. doi: 10.1021/bi200785s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Scales SJ, Yoo BY, Scheller RH. The ionic layer is required for efficient dissociation of the SNARE complex by alpha-SNAP and NSF. Proc Natl Acad Sci USA. 2001;98:14262–14267. doi: 10.1073/pnas.251547598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Chen J, Maynard J, Zhang B, Quiocho F. A novel all helix fold of the AP180 amino-terminal domain for phosphoinositide binding and clathrin assembly in synaptic vesicle endocytosis. Cell. 2001;104:433–440. doi: 10.1016/s0092-8674(01)00230-6. [DOI] [PubMed] [Google Scholar]
- Hartmann MD, Ridderbusch O, Zeth K, Albrecht R, Testa O, Woolfson DN, Sauer G, Dunin-Horkawicz S, Lupas AN, Alvarez BH. A coiled-coil motif that sequesters ions to the hydrophobic core. Proc Natl Acad Sci USA. 2009;106:16950–16955. doi: 10.1073/pnas.0907256106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olia AS, Casjens S, Cingolani G. Structural plasticity of the phage P22 tail needle gp26 probed with xenon gas. Protein Sci. 2009;18:537–548. doi: 10.1002/pro.53. [DOI] [PMC free article] [PubMed] [Google Scholar]