

Abstract
Chameleon sequences (ChSeqs) refer to sequence strings of identical amino acids that can adopt different conformations in protein structures. Researchers have detected and studied ChSeqs to understand the interplay between local and global interactions in protein structure formation. The different secondary structures adopted by one ChSeq challenge sequence-based secondary structure predictors. With increasing numbers of available Protein Data Bank structures, we here identify a large set of ChSeqs ranging from 6 to 10 residues in length. The homologous ChSeqs discovered highlight the structural plasticity involved in biological function. When compared with previous studies, the set of unrelated ChSeqs found represents an about 20-fold increase in the number of detected sequences, as well as an increase in the longest ChSeq length from 8 to 10 residues. We applied secondary structure predictors on our ChSeqs and found that methods based on a sequence profile outperformed methods based on a single sequence. For the unrelated ChSeqs, the evolutionary information provided by the sequence profile typically allows successful prediction of the prevailing secondary structure adopted in each protein family. Our dataset will facilitate future studies of ChSeqs, as well as interpretations of the interplay between local and nonlocal interactions. A user-friendly web interface for this ChSeq database is available at prodata.swmed.edu/chseq.
Keywords: chameleon sequence, secondary structure, secondary structure prediction, conformational change, structural plasticity, sequence profile, ChSeq, biological function
Introduction
Protein secondary structure elements have been viewed as the fundamental building blocks of protein tertiary structures.1–3 The formation of α-helical and β-strand elements is induced by the interplay between local amino acid propensities and global interactions.4–6 To investigate the influence of global interactions on the formation of secondary structures, researchers have discovered stretches of identical amino acid sequences that adopt distinct conformations, also called as chameleon sequences (ChSeqs).7 Further studies8 revealed the importance of such structural ambiguity in ChSeqs for a better understanding of amyloid diseases,9–11 where native proteins can refold into β-strands to stabilize the pathogenic assemblies. Additionally, ChSeqs are reported to contribute to functional diversity described in alternatively spliced isoforms.12
The first search for ChSeqs in proteins was carried out by Kabsch and Sander.13 They reported 25 chameleon pentapeptides from 62 protein structures. From then on, researchers have shown increased interest in the detection of ChSeqs.12,14–19 Besides analyzing the amino acid properties of ChSeqs, scientists have used ChSeqs to evaluate the performance of secondary structure predictors.12,20,21 Collectively, such evaluation studies showed that methods based on sequence profiles outperformed methods based on single sequences.22 Surprisingly, the evaluations of neural network-based secondary structure predictors have shown that profile-based methods predict ChSeqs with similar efficiency as on sequences where alternative conformations are never observed.21,23
To better understand the principles of protein structure changes, aided with increasing numbers of available Protein Data Bank (PDB)24 structures, we searched for ChSeqs and identified a large set ranging from 6 to 10 in residue length. ChSeqs found in homologous structures tend to reveal conformational changes involved in switching protein functional states. Alternatively, the different environments surrounding ChSeqs from unrelated structures tend to dictate their conformation. We found that the evolutionary information provided by the sequence profiles can successfully predict the secondary structure feature that prevails in a given protein family. We present our dataset in a user-friendly web interface available at prodata.swmed.edu/chseq, as well as in csv format at http://prodata.swmed.edu/chseq/downloads/.
Results and Discussion
Our comprehensive search for ChSeqs identified 19,603 (20 homologous and 19,583 unrelated) ChSeqs of entirely helix-to-strand transitions (Fig. 1) in the current nonredundant PDB database. For a fair comparison with the latest study,18 which detected ChSeqs with any secondary structure difference in the sequence strings, we also loosened our criteria and detected 128,703 ChSeqs in unrelated proteins with any helix-to-strand transition in the middle two residues of the sequence strings.
Figure 1.
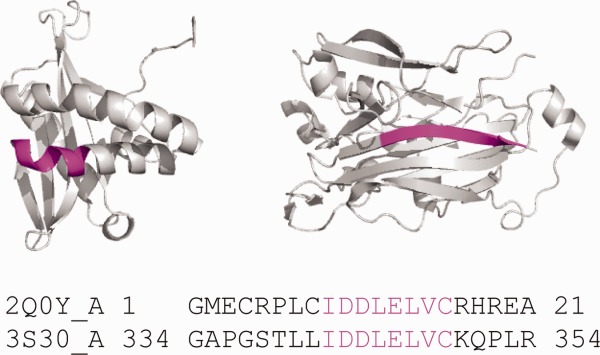
Chameleon sequences (ChSeqs) and their distributions in homologous and unrelated proteins. A ChSeq adopting different conformations. The pdb codes are 2Q0Y (left) and 3S30 (right), respectively. ChSeqs are colored magenta in both the structure and sequence.
ChSeqs in homologous structures highlight dramatic conformational changes
We detected 20 ChSeqs that undergo complete helix-to-strand transitions in homologous structures. We found 12 of the 20 ChSeqs to be associated with biological functions (Table1). Based on their experimental studies, the biological processes of the 12 ChSeqs can be classified into four types. First, the conformational changes upon activation (6 ChSeqs); these include the fusion protein of respiratory syncytial virus (2 ChSeqs),25–27 the fusion protein of paramyxovirus (2 ChSeqs),28,29 the 50S ribosomal protein L24,30,31 and a cysteine proteinase.32,33 Second, the changes upon substrate binding (3 ChSeqs); these include the transcription factor Rfah (2 ChSeqs)34,35 and the 4Fe–4S cluster domain of human DNA primase.36,37 Third, the changes resulting from cleavage or insertion of a peptide (2 ChSeqs); these include the serine protease inhibitor ovalbumin38,39 and the cell surface adhesion molecule neurexin 1β.40,41 Fourth, the changes upon oligomerization (1 ChSeq); this includes a tubulin acetyltransferase.42,43
Table 1.
ChSeqs in Homologous Proteins
| Sequence | pdb1 | length1 | pdb2 | length2 | Alignment lengtha | Alignment fractionb (%) | Annotationc | Protein named |
|---|---|---|---|---|---|---|---|---|
| VSVLTSKVLD | 4jhF | 498 | 3rkiA | 528 | 454 | 86 | Functional | Fusion protein of respiratory syncytial virus |
| MDSKLRCVFE | 3ikkA | 127 | 2mdkA | 125 | 124 | 98 | Unpublished | hVAPB MSP domain |
| IKASQELV | 3n4pA | 279 | 2kn8A | 68 | 68 | 24 | Fragment | Human cytomegalovirus terminase nuclease domain |
| SAEAGVDAe | 1jtiA | 385 | 1ovaD | 386 | 383 | 99 | Functional | Serine protease inhibitor ovalbumin |
| AKEEAIKE | 2kdmA | 56 | 2jwsA | 56 | 51 | 91 | Engineer | GA95 and GB95 |
| VKYKAKLIe | 1p3rA | 160 | 2lswA | 40 | 26 | 16 | Fragment | Phosphotyrosin binding domain (Ptb) of mouse disabled 2 |
| EIKHSVK | 2lclA | 66 | 2ougA | 162 | 62 | 38 | Functional | Transcription factor Rfah |
| RSMLLLN | 2lclA | 66 | 2ougA | 162 | 62 | 38 | Functional | Transcription factor Rfah |
| LGRVVDE | 3mw3A | 208 | 2r1bA | 220 | 168 | 76 | Functional | Cell surface adhesion molecule neurexin 1β |
| LDPLEVH | 3lruA | 160 | 4jkeA | 222 | 160 | 72 | Unpublished | Human Prp8 Rnase H-like domain |
| QSLGTAVe | 4gipD | 409 | 1svfA | 64 | 63 | 15 | Functional | Fusion protein of paramyxovirus |
| FKKIKVL | 2rfeA | 324 | 1z9iA | 53 | 20 | 6 | Fragment | Epidermal growth factor receptor |
| KILVQAe | 1p3hA | 99 | 1p82A | 25 | 24 | 24 | Fragment | Mycobacterium tuberculosis chaperonin 10 |
| RLFQVK | 3ffnA | 782 | 1solA | 20 | 20 | 3 | Fragment | Calcium-free human gelsolin |
| VADVVQe | 4gipD | 409 | 1svfA | 64 | 63 | 15 | Functional | Fusion protein of paramyxovirus |
| KKVRFF | 3r8sU | 102 | 2gyaS | 99 | 99 | 97 | Functional | 50S ribosomal protein L24 |
| LIEYFR | 3ly6A | 697 | 2q3zA | 687 | 683 | 98 | Functional | Cysteine proteinase |
| SYNIRH | 3l9qA | 195 | 3q36A | 192 | 186 | 95 | Functional | 4Fe–4S cluster domain of human DNA primase |
| KAVVSL | 4jhwF | 498 | 3rkiA | 528 | 454 | 86 | Functional | Fusion protein of respiratory syncytial virus |
| TVIDEL | 4h6zA | 190 | 4hkfA | 191 | 186 | 97 | Functional | Tubulin acetyltransferase |
Alignment length: the length of the alignments between pdb1 and pdb2.
Alignment fraction: the alignment length divided by the maximum of length1 and length2.
Annotation: categorization of conformational differences in ChSeqs, including conformational changes (i) with associated function (functional), (ii) in protein of diverse lengths (fragment), (iii) involving unpublished structures (unpublished), and (iv) in engineered proteins (engineer).
Protein name: the protein name summarized from the pdb entries.
One structure of the ChSeqs recorded in the DynDom database.
Both structures of the ChSeqs recorded in the DynDom database.
The fusion protein in respiratory syncytial virus25–27 contains one of the longest ChSeqs (10 residues), as well as another ChSeq of six residues (Fig. 2). In the prefusion structure (pdb: 4jhw, Chain F), the two ChSeqs together form a β3176–181/β4185–194 hairpin that packs against the “fusion peptide.”27 In the profusion structure (pdb: 3rki, Chain A), each of the ChSeq strands transforms into a helical conformation, extending the “fusion peptide” helix and packing with the C-terminal helix to form a coiled coil stalk for membrane insertion.26 As illustrated in this example, the ChSeqs undergo dramatic conformational changes and participate in the transition between the protein's inactive and active states.
Figure 2.
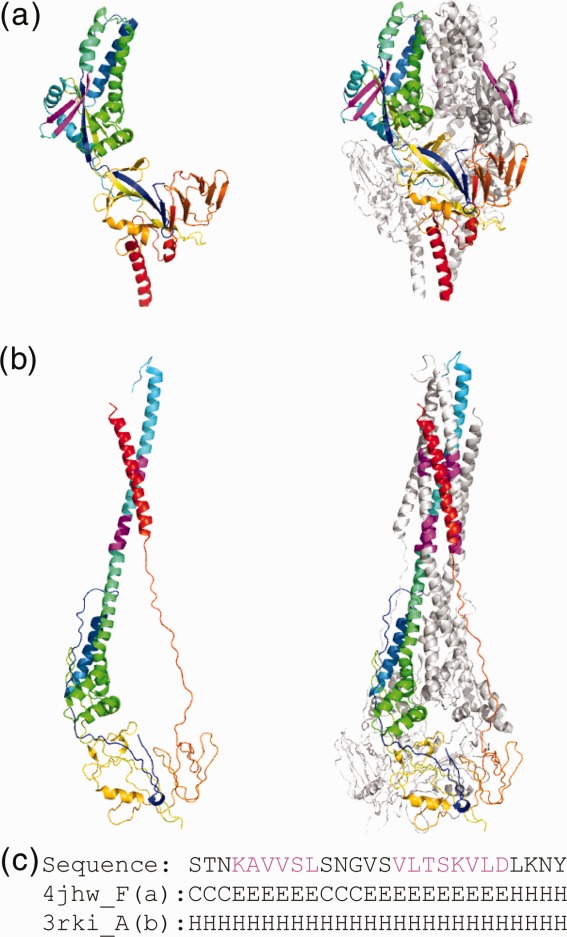
Conformational changes in Type I fusion protein of respiratory syncytial virus. (a) ChSeqs (colored magenta) between residues 185–194 and 176–181 (pdb: 4jhw, Chain F) form a β-hairpin in the prefusion complex (pdb: 4jhw) illustrated in rainbow as monomeric (left panel) and trimeric (right panel). (b) The ChSeqs form helical conformations in the profusion complex (pdb: 3rki, Chain A) illustrated as above. (c) The sequence and the corresponding secondary structures of the ChSeq segments in prefusion (Line 2: 4jhw) and profusion (Line 3: 3rki) complexes.
The remaining eight ChSeq examples lack experimentally verified functions. Five of them come from structures of substantially different lengths (Table1). The longer length structures form complete protein domains (determined by X-ray crystallography), whereas the shorter length structures are limited to several secondary structure elements (solved by NMR). As exemplified by the DH domains of Dab2 (illustrated in Fig. 3),44,45 we found that all the ChSeqs from truncated structures exhibit helical conformation. Alternately, the ChSeqs from the complete domains form β-strands. For example, in the complete DH domain (Fig. 3, pdb: 1p3r), the ChSeq β-strand (magenta) integrates into the center of an open β-barrel, forming a hydrogen bonding network with two neighboring β-strands (residues 92–97 and 145–151) that are missing in the truncated structure (Fig. 3, pdb: 2lsw). In the absence of the stabilizing hydrogen bonding network provided by the β-barrel, the single β-strand transforms into an α-helix in the shorter length structures. All five ChSeqs from truncated domains exhibit similar conformational transitions, suggesting that the helical conformations resulting from truncations are nonphysiological and caused by the lack of sufficient hydrogen bonding networks.
Figure 3.
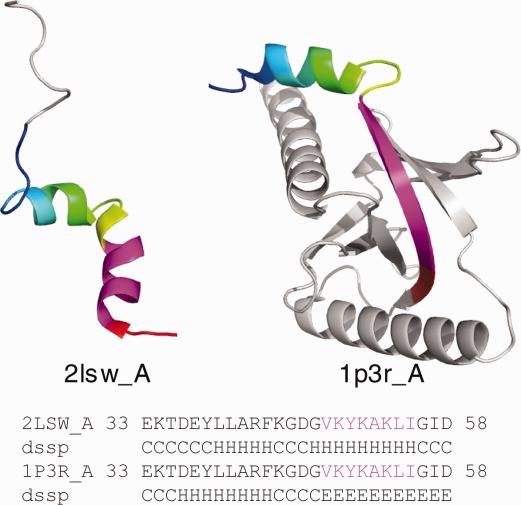
ChSeqs in proteins of different lengths. The region of identical sequences is shown in the alignment and colored rainbow in the structures. ChSeqs are colored magenta.
Two of the remaining three ChSeq examples include unpublished structures. For one, an unpublished NMR structure (pdb: 2mdk) of a human major sperm protein (MSP) domain contains an α-helix, whereas the crystal structure (pdb: 3ikk)46 contains a β-strand. For the other, an unpublished crystal structure (pdb: 3lru) of a truncated human pre-mRNA processing factor 8 (Prp8) RNase H-like domain47 exhibits a β-strand in a sheet formed by a swapped dimer, whereas a crystal structure of the complete domain (pdb: 4jke) has an α-helix. The last homologous ChSeq (sequence: AKEEAIKE) is from two engineered proteins designed to explore the mutation pathways for a single mutation to switch from an IgG-binding fold (α + β topology) into an albumin-binding fold (all-α topology).48,49
Previous searches for ChSeqs either did not distinguish homologies of the ChSeqs12,16 or focused their searches on unrelated ChSeqs.13,15–19 However, some studies have investigated conformational diversity and structural motions present in the structures.50–56 We examined whether our ChSeqs are also present in these studies. Although these studies collected redundant chains of close homologs (and we removed redundancy), five of the homologous ChSeqs we identified have been recorded in the “dynamic domains” (DynDom) database.54 Recently, the database of conformational diversity in the native state of proteins (CoDNaS)56 characterized structures of 100% sequence identity. The database for protein structural change upon ligand binding (PSCDB)55 concentrated on the conformational changes on binding small molecules. As we used nonredundant structures and no conformational changes induced by binding small molecule were detected, none of our ChSeqs were reported in these two most recent databases. We attempted to compare our ChSeqs with the database of protein conformational diversity (PCDB)50; however, the dataset seems to be no longer accessible through its website.
ChSeqs in unrelated structures illustrate the interplay between local and nonlocal interactions
We detected ChSeqs in unrelated structures using two different criteria. The more stringent search aims to detect entirely helix-to-strand transitions and detected 19,583 ChSeqs. However, the results using this set of criteria are not suitable for direct comparison with previous works. Therefore, we also searched with a looser criteria that allows shorter secondary structural element transitions; the detected ChSeqs increased to 128,703. When compared with previous studies (see Table2), this search identified approximately 20-fold more ChSeqs. This increase corresponds well with the approximately 20-fold growth in the data size of nonredundant PDB structures (from 3214 to 67,589). The large number of hexamers detected is more than double the pentamer count in the most recent study.18 We also increase the length of the longest ChSeqs identified from 8 to 10 (with four 10-mers seen here).18
Table 2.
Comparison of studies searching for ChSeqs
| Authors | Year | Number of proteins | 5mer | 8mer | >8mer |
|---|---|---|---|---|---|
| Kabsch and Sander13 | 1984 | 62 | 25 | — | — |
| Sudharsanam57 | 1998 | 828 | — | (4) | — |
| Casadio and coworkers21 | 2000 | 822 | 2452 | — | — |
| Rackovsky and Kuznetsov17 | 2003 | 1647 | 45,391 | 15 | — |
| Saravanan and Selvaraj58 | 2011 | 3124 | 61,821 | 30 | — |
| Grishin and coworkers59 | 2014 | 67,589 | 118,833 (6mer)a | 516 | 40 |
This table is generated based on the numbers in Table1 of Ref.18. For a list of ChSeqs with more than eight residues, please visit http://prodata.swmed.edu/wenlin/pdb_survey2/index.cgi/pages/unrelated/middle-match.
As our lower limit for ChSeq length is six, we assign the number of 6mers in the column of 5mers for our study.
ChSeqs that form different secondary structures in unrelated proteins were used to analyze the interplay between local and nonlocal interactions.16–18 Such interactions can be illustrated in one of the 10-residue ChSeqs detected by loose criterion (Fig. 4). This ChSeq (sequence: QGTAVVVSAA) is found in an immunoglobulin fold (ECOD domain ID: e4jb9H4) and a Rossmann fold (ECOD domain ID: e1vl6A1). In the immunoglobulin structure (pdb: 4jb9), the ChSeq forms a β-strand (residues 105–114) embedded in a β-sandwich; in the Rossmann-fold structure (pdb: 1v16), it forms a helix (residues 157–166). In this example, the ChSeq sequence includes a number of strong α-helix formers (e.g., A) and strong β-strand formers (e.g., V), as measured by Chou-Fasman parameters.60 This mix of strong but ambiguous α-helical and β-strand propensities is similar to that observed in a previous study of helix-to-strand transitions.16 In the immunoglobulin structure, nearby β-strands form a hydrogen-bonding network with the ChSeq to stabilize the extended conformation; in the Rossmann fold, the lack of surrounding hydrogen bonding partners allows the ChSeq to form a helix induced by strong α-helix propensity of its sequence [Fig. 4(b)]. Therefore, in this example, the global interactions impose constraints on the sequences of ambiguous secondary structure propensity, guiding local interactions to stabilize the respective secondary structures.
Figure 4.
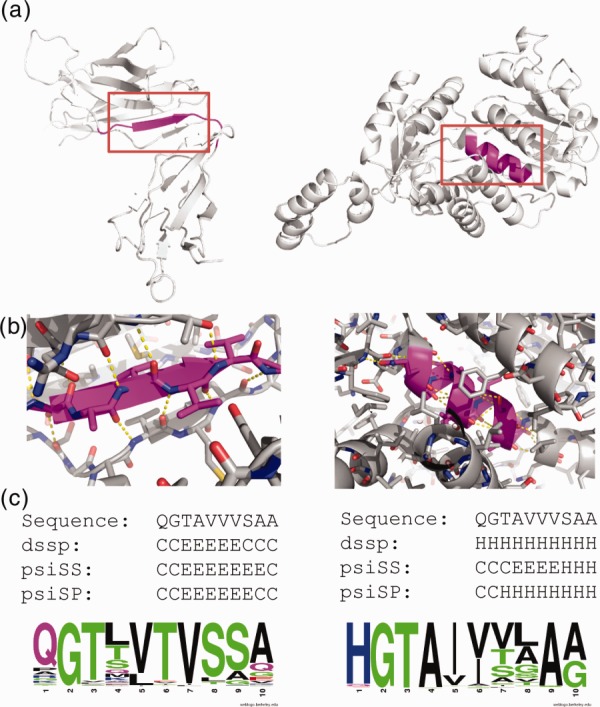
Example of a 10-residue ChSeq in unrelated proteins. (a) ChSeqs (magenta) in the structures 4JB9 (left) and 1VL6 (right). (b) Close-ups of red box regions of panel (a) with some backbone hydrogen bonds (dashed yellow lines) shown. (c) Sequence, observed secondary structure, and psiS- and psiP-predicted secondary structure are shown along with weblogo pictures visualizing the sequence profiles in each protein family.
In the above example (Fig. 4), the ChSeq has a mixture of amino acids with ambiguous secondary structure preferences. We compared the amino acid frequencies of all detected ChSeqs (under the stringent criterion) with the amino acid frequencies of proteins in the Swiss-Prot database (Fig. 5). When compared with the frequencies in Swiss-Prot (green line in Fig. 5), the residues Ile, Val, Ala, and Leu are overrepresented in ChSeqs. As pointed out in previous analyses,12,16 these residues have strong propensities in forming either α-helix (residues) or β-strand (residues). Alternately, Pro is underrepresented in ChSeqs consistent with its tendency to be both a helix and a strand breaker. Other residues with low Chou-Fasman60 helical or strand propensities, that is, Gly, Ser, Asp, and Asn, also show low frequencies in ChSeqs. The low frequency of Cys can be explained by its potential to reduce structural flexibility through forming disulfide bonds.12,16 The low frequencies of Trp, His, Met, and Gln were also observed previously.12,16
Figure 5.
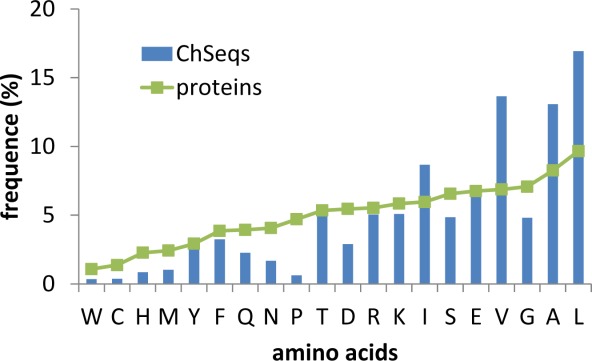
Amino acid composition of ChSeqs. Amino acid frequencies in ChSeqs (blue) are compared with the frequencies seen in proteins from the Swiss-Prot database (green).
As has been noted15 and was seen in the examples in Figure 4, ChSeqs tend to be largely buried in the protein core, forming interactions with surrounding secondary structure elements. To study the solvent exposure of residues in ChSeqs, we calculated the relative solvent accessibility (RSA), which indicates the percentage of surface area exposed to the solvent for a residue (Fig. 6). In general, when compared with residues in proteins, the distribution of RSAs in ChSeqs shows more fully buried residues (<5% RSA) and many fewer highly exposed residues (>85% RSA). However, when compared with residues contained in β-strands and α-helices, the distribution of RSAs in ChSeqs is comparable (green), indicating that the RSA decrease may be simply a result of the constraints of being in secondary structures.
Figure 6.
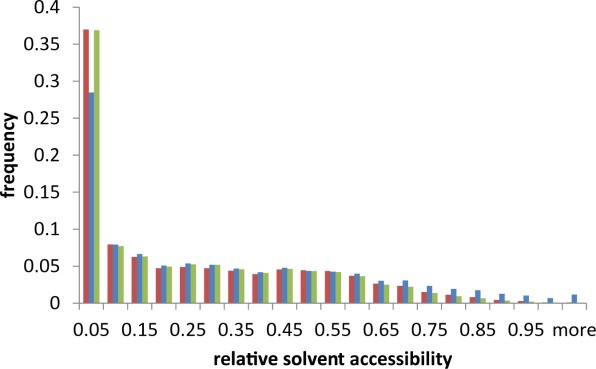
ChSeqs are similarly buried as residues in strands and helices. Histogram of the RSA distribution of residues in “stringent” ChSeqs (red), in a set of 1000 random proteins (blue), and in a set of “random” β-strands and α-helices (green).
Evaluation of secondary structure predictors on ChSeqs highlights the advantage of profile-based predictors
ChSeqs may be the most stringent test set for secondary structure predictors.20,21 Previous studies have applied profile-based secondary structure prediction methods to unrelated ChSeqs and have shown their high accuracy in predicting ChSeq secondary structures.12,21,23 To study the influence of the evolutionary information on the success of profile-based predictors, we applied both a profile-based predictor, here called psiP (for PSIPRED using sequence profile), and a single sequence-based predictor, here called psiS (for PSIPRED using single sequence), to the set of 655 ChSeqs with more than six residues. Consistent with previous evaluations, for the overwhelming majority, 92% (605/655) of ChSeqs, the profile-based psiP predicted correct secondary structures for both forms. Influenced by flanking residues, single-sequence-based psiS is in principle able to produce distinct predictions for sequences in a ChSeq pair; however, correct psiS secondary structure predictions for both forms are obtained for fewer than half, 42% (274/655), of the ChSeqs. Among the 58% ChSeqs that had incorrect predictions, for 96% (i.e., 56% of the 655 ChSeqs), the correct secondary structure is obtained for one of the families but not the other.
As was seen for the example ChSeq shown in Figure 4, psiS produced mainly β-strand predictions for both structures, whereas psiP could successfully distinguish the secondary structures from different protein structures. As shown in the secondary structure predictions for the ChSeq helix in the Rossmann fold [Fig. 4(c)], psiS predicts the “AVVV” stretch as a strand. However, the family profile includes alternate residues that allow psiP to correctly predict the AVVV as a helix. To quantify the prevalence of this type of alternate single-sequence-based prediction, we computed a prediction P-value (PPV) to indicate the probability of observing a given psiS prediction based on the psiS predictions carried out for every sequence in a given protein family. A lower PPV means the single-sequence prediction is more dissimilar to the prevailing psiS prediction among members of a protein family. The PPV distribution of incorrect psiS predictions for ChSeqs is different from the distribution of psiS predictions for random sequences without observed helix-to-strand transitions (green line in Fig. 7). For incorrect psiS ChSeq predictions [blue bars in Fig. 7(a)], about one-third of the PPVs are below 0.05, indicating that the predictions significantly deviate from the prevailing predictions of family members. On the other hand, the distribution of ChSeqs with correct psiS prediction closely approximates the random distribution except at PPVs < 0.15 [Fig. 7(b)].
Figure 7.
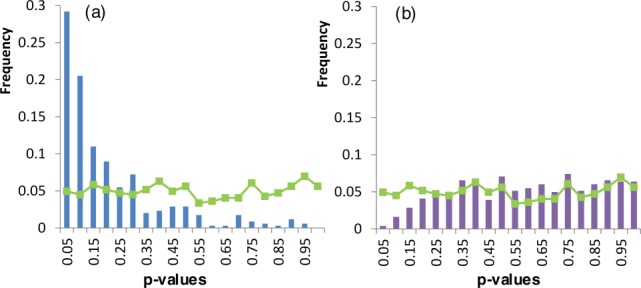
Histograms of prediction P-values (PPVs) for ChSeqs with (a) incorrect psiS predictions and (b) correct psiS predictions. Green lines represent the PPVs for controls computed from a random sequence from the family.
To study the influence of secondary structure type on the PPV distributions, we separately analyzed the helix and strand conformations. The PPV distributions [Fig. 8(a)] show that ChSeqs adopting strands have significantly lower PPVs than ChSeqs adopting helices, with a two-sided Kolmogorov–Smirnov (K-S) test P-value of 1.36 e − 06. This indicates that psiS predictions for β-strands tend to deviate more from their prevailing family predictions than do the predictions for α-helices. This explains a clear asymmetry in the predictability of helices and strands in that, among all the ChSeqs, 42% had both α-helices and β-strands predicted correctly, 40% had only the α-helix predicted correctly, 16% had only the β-strand predicted correctly, and 2% had neither predicted correctly. Interestingly, if we further divide each conformation into those having correct versus incorrect psiS predictions, the PPV distributions are not distinguishable for either the correctly [Fig. 8(b)] or the incorrectly [Fig. 8(c)] predicted ChSeqs, with the K-S test P-values to be 0.21 and 0.39, respectively. Correctly predicted ChSeqs of both conformations tend to have higher PPVs [Fig. 8(b)], and incorrectly predicted ChSeqs of both conformations show a trend for lower PPVs [Fig. 8(c)]. Therefore, psiS predictions from α-helices tend to match the prevailing family prediction more than β-strands, consistent with the higher fraction of correct predictions for α-helices.
Figure 8.
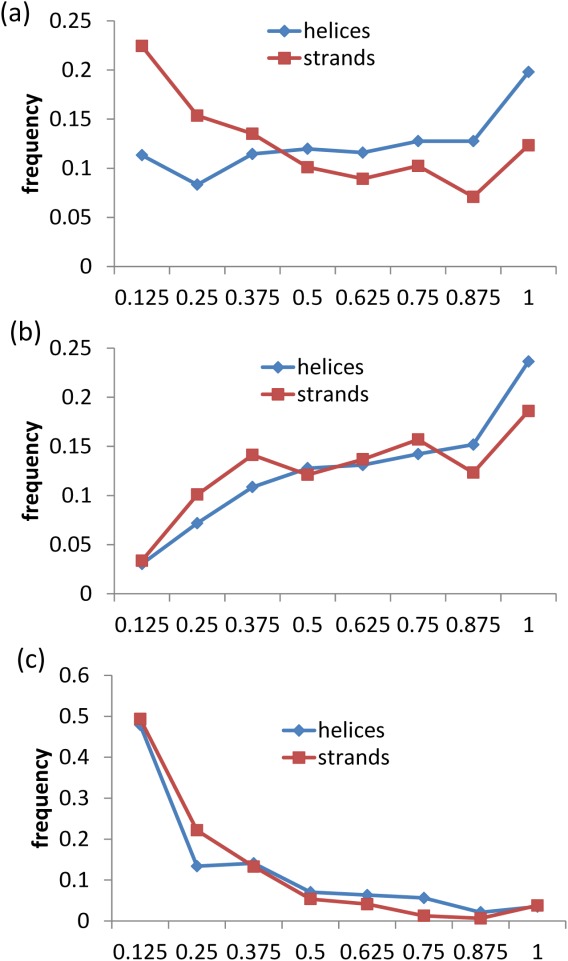
Histograms of PPVs for ChSeqs with helical (red) and stranded (blue) conformations. All studied ChSeqs (a) are further divided into those with correct psiS predictions (b) and incorrect predictions (c).
Cross-validation of homologies by ECOD identified ChSeqs in unrelated regions of homologous protein folds
ECOD is an evolutionary classification of protein domains based on structural and sequence similarity, where structures within the same H-group are considered homologs.59 As a cross-check of our homology assignments, we applied the ECOD classification to our BLAST-based ChSeq homologs. ECOD allowed us to correct classifications of three ChSeqs that are falsely found as homologs by BLAST due to multidomain problem (Supporting Information Table S1).61 Additionally, ECOD helped us to filter 65 ChSeqs that were in homologous proteins but did not represent homologous parts of the proteins (Supporting Information Table S1, recorded as unrelated ChSeqs in the final dataset). For example, the ChSeq shown in Figure 9 (with sequence: AIVLSKY) is from two structures classified by ECOD as homologous Rossmann folds (pdb: 3id6 and 4lg1); however, the ChSeq is in the N-terminal helix in one structure (4lg1) but in the C-terminal strand in another structure (3id6). The pairwise alignment of these two structure sequences is only limited to the ChSeq region (E-value 0.12), which is not sufficient to support their homology. Examples of unrelated ChSeqs in homologous folds are mainly concentrated in three large H-groups: the Rossmann fold (20 ChSeqs), the TIM barrel (16 ChSeqs), and the P-loop domain (8 ChSeqs).
Figure 9.
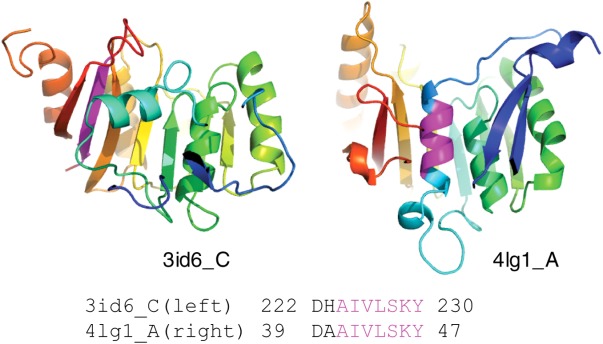
Nonhomologous ChSeq in homologous proteins. The ChSeq (purple) is highlighted in the two ribbon diagrams, and the BLAST alignment is shown.
We did not include 400 ChSeqs (1.8% of total ChSeqs) in our final dataset, as they could not be mapped to current ECOD domains. Those sequences include (i) 257 ChSeqs mapped to ECOD as peptides, coiled coils, fragments, and artificial sequences, for which homology cannot be inferred with confidence; and (ii) 143 ChSeqs mapped to the protein regions not covered by ECOD domains due to ECOD domain parsing limitations. These 400 ChSeqs are available at http://prodata.swmed.edu/wenlin/pdb_survey2/index.cgi/artifacts/.
A user-friendly web interface to the ChSeq database integrates a wide range of relevant information
For making this information accessible, we imported our dataset into a web interface (Fig. 10) that integrates structural and sequence information relevant for a ChSeq analysis. For efficiency, the default display includes only a single representative PDB entry for each form of a ChSeq, with a “show all PDB chains for this group” option to display all relevant PDB entries. Cross-database information, including protein names from PDB24 and H-groups from ECOD,59 is provided at the top of each panel. For more in-depth study of the structures, one can load the structure in JSmol62 or download PyMol63 session files (having a white protein chain with magenta ChSeq). In addition, below each image, the secondary structure (from PDB and psiS and psiP predictions) and sequence information (including gap fraction) are given along with a weblogo64 visualization of the sequence profile of the ChSeq region. The full alignment of the protein family is accessible via the link on the right of the weblogo image. This web interface to the ChSeq database is available through a portal at prodata.swmed.edu/chseq.
Figure 10.
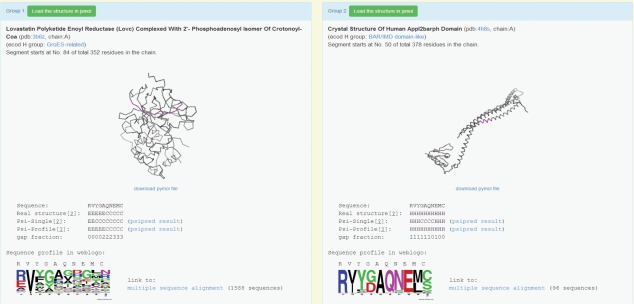
An example web interface. This shows a ChSeq that occurs in unrelated proteins (accessible at http://prodata.swmed.edu/wenlin/pdb_survey2/index.cgi/new_dssp/middle-match/RVYGAQNEMC/).
Conclusions
We have developed a rather comprehensive, updated dataset of ChSeqs. Interestingly, among the 20 examples of homologous ChSeqs that undergo helix-to-strand conformational changes, 12 were found to be involved in biological function. When compared with the most comprehensive previous study, we achieved a roughly 20-fold increase in detected unrelated ChSeqs (similar to the growth of the nonredundant PDB database in the relevant timeframe) and increased the length of the longest ChSeq from 8 to 10 residues. We find that for the ∼56% of ChSeqs, for which a prediction based on single sequences is correct for only one of the families, there is a strong tendency for the sequence to be an “outlier” sequence for the other family. Its presence as a minority type of sequence in the family explains why it does not negatively impact the success of profile-based secondary structure predictions, which effectively capture the information present in the prevailing sequence patterns present in the family. A user-friendly web interface to the ChSeq database (available at prodata.swmed.edu/chseq) will facilitate future studies of ChSeqs and the gleaning of insights they can provide into the interplay between the influences of local and nonlocal interactions on protein structures.
Materials and Methods
Detection of ChSeqs
The nonredundant PDB database, which combines structures of an identical sequence into one record, was downloaded on February 14, 2014, from ftp://ftp.ncbi.nih.gov/blast/db/FASTA/pdbaa.gz. The structures with Cα-atoms only were filtered. To select representative structures for each record, we prioritized crystal structures with the best resolution, followed by NMR structures, and then EM structures. We used a sliding window ranging from 6 to 40 to detect identical sequence strings. We further filtered out sequence strings contained in a longer sequence. The DSSP software65 was used to define ChSeq secondary structures from representative PDBs. We followed the DSSP nomenclature66 and reduced the eight DSSP secondary structure states into three: (1) “H,” “G,” and “I” as “H,” (2) “E” and “B” as “E,” and (3) others as “C.” As a stringent criterion, we define ChSeqs1 as sequence strings with transitions between α-helices (H) and β-strands (E) in every position. To make our statistics comparable with previous studies, we also applied a looser criterion to define ChSeqs2 as segments for which helix-to-strand transitions occurred for the middle two residues of identical segments from unrelated proteins (for how relatedness was defined, see the next section).
Classification of ChSeqs by protein homology
We ran BLAST against the nonredundant PDB database to identify homologs for each structure. BLAST hits with an E-value better than 1 e−5 were considered homologs. As a cross-check, we also applied the ECOD59 classification to our dataset using H-groups (similar to SCOP67 superfamily) to define homologs. We manually inspected all the homologous ChSeqs detected by BLAST and ECOD to make sure that (1) structures of a homologous ChSeq are from only one ECOD H-group and that (2) homologous ChSeqs are aligned in the BLAST alignment with confident statistics.
Evaluation of PSIPRED prediction on ChSeqs
By default, the PSIPRED68 program runs PSIBLAST69 and uses the statistics from the sequence profile to perform prediction (denoted as psiP for “Profile”). To study the influence of the sequence profile, we tweaked PSIPRED to use the statistics from the input sequence alone without running PSIBLAST (denoted as psiS for “Single” sequence). To evaluate the performance of psiP and psiS, we compared the secondary structure prediction with that found in the representative structures. The DSSP program has relatively strict criteria in defining α-helices and β-strands. As “C” might contain atypical helices or strands, we allowed mismatches against Cs and only penalized incorrect predictions between Es and Hs. We also allowed errors in defining the secondary structure boundary and only penalized the E and H mismatches in the middle four residues of a ChSeq. Therefore, a correct prediction is defined as a prediction with no H versus E mismatches in the middle four residues of a ChSeq. To quantify the magnitude of the difference between the psiS and psiP predictions for a given sequence, we extracted the multiple sequence alignments (MSAs) used in psiP and calculated the prediction distance (Dp) for each sequence in the MSA using the following equation:
where n is the length of the ChSeq, || || is the operator to calculate a Euclidean distance, and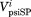 and
and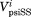 are the probability vectors of secondary structure predictions for position i from psiP and psiS, respectively.
are the probability vectors of secondary structure predictions for position i from psiP and psiS, respectively.
To indicate the extent to which the psiS of a sequence diverges from those that would be predicted by single sequences within its family, we estimated a PPV using the following equation:
where Ntail is the number of Dps larger than the Dp of the sequence, and Nall is the number of proteins in the MSA. To ensure the statistical significance of the PPVs, we filtered out protein families with Nall < 150.
Calculation of amino acid frequency and solvent accessibility
For the sequences of unrelated ChSeqs1 (i.e., those stringently defined), we calculated the frequencies of the 20 amino acid types. A set of reference frequencies of amino acids was obtained by the amino acid frequencies of proteins in the Swiss-Prot70 database available at http://web.expasy.org/protscale/pscale/A.A.Swiss-Prot.html. RSA was calculated as dividing the solvent accessibility (in Å2) observed for each residue in a protein of interest (from DSSP) by the total surface area of the residue.71 To estimate the RSA distribution in proteins, we sampled 1000 proteins from ChSeq-containing structures and calculated the RSA for every residue. To estimate the RSA distribution of α-helices and β-strands of length N (for comparison with ChSeqs of length N), we randomly selected a segment of N residues from the secondary structure elements (excluding coils) of ChSeq-containing structures and calculated the RSA for every residue.
Filtering ambiguous and non-native sequences
We used the PDBx/mmCIF file of each structure in the PDB database to convert modified residues to their original (parent) residues. After our conversion, sequences containing unknown residues remained (e.g., the unknown residues in Chain D of pdb: 4hu6), which hindered our definition of identical sequence strings. Additionally, we detected protein expression tags near the termini by checking sequence conservation. Homologous sequences were retrieved by PSI-BLAST with three iterations against the UniRef90 database. The results were filtered to include sequences with E-value better than 0.001, identity larger than 30%, and gap positions smaller than 50% of the sequence length. The resulting positional gap fractions were calculated and rescaled to 0–9 (9 is more gapped). If positions within 20 residues of either terminus had an average positional gap fraction larger than 6, we categorized the termini as protein expression tags. These ambiguous and non-native sequences (8.5% of total ChSeqs) can be found at http://prodata.swmed.edu/wenlin/pdb_survey2/index.cgi/artifacts/.
Preparation of the web interface
To reduce redundancy for web visualization, we clustered the ChSeqs by their secondary structure elements such that each cluster contains ChSeqs of identical secondary structures. For unrelated ChSeqs, these clusters were further split according to ECOD H-groups. By default, we show the most diverse representative pair on top. In the downloadable PyMol63 sessions of the structures, we limit to unique chains containing ChSeqs to reduce the file size. The MSAs used in detecting protein expression tags are included in the web interface.
Acknowledgments
The authors thank Dr. Dustin Schaeffer and Yuxing Liao for PDB structure parsing and ECOD domain parsing.
Supporting Information
Additional Supporting Information may be found in the online version of this article
Supporting Information
References
- Ballew RM, Sabelko J, Gruebele M. Direct observation of fast protein folding: the initial collapse of apomyoglobin. Proc Natl Acad Sci USA. 1996;93:5759–5764. doi: 10.1073/pnas.93.12.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund SM, Wong KB, Fersht AR. Initiation sites of protein folding by NMR analysis. Proc Natl Acad Sci USA. 1996;93:10600–10603. doi: 10.1073/pnas.93.20.10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KF, Baker D. Global properties of the mapping between local amino acid sequence and local structure in proteins. Proc Natl Acad Sci USA. 1996;93:5814–5818. doi: 10.1073/pnas.93.12.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socci ND, Onuchic JN, Wolynes PG. Protein folding mechanisms and the multidimensional folding funnel. Proteins. 1998;32:136–158. [PubMed] [Google Scholar]
- Dill KA. Polymer principles and protein folding. Protein Sci. 1999;8:1166–1180. doi: 10.1110/ps.8.6.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross M. Protein folding: think globally, (inter) act locally. Curr Biol. 1998;8:R308–R309. doi: 10.1016/s0960-9822(98)70194-0. [DOI] [PubMed] [Google Scholar]
- Minor DL, Kim PS. Context-dependent secondary structure formation of a designed protein sequence. Nature. 1996;380:730–734. doi: 10.1038/380730a0. [DOI] [PubMed] [Google Scholar]
- Gendoo DMA, Harrison PM. Discordant and chameleon sequences: their distribution and implications for amyloidogenicity. Protein Sci. 2011;20:567–579. doi: 10.1002/pro.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol. 2008;3:11–40. doi: 10.1146/annurev.pathmechdis.3.121806.154326. [DOI] [PubMed] [Google Scholar]
- Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Guo J-T, Jaromczyk JW, Xu Y. Analysis of chameleon sequences and their implications in biological processes. Proteins. 2007;67:548–558. doi: 10.1002/prot.21285. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C. On the use of sequence homologies to predict protein structure: identical pentapeptides can have completely different conformations. Proc Natl Acad Sci USA. 1984;81:1075–1078. doi: 10.1073/pnas.81.4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IA, Haft DH, Getzoff ED, Tainer JA, Lerner RA, Brenner S. Identical short peptide sequences in unrelated proteins can have different conformations: a testing ground for theories of immune recognition. Proc Natl Acad Sci USA. 1985;82:5255–5259. doi: 10.1073/pnas.82.16.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen BI, Presnell SR, Cohen FE. Origins of structural diversity within sequentially identical hexapeptides. Protein Sci. 1993;2:2134–2145. doi: 10.1002/pro.5560021213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Alber F, Folkers G, Gonnet GH, Chelvanayagam G. An analysis of the helix-to-strand transition between peptides with identical sequence. Proteins. 2000;41:248–256. doi: 10.1002/1097-0134(20001101)41:2<248::aid-prot90>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Kuznetsov IB, Rackovsky S. On the properties and sequence context of structurally ambivalent fragments in proteins. Protein Sci. 2003;12:2420–2433. doi: 10.1110/ps.03209703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravanan KM, Selvaraj S. Search for identical octapeptides in unrelated proteins: structural plasticity revisited. Biopolymers. 2012;98:11–26. doi: 10.1002/bip.21676. [DOI] [PubMed] [Google Scholar]
- Ghozlane A, Joseph AP, Bornot A, de Brevern AG. Analysis of protein chameleon sequence characteristics. Bioinformation. 2009;3:367–369. doi: 10.6026/97320630003367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravanan KM, Selvaraj S. Performance of secondary structure prediction methods on proteins containing structurally ambivalent sequence fragments. Biopolymers. 2013;100:148–153. doi: 10.1002/bip.22178. [DOI] [PubMed] [Google Scholar]
- Jacoboni I, Martelli PL, Fariselli P, Compiani M, Casadio R. Predictions of protein segments with the same aminoacid sequence and different secondary structure: a benchmark for predictive methods. Proteins. 2000;41:535–544. doi: 10.1002/1097-0134(20001201)41:4<535::aid-prot100>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Rost B, Sander C. Third generation prediction of secondary structures. Methods Mol Biol. 2000;143:71–95. doi: 10.1385/1-59259-368-2:71. [DOI] [PubMed] [Google Scholar]
- Rost B. Review: protein secondary structure prediction continues to rise. J Struct Biol. 2001;134:204–218. doi: 10.1006/jsbi.2001.4336. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerens AT, Marshall DD, Bose CL. Nosocomial respiratory syncytial virus: a threat in the modern neonatal intensive care unit. J Perinatol. 2002;22:306–307. doi: 10.1038/sj.jp.7210696. [DOI] [PubMed] [Google Scholar]
- Swanson KA, Settembre EC, Shaw CA, Dey AK, Rappuoli R, Mandl CW, Dormitzer PR, Carfi A. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc Natl Acad Sci USA. 2011;108:9619–9624. doi: 10.1073/pnas.1106536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, Graham BS. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science. 2013;340:1113–1117. doi: 10.1126/science.1234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KA, Dutch RE, Lamb RA, Jardetzky TS. Structural basis for paramyxovirus-mediated membrane fusion. Mol Cell. 1999;3:309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- Yin H-S, Wen X, Paterson RG, Lamb RA, Jardetzky TS. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Schaffitzel C, Fabiola F, Chapman MS, Ban N, Frank J. Elongation arrest by SecM via a cascade of ribosomal RNA rearrangements. Mol Cell. 2006;22:533–543. doi: 10.1016/j.molcel.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, Kapral GJ, Noeske J, Richardson JS, Blanchard SC, Cate JHD. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science. 2011;332:981–984. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B-G, Cho J-W, Cho YD, Jeong K-C, Kim S-Y, Lee BI. Crystal structure of human transglutaminase 2 in complex with adenosine triphosphate. Int J Biol Macromol. 2010;47:190–195. doi: 10.1016/j.ijbiomac.2010.04.023. [DOI] [PubMed] [Google Scholar]
- Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmann BM, Knauer SH, Sevostyanova A, Schweimer K, Mooney RA, Landick R, Artsimovitch I, Rösch P. An α helix to β barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell. 2012;150:291–303. doi: 10.1016/j.cell.2012.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogurov GA, Vassylyeva MN, Svetlov V, Klyuyev S, Grishin NV, Vassylyev DG, Artsimovitch I. Structural basis for converting a general transcription factor into an operon-specific virulence regulator. Mol Cell. 2007;26:117–129. doi: 10.1016/j.molcel.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarkar VB, Babayeva ND, Pavlov YI, Tahirov TH. Crystal structure of the C-terminal domain of human DNA primase large subunit: implications for the mechanism of the primase-polymerase α switch. Cell Cycle. 2011;10:926–931. doi: 10.4161/cc.10.6.15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaithiyalingam S, Warren EM, Eichman BF, Chazin WJ. Insights into eukaryotic DNA priming from the structure and functional interactions of the 4Fe–4S cluster domain of human DNA primase. Proc Natl Acad Sci USA. 2010;107:13684–13689. doi: 10.1073/pnas.1002009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Arii Y, Mikami B, Hirose M. Loop-inserted and thermostabilized structure of P1–P1′ cleaved ovalbumin mutant R339T. J Mol Biol. 2002;315:113–120. doi: 10.1006/jmbi.2001.5056. [DOI] [PubMed] [Google Scholar]
- Stein PE, Leslie AG, Finch JT, Carrell RW. Crystal structure of uncleaved ovalbumin at 1.95 A resolution. J Mol Biol. 1991;221:941–959. doi: 10.1016/0022-2836(91)80185-w. [DOI] [PubMed] [Google Scholar]
- Shen KC, Kuczynska DA, Wu IJ, Murray BH, Sheckler LR, Rudenko G. Regulation of neurexin 1β tertiary structure and ligand binding through alternative splicing. Structure. 2008;16:422–431. doi: 10.1016/j.str.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J, Katsamba PS, Ahlsen G, Bahna F, Vendome J, Honig B, Shapiro L, Jin X. Splice form dependence of β-neurexin/neuroligin binding interactions. Neuron. 2010;67:61–74. doi: 10.1016/j.neuron.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kormendi V, Szyk A, Piszczek G, Roll-Mecak A. Crystal structures of tubulin acetyltransferase reveal a conserved catalytic core and the plasticity of the essential N terminus. J Biol Chem. 2012;287:41569–41575. doi: 10.1074/jbc.C112.421222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhong C, Li L, Sun B, Wang W, Xu S, Zhang T, Wang C, Bao L, Ding J. Molecular basis of the acetyltransferase activity of MEC-17 towards α-tubulin. Cell Res. 2012;22:1707–1711. doi: 10.1038/cr.2012.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M, Keshvara L, Park C-G, Zhang Y-M, Dickerson JB, Zheng J, Rock CO, Curran T, Park H-W. Crystal structures of the Dab homology domains of mouse disabled 1 and 2. J Biol Chem. 2003;278:36572–36581. doi: 10.1074/jbc.M304384200. [DOI] [PubMed] [Google Scholar]
- Xiao S, Charonko JJ, Fu X, Salmanzadeh A, Davalos RV, Vlachos PP, Finkielstein CV, Capelluto DGS. Structure, sulfatide binding properties, and inhibition of platelet aggregation by a disabled-2 protein-derived peptide. J Biol Chem. 2012;287:37691–37702. doi: 10.1074/jbc.M112.385609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Lua S, Tong JS, Song J. Elimination of the native structure and solubility of the hVAPB MSP domain by the Pro56Ser mutation that causes amyotrophic lateral sclerosis. Biochemistry. 2010;49:3887–3897. doi: 10.1021/bi902057a. [DOI] [PubMed] [Google Scholar]
- Schellenberg MJ, Wu T, Ritchie DB, Fica S, Staley JP, Atta KA, LaPointe P, MacMillan AM. A conformational switch in PRP8 mediates metal ion coordination that promotes pre-mRNA exon ligation. Nat Struct Mol Biol. 2013;20:728–734. doi: 10.1038/nsmb.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander PA, He Y, Chen Y, Orban J, Bryan PN. A minimal sequence code for switching protein structure and function. Proc Natl Acad Sci USA. 2009;106:21149–21154. doi: 10.1073/pnas.0906408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Chen Y, Alexander P, Bryan PN, Orban J. NMR structures of two designed proteins with high sequence identity but different fold and function. Proc Natl Acad Sci USA. 2008;105:14412–14417. doi: 10.1073/pnas.0805857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juritz EI, Alberti SF, Parisi GD. PCDB: a database of protein conformational diversity. Nucleic Acids Res. 2011;39:D475–D479. doi: 10.1093/nar/gkq1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M, Krebs W. A database of macromolecular motions. Nucleic Acids Res. 1998;26:4280–4290. doi: 10.1093/nar/26.18.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores S, Echols N, Milburn D, Hespenheide B, Keating K, Lu J, Wells S, Yu EZ, Thorpe M, Gerstein M. The Database of Macromolecular Motions: new features added at the decade mark. Nucleic Acids Res. 2006;34:D296–D301. doi: 10.1093/nar/gkj046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RA, Razaz M, Hayward S. The DynDom database of protein domain motions. Bioinformatics. 2003;19:1290–1291. doi: 10.1093/bioinformatics/btg137. [DOI] [PubMed] [Google Scholar]
- Qi G, Lee R, Hayward S. A comprehensive and non-redundant database of protein domain movements. Bioinformatics. 2005;21:2832–2838. doi: 10.1093/bioinformatics/bti420. [DOI] [PubMed] [Google Scholar]
- Amemiya T, Koike R, Kidera A, Ota M. PSCDB: a database for protein structural change upon ligand binding. Nucleic Acids Res. 2012;40:D554–D558. doi: 10.1093/nar/gkr966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzon AM, Juritz E, Fornasari MS, Parisi G. CoDNaS: a database of conformational diversity in the native state of proteins. Bioinformatics. 2013;29:2512–2514. doi: 10.1093/bioinformatics/btt405. [DOI] [PubMed] [Google Scholar]
- Sudharsanam. http://www.ncbi.nlm.nih.gov/pubmed/9517538.
- Saravanan and Selvaraj. http://www.ncbi.nlm.nih.gov/pubmed/23325556.
- Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, Kim B-H, Grishin NV. ECOD: an evolutionary classification of protein domains. PLoS Comput Biol. 2014;10:e1003926. doi: 10.1371/journal.pcbi.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- Kim B-H, Cong Q, Grishin NV. HangOut: generating clean PSI-BLAST profiles for domains with long insertions. Bioinformatics. 2010;26:1564–1565. doi: 10.1093/bioinformatics/btq208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JSmol: an open-source HTML5 viewer for chemical structures in 3D. http://wiki.jmol.org/index.php/JSmol#JSmol.
- The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC; http://www.ncbi.nlm.nih.gov/pubmed/15173120. [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Eyrich VA, Przybylski D, Koh IYY, Grana O, Pazos F, Valencia A, Rost B. CAFASP3 in the spotlight of EVA. Proteins. 2003;53(Suppl 6):548–560. doi: 10.1002/prot.10534. [DOI] [PubMed] [Google Scholar]
- Lo Conte L, Ailey B, Hubbard TJ, Brenner SE, Murzin AG, Chothia C. SCOP: a structural classification of proteins database. Nucleic Acids Res. 2000;28:257–259. doi: 10.1093/nar/28.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A. UniProtKB/Swiss-Prot. Methods Mol Biol. 2007;406:89–112. doi: 10.1007/978-1-59745-535-0_4. [DOI] [PubMed] [Google Scholar]
- Zamyatnin AA. Protein volume in solution. Prog Biophys Mol Biol. 1972;24:107–123. doi: 10.1016/0079-6107(72)90005-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information