

Abstract
Rev-erbα and β are nuclear receptors that function as transcriptional repressors of genes involved in regulating circadian rhythms, glucose, and cholesterol metabolism and the inflammatory response. Given these key functions, Rev-erbs are important drug targets for treatment of a number of human pathologies, including cancer, heart disease, and type II diabetes. Transcriptional repression by the Rev-erbs involves direct competition with transcriptional activators for target sites, but also recruitment by the Rev-erbs of the NCoR corepressor protein. Interestingly, Rev-erbs do not appear to interact functionally with a very similar corepressor, Smrt. Transcriptional repression by Rev-erbs is thought to occur in response to the binding of heme, although structural, and ligand binding studies in vitro show that heme and corepressor binding are antagonistic. We carried out systematic studies of the ligand and corepressor interactions to address the molecular basis for corepressor specificity and the energetic consequences of ligand binding using a variety of biophysical approaches. Highly quantitative fluorescence anisotropy assays in competition mode revealed that the Rev-erb specificity for the NCoR corepressor lies in the first two residues of the β-strand in Interaction Domain 1 of NCoR. Our studies confirmed and quantitated the strong antagonism of heme and corepressor binding and significant stabilization of the corepressor complex by a synthetic ligand in vitro. We propose a model which reconciles the contradictory observations concerning the effects of heme binding in vitro and in live cells.
Keywords: Rev-erb, corepressor, heme, binding, anisotropy
Introduction
Rev-erbα and Rev-erbβ belong to the Nuclear Receptor (NR) superfamily1 of ligand responsive transcription factors (TF). Rev-erbs are implicated in a number of human pathologies, including type 2 diabetes, inflammation and heart disease, amongst many others.2 They exert their control via interaction with DNA targets, the RORE (retinoid related orphan receptors response elements) and RevDR2 sequences, found upstream of a wide variety of genes implicated in key cellular processes.3–5 For example, Rev-erb regulates expression of Bmal1 and clock,6–10 which control circadian rhythms, Ucp1,11 which regulates body temperature cycles, PEPCK and G6P, key enzymes in gluconeogenesis,9 ApoA1 and ApoCIII which regulate cholesterol metabolism,12–14 and IkBα and other targets involved in inflammation.15
Members of the NR family of TF's bind lipophilic molecules in structurally homologs ligand binding domains (LBD) found in their C-termini. They contain as well, a central structurally homologous DNA binding domain, and an N-terminal domain that is relatively unstructured and of low homology between family members.16,17 Ligand dependent regulation of transcription by the NRs occurs primarily via conformational changes in their LBD upon ligand binding, leading to recruitment of chromatin modifying enzymes that in turn, either enhance or inhibit recruitment of the general transcription machinery.18
Rev-erbs function generally as transcriptional repressors. Binding of Rev-erbs to most of their target sites in the genome inhibits transcriptional activation by RORs via direct competition for these sites. In addition, Rev-erb lacks the C-terminal helix 12 (activation function 2, or AF2 domain) in its LBD, which for other NRs, is essential for the ligand dependent recruitment of coactivators, such as Tif2 or Src1.18 Instead, Rev-erb interacts with the corepressor, NCoR, to downregulate expression of its target genes.19–23 Interestingly, the corepressor, Smrt, which functions with retinoid and thyroid receptors (RAR and TR)24 does not appear to interact functionally with Rev-erb.19,21–23
The corepressors, NCoR, and Smrt interact with their respective NR LBDs via short interacting domains (ID), three in NCoR and two or three in Smrt, depending upon the splice variant,25 which exhibit a conserved α-helical motif (I/LxxIIxxxI) or CoRNR box. Rev-erb exhibits a higher affinity for ID1 of NCoR,19 as compared to the remaining ID2 and ID3 domains. Structural studies of complexes between peptides derived from the ID1 motifs of NCoR and Smrt with their partners, Rev-erbα and RAR have shown extensive interactions beyond those of the CoRNR box motif.26,27 Indeed an additional N-terminal turn of the helix positions a leucine residue (2051), and a C-terminal turn of the helix similarly positions phenylalanine (2064) along the same hydrophobic surface. Moreover, histidine 2054 of NCoR forms a hydrogen bond with threonine 444 of Rev-erb helix 3, completing an extensive network of contacts between the NCoR helix and the Rev-erbα LBD. In addition to this extended helix, a two stranded β-sheet is formed between an N-terminal β-strand of NCoR and a short sequence in the C-terminus of helix 11 of the LBD26,27 [Fig. 1(C)]. The molecular basis for corepressor specificity has been linked to this N-terminal β-strand,21 although quantitative energetic information concerning these complexes remains to be obtained. Interestingly, a very similar structural arrangement including both the strand and comparable hydrophobic interactions of the helix was observed for the Smrt–RAR interaction.26 Given the apparently analogous interaction modes, the molecular basis for the preference of Rev-erb for NCoR over Smrt is not readily apparent.
Figure 1.
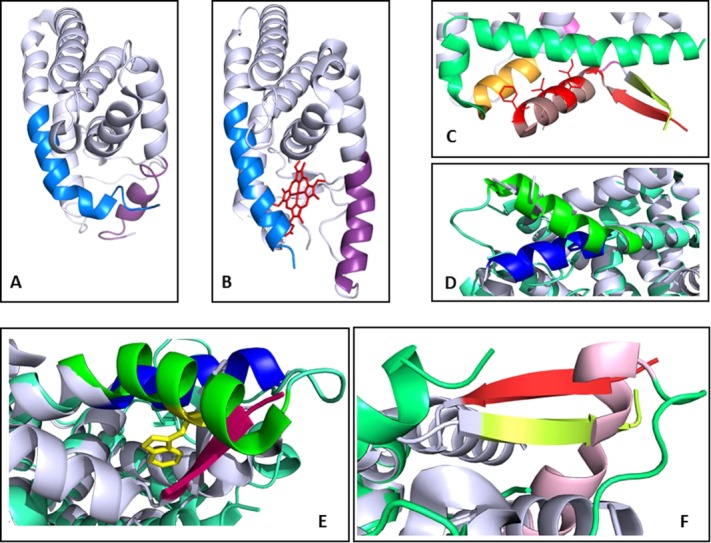
Structural comparison of NCoRID1 and heme binding to Rev-erb LBD. (A) Structure of the unliganded Rev-erbβ LBD31 with helices 11 and 3 in blue and lavender, respectively, (B) structure of Rev-erbβ LBD liganded by heme30 (red), helices 3 and 11 are colored as in (A); (C) a zoom of the Rev-erbα LBD/NCoRID1 peptide complex27 showing the β-sheet with the peptide β-strand in orange and the Rev-erbα LBD β-strand in yellow. The peptide helix is in brown with the interacting residues in red stick representation. Helix 3 of Rev-erbα LBD is in green; (D) Alignment of the Rev-erbα LBD/NCoRID1 and Rev-erbβ LBD/heme structures with a zoom of the C-terminal end of helix 3 from the structure in complex with the NCoRID1 peptide (blue) and from the Rev-erbβ LBD heme-bound structure (green); (E) Another view of D, with the β-strand of the NCoRID1 peptide (red) in steric clash with the end of helix 3 from the Rev-erbβ LBD from the heme-bound structure (green); (F) the same alignment showing the β-strand from the C-terminus of Rev-erbα LBD (yellow) and the β-strand of the NCoRID1 peptide (red) in steric clash with helix 3 (pink) from the heme-bound structure.
The Ecdysone-induced protein E75, a close homologue of Rev-erbα and Rev-erbβ, is a heme-binding NR implicated in Drosophila development.28 Rev-erbα and Rev-erbβ have been shown to bind heme, as well,9,10,29 although neither has been purified from natural sources in a heme-bound form. In live cells, heme binding leads to increased NCoR recruitment and hence increased repression of gene expression.10 In apparent contradiction to the in vivo effects of heme, in vitro studies using constructs of Rev-erb LBDs have shown that heme binding antagonizes Rev-erb–NCoR interactions.29,30 Moreover, the heme iron can exist in either the oxidized or reduced state with different coordination numbers. In the reduced state, gas molecules such as NO can bind, and this relieves repression,30 leading to the hypothesis that Rev-erbs are gas responsive NRs. These multiple physical states of the heme add to the complexity of the apparently contradictory relations between corepressor and heme binding to Rev-erb.
Comparison of the structures of the free31 and heme-bound Rev-erbβ LBD30 reveal important rearrangements of helices 3 and 11 [Fig. 1(A,B)]. This structural re-arrangement also entails the ejection of a number of hydrophobic side chains from the ligand binding pocket to accommodate the heme. As previously pointed out,27 comparison of the structure of the Rev-erbα/NCoRID1 complex with the heme-bound structure of Rev-erbβ reveals significant structural differences. We note here a large and important difference in the orientation of helix 3 [Fig. 1(D)]. Indeed, strong steric clashes between tryptophan 402 and the C-terminus of helix 3 of Rev-erbα LBD with the β-strand of the NCoR peptide are apparent in the alignment of the two structures [Fig. 1(E,F)]. Moreover, the histidine residue (residue 568 in Rev-erbβ and 602 in Rev-erbα) necessary for heme binding is orientated away from the ligand binding pocket in the corepressor bound structure. The results of this comparison are consistent with the in vitro measurements demonstrating antagonism between NCoR and heme binding,29 and leave open the question of the molecular mechanism for heme-mediated transcriptional repression in vivo.32
Given the implication of Rev-erbs in human health and disease, a detailed mechanistic understanding of the structure-function relations underlying ligand effects on function is highly desirable. We set out to lay the quantitative energetic foundations for addressing the dual questions of the molecular mechanisms underpinning corepressor selectivity by the Rev-erbs and secondly, the structure–function relationships involved in the heme-dependence of corepressor binding. Accordingly, we have carried out a systematic determination of the binding affinities in vitro between the Rev-erbα LBD and a series of peptides derived from the NCoR and Smrt corepressors using a variety of biophysical techniques; fluorescence anisotropy, surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), and mass spectrometry (MS). Furthermore, we tested the effects of heme binding, as well as the binding of two synthetic ligands, SR3335 (SD7),33 and GSK4112 (SGN)34 on Rev-erb/CoR peptide interactions.
We found that Rev-erbα exhibited the highest affinity for a peptide comprising the entire length of the NCoR ID1 (β-strand and α-helix) present in the reported structure of the complex.27 The equivalent peptide derived from Smrt ID1 bound with 3.5-fold lower affinity. Interestingly, deletion of the first two residues of the β-strand in the NCoRID1 peptide led to a 10-fold loss in affinity, whereas a similar deletion in the Smrt peptide had little effect. The NCoRID2 and ID3 peptides exhibited ≥ 10-fold lower affinity compared to NCoRID1, whereas no difference was observed between Smrt ID1 and ID2 binding. We confirm and quantify, previous in vitro observations29,30 of strong antagonism between heme and CoR binding in vitro. The synthetic ligand SD7 was mildly antagonistic also to CoR recruitment by Rev-erb, whereas the interaction was significantly enhanced by the SGN ligand. These quantitative results are interpreted in light of known structure-function relationships of NRs.
Results
Molecular basis for peptide selectivity
We used anisotropy-based competition titrations to compare the affinities of the various NCoR and Smrt ID motifs (Table1) for the Rev-erbα LBD construct 281–614 (Δ324–42227. We chose to use peptides in these studies because the full-length NCoR and Smrt proteins are very large and difficult to obtain in purified form. We selected the sequences of the peptides in Table1 by aligning them based on the NCoRID1 and SmrtID interactions with Rev-erbα and RAR, respectively observed in the two crystal structures.26,27 The SmrtID1 peptide from the structure of its complex with RAR,26 labeled with rhodamine at its N-terminus (SmrtID1B-2-Rho), was used to measure its interaction with Rev-erbα LBD by fluorescence anisotropy [Fig. 2(A,C), pink circles]. The affinity of the labeled peptide was found to be 0.2 μM. Next, this labeled peptide was used in competition experiments with unlabeled competitor peptides to extract the affinities of full-length peptides of NCoRID1 (NCoRID1C) [Fig. 2(A)] and SmrtID1 (SmrtID1C) [Fig. 2(C)]. Competition assays eliminate any contribution from the fluorophore to the apparent affinity, and moreover circumvent the necessity of obtaining labeled peptides for all sequences tested. The NCoR and Smrt peptides designated here as full-length encompass the entire putative β-strand and α-helix (2045–2066 for NCoRID1) and the corollary residues for NCoRID2 and 3 and Smrt ID1 and ID2, which include an extra two N-terminal residues with respect to the reported crystal structure of the complex of RAR with a Smrt ID1 peptide.26 Competition binding curves were fit for the free energies of interaction between Rev-erbα LBD and the unlabeled peptides, using the previously determined affinity for the labeled SmrtID1B-2Rho peptide as a constant. Then, the data and the fits of all experiments were normalized for comparison.
Table 1.
Sequence of the Peptides Used in This Study with Their Determined Kd and ΔG Values
| (Putative) β-strand | α-Helix | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | Kd (μM) | −ΔG (kcal/mol) | ||
| NCoR | T | H | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | 0.4 ± 0.02 | 8.6 ± 0.03 | |
| ID1C | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| NCoR | T | H | R | L | I | T | L | A | D | H | I | A | Q | I | I | T | Q | D | F | A | R | 0.36 ± 0.02 | 8.8 ± 0.1 | |
| ID1C | ||||||||||||||||||||||||
| ITC | ||||||||||||||||||||||||
| NCoR | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | 5.1 ± 0.82 | 7.2 ± 0.1 | |||
| ID1B-2 | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| NCoR | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | 0.6 ± 0.2 | 8.5 ± 0.2 | |||
| ID1B-2 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| NCoR | S | F | A | D | P | A | S | N | L | G | L | E | D | I | I | R | K | A | L | M | G | 3.5 ± 2.4 | 7.4 ± 0.4 | |
| ID2C | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| NCoR | D | P | A | S | N | L | G | L | E | D | I | I | R | K | A | L | M | G | SFD | 10.7 ± 6.3 | 6.8 ± 0.4 | |||
| ID2B-3 | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| NCoR | D | P | A | S | N | L | G | L | E | D | I | I | R | K | A | L | M | G | SFD | 3.8 ± 0.04 | 7.31 ± 0.01 | |||
| ID2B-3 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| NCoR | K | T | T | I | T | A | A | N | F | I | D | V | I | I | T | R | Q | I | A | S | DKDAR | 14.8 ± 3.7 | 6.5 ± 0.15 | |
| ID3B-1 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| NCoR | K | T | T | I | T | A | A | N | F | I | D | V | I | I | T | R | Q | I | A | S | DKDAR | 91 ± 43 | 5.5 ± 0.35 | |
| ID3B-1 | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| NCoR5 | T | H | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | ……………. | 0.5 ± 0 | 8.4 ± 0 |
| S | F | A | D | P | A | S | N | L | G | L | E | D | I | I | R | K | A | L | M | G | ||||
| Smrt | H | Q | R | V | V | T | L | A | Q | H | I | S | E | V | I | T | Q | D | Y | T | R | 1.4 ± 0.5 | 7.9 ± 0.2 | |
| ID1C | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| Smrt | R | V | V | T | L | A | Q | H | I | S | E | V | I | T | Q | D | Y | T | R | 1.7 ± 0.6 | 7.8 ± 0.2 | |||
| ID1B-2 | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| Smrt | R | V | V | T | L | A | Q | H | I | S | E | V | I | T | Q | D | Y | T | R | 2.4 ± 0.5 | 7.7 ± 0.1 | |||
| ID1B-2 | ||||||||||||||||||||||||
| ITC | ||||||||||||||||||||||||
| Smrt | R | V | V | T | L | A | Q | H | I | S | E | V | I | T | Q | D | Y | T | R | 0.2 ± 0.06 | 9.1 ± 0.2 | |||
| ID1B-2 | ||||||||||||||||||||||||
| Rho | ||||||||||||||||||||||||
| Smrt | V | Q | E | H | A | S | T | N | M | G | L | E | A | I | I | R | K | A | L | M | G | 2.3 ± 1.4 | 7.7 ± 0.4 | |
| ID2C | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| Smrt | E | H | A | S | T | N | M | G | L | E | A | I | I | R | K | A | L | M | G | 1.2 ± 0.45 | 8.0 ± 0.1 | |||
| ID2C | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| Smrt | T | N | M | G | L | E | A | I | I | R | K | A | L | M | G | KYDQ WEE | 7.1 ± 2.2 | 6.9 ± 0.2 | ||||||
| ID2B-6 | ||||||||||||||||||||||||
| NM | ||||||||||||||||||||||||
| Smrt | T | N | M | G | L | E | A | I | I | R | K | A | L | M | G | KYDQ WEE | 1.15 ± 0.4 | 8.0 ± 0.2 | ||||||
| ID2B-6 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
Estimated errors are standard deviations from 3 or more experiments. Labeled peptides were used at a concentration of 4 nM. The sequences ID1 and ID2 motifs of the NCoR5 construct are given here. The complete 266 residue sequence can be found in the Materials and Methods. The secondary structures of the corresponding residues in the crystal structures are indicated in yellow for the putative β-strand in yellow and the α-helix in green in the top line of the table. Red indicates that the residues at these positions have been deleted for the corresponding peptide. FITC and Rho refer to N-terminal labeling of the peptides with either fluorescein or rhodamine. The peptides are ordered beginning with the NCoR variants and then the Smrt variants, and in order of ID1, ID2, and ID3, with N-terminal deletions appearing in order of increasing deletions. ITC refers to results from the titrations by isothermal titration calorimetry. The letter C following the peptide identification indicates that the peptide is complete, or full-length, with respect to the crystal structure interactions. NM refers to not marked or not labeled.
Figure 2.
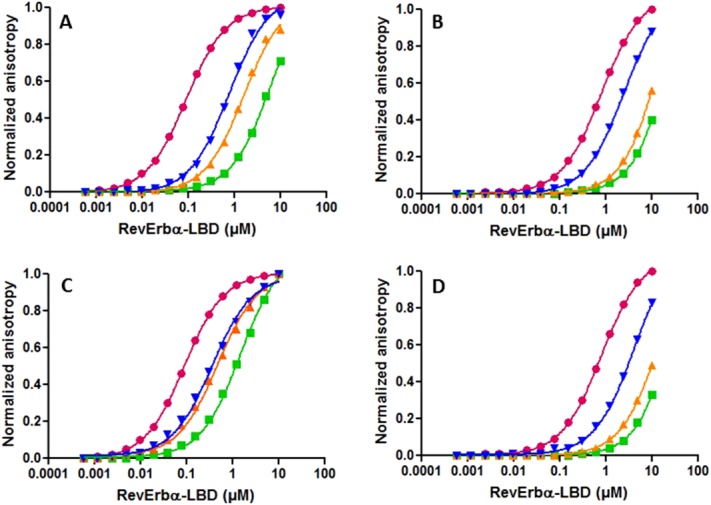
Anisotropy competition assays for Rev-erbα LBD corepressor selectivity. Rhodamine-labeled SmrtID1B-2 (A and C) or fluorescein labeled SmrtID2B-6 (B and D) were titrated with Rev-erbα-LBD (281–614 Δ324–422) in the absence (pink circles) and in the presence of (A) full-length NCorID1C (0, 2.5, 5, 10 μM); (B) full-length NCorID2C (0, 10, 50, 100 μM); (C) full-length SmrtID1C (0, 2.5, 5, 10 μM); and (D) full-length SmrtID2C (0, 10, 50, 100 μM). Increasing concentrations of unlabeled competitor peptide are blue, yellow and green. Lines through the points represent the fits of the data.
The NCoR peptide, at identical concentrations, exhibits stronger competition than the Smrt peptide. The affinities obtained from nonlinear least squares fits of the competition curves reveal 3.5-fold higher affinity for the NCoRID1C peptide compared to the equivalent peptide derived from Smrt (SmrtID1C, Table1). Both unlabeled peptides present a lower affinity than the fluorescently labeled peptide, underscoring the importance of measurement via competition assays in order to avoid artifactual contributions from the hydrophobic fluorescent dyes used in such measurements. Identical affinity for the Rev-erbα LBD construct was observed for the NCoRID1C peptide by ITC [Fig. 3(A); Table1]. We note that the peptide used in the ITC measurements had a Cys to Ala at position 12 substitution for increased solubility, but this had no effect on the affinity. The Rev-erbα LBD complex with NCoRID1C is enthalpically favored primarily, with a small favorable entropic contribution. However, we observed using SPR that sufficient concentrations of the SmrtID1B-2 peptide could compete for the NCoR-Rev-erbα interaction [Fig. 4(A)].
Figure 3.
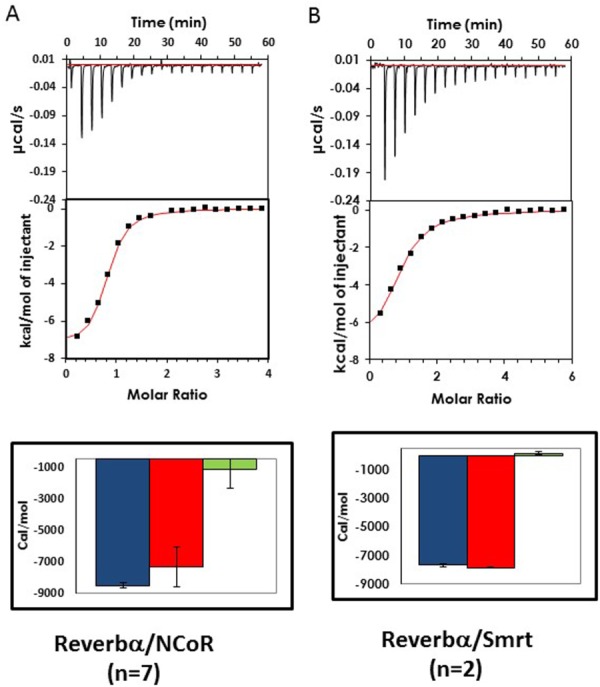
ITC experiments of Rev-erbα LBD interactions with corepressor peptides. Images of representative thermograms obtained with (A) full-length NCoRID1C (mutated at positions T21R C12A to increase solubility) and (B) SmrtID1B-2 binding to Rev-erbα LBD (281–614 Δ324–422). In all cases the heat signals (in µcal/s) as a response by the release of the ligand into the protein solution is shown over the course of the experiment along with the integrated heat signals of the injections (kcal/mol). Thermodynamic parameters (ΔG° in blue, ΔH° in red, −TΔS° in green in cal/mol) determined by direct ITC titrations. The estimated standard deviations, calculated from at least duplicate measurements, are indicated by the black error bars.
Figure 4.
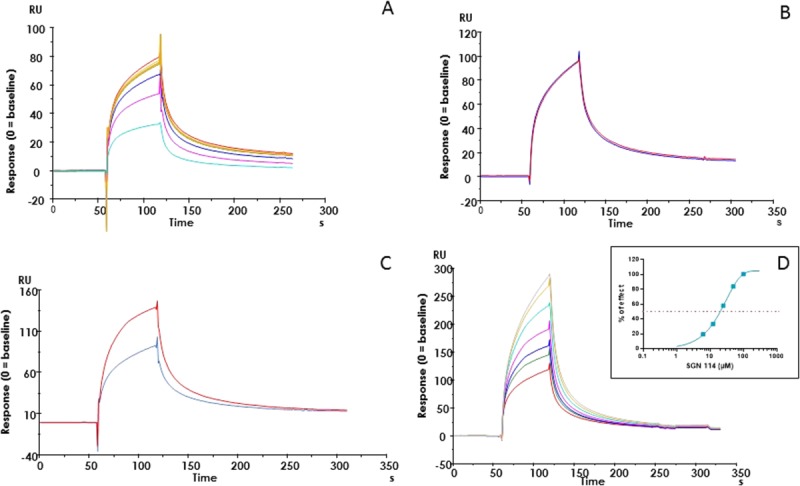
SPR measurements of Rev-erbα LBD interactions. SPR response (expressed as RU) of different modulators on Rev-erbα LBD (313 nM) binding to biotinylated-NCoRID1 immobilized on a streptavidin chip. (A) Competition of NCoRID1 binding to Rev-erbα LBD by addition of SmrtID1 (Rev-erb alone: yellow brown; 0.1—red, 0.3—green, 1.0—blue, 3.0—pink, and 10—cyan µM SmrtID1); (B) effect of the ligand SD7 (25 µM) on NCoRID1/Rev-erbα LBD interaction (no ligand—blue; 25 µM SD7—red); (C) effect of the ligand SGN (12.5 µM) on NCoR/Rev-erbα LBD interactions (no ligand—blue, 12.5 µM SGN—red) ; (C) Dose–effect response of the ligand SGN on NCoRID1/Rev-erbα LBD interactions (no ligand—red, 3.12 µM SGN—green; 6.25 µM—blue; 12.5 µM—pink; 25 µM—cyan; 50 µM—yellow brown; 100 µM gray). EC50 determination in the insert (EC50 = 20 µM). The percentage of activation of Rev-erbα LBD (313 nM) binding to immobilized NCoRD1 in the presence of SGN is reported in the insert and this allowed EC50 determination (EC50 = 20 µM). All experiments were performed in duplicate. Experiments were analyzed in PRISM® software using a one phase exponential association fitting method.
Using this anisotropy competition approach with a fluorescein labeled SmrtID2B-2 construct (Kd = 1.15 μM), we found that the unlabeled full-length NCoRID2C peptide had a tenfold lower affinity for Rev-erbα LBD, than the NCoRID1C peptide, whereas the affinities of the unlabeled SmrtID1C and ID2C full-length peptides were similar and 1.5–2 to fold higher than that of NCoRID2C [Fig. 2(B,D); Table1]. An unlabeled NCoRID3 peptide (NCoRID3B-1), missing only the N-terminal residue with respect to the NCoRID1C peptide, had over 200-fold lower affinity than the NCoRID1C peptide (Table1), indicating a hierarchy of ∼10-fold each between NCoRID1, NCoRID2, and NCoRID3 motifs. The ID motifs can be defined following a different alignment with respect to ID1. The LEDII of NCoRID2 can be aligned with the LADHI of NCoRID1. To eliminate any bias in the alignments we used a 266 residue peptide bearing both the NCoRID1 and NCoRID2 motifs (NCoR5). It bound with the same affinity as the NCoRID1C motif alone (Table1). Identical affinity for these two peptides is understandable, since Rev-erbα LBD is a monomer in solution and would preferentially bind to the high affinity ID1 motif. Addition of the second motif in the NCoR5 peptide would not be expected to increase affinity. Moreover, since this long peptide allows for either of the alternative alignments, with 35 residues C-terminal to our aligned ID2 motif and nearly 100 residues N-terminal, strong interactions from residues not present in the short peptides above should have led to increased affinity with respect to NCoRID1C. This was not the case. A comparison of free energies of interaction for the different Smrt and NCoR constructs is given in Figure 5(A).
Figure 5.
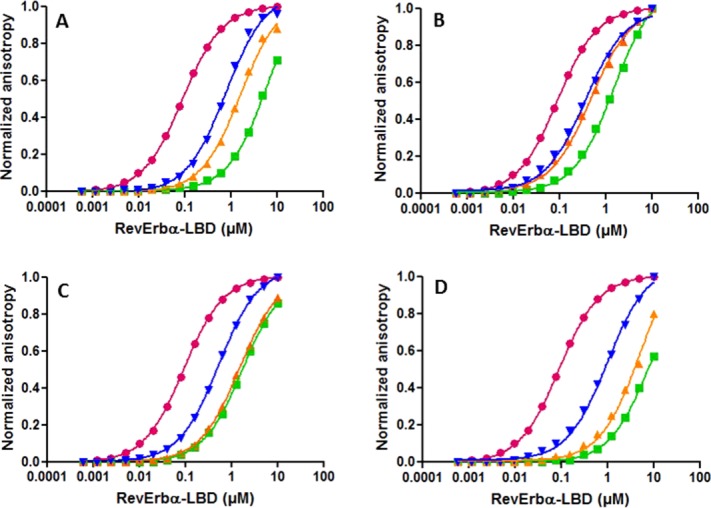
Comparison of the interaction free energies between the repressor peptide constructs and Rev-erbα LBD. (A) ΔGd for the different ID sequences from NCoR and Smrt, and (B) ΔGd the N-terminal deletion variants compared to the full-length peptides. Free energy values are those reported in Table1, and are given as dissociation free energies so as to be >0.
To characterize the energetic contribution of the putative N-terminal β-strand to the interactions of the NCoR and Smrt peptides with Rev-erbα, we measured by competition assays the effect of deleting one or more residues at the N-terminus of the peptides. Compared to the full-length NCoRID1C peptide, the NCoRID1B-2 peptide, missing the capping residue and the first residue in the putative β-strand, exhibited 12.5-fold lower affinity [Fig. 6(A,C); Table1], making it less likely to bind than the SmrtID1C full-length peptide or the SmrtID1B-2 peptide since, deleting the corresponding residues from the SmrtID1 peptide had no effect [Fig. 2(B,D)]. Similar results for the SmrtID1B-2 peptide were obtained by ITC [Fig. 3(B); Table1], which was entirely an enthalpically driven interaction. Deletion of either the first 3 residues of NCoRID2 (NCoRID2B-3) or the entire putative β-strand from SmrtID2 (SmrtID2B-6) both led to a similar 2.5–3-fold loss in affinity. These results underscore the importance of the residues in the putative β-strand for the stability of the Rev-erbα LBD/NCoRID1 complex. The modest loss of affinity upon deleting three N-terminal residues in NCoRID2 with respect to an over 10-fold difference deleting only two residues from NCoRID1, in addition to the very low affinity for NCoRID3, suggests that the β-sheet motif may be stably formed only in the case of the Rev-erbα LBD/NCoRID1 complex. A comparison of free energies of interaction for the different N-terminal deletions and full-length Smrt and NCoR constructs is given in Figure 5(B).
Figure 6.
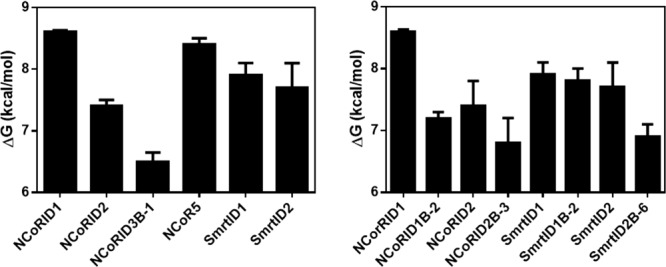
Effect of the N-terminus of the β-strand on corepressor affinity for Rev-erbα LBD. SmrtID1B-2-rhodamine competed with increasing concentrations of full-length NcorID1C (0, 2.5, 5, 10 μM); (B) full-length SmrtID1C (0, 2.5, 5, 10 μM); (C) NCoRD1B-2 (0, 10, 50, 100 μM); (D) SmrtID1B-2 (0, 10, 50, 100 μM). Graphs in (A) and (C) are redrawn from Figure 2 for comparison. Titration of the labeled peptide by Rev-erbα LBD in the absence of competitor peptide is in pink. Increasing concentrations of competitor peptide are blue, yellow and green, respectively.
Effect of ligands on Rev-erb/NCoR interactions
To begin to understand the contradictory structure-function relationships concerning the effects of heme binding on Rev-erb/corepressor interactions in vitro and in vivo, we measured Rev-erbα LBD binding to our labeled peptides (NCoRID1B-2-FITC and SmrtID1B-2-Rho) in the presence of heme. We also tested the effect of heme on Rev-erbα binding to the NCoR5 peptide (bearing both complete ID1 and ID2 motifs) labeled with the Atto647 dye (NCoR5-Atto). Unlike the significant stabilizing effects of the rhodamine and fluorescein labels in the B-2 peptide constructs, the Atto dye in the NCoR5 context had only a marginal effect on Rev-erbα LBD/corepressor peptide affinity. This is likely due to the fact that the capping residue and the first residue of the β-strand are present in the ID1 motif of the NCoR5 peptide. The higher affinity between Rev-erbα and the fluorescently labeled B-2 peptides compared to their unlabeled counterparts may arise from substitution by the dye for some of the missing contacts in the β-sheet, as the hydrophobic dye moieties could enhance nonspecific interactions between the fluorophore-labelled peptide and Rev-erbα LBD.
Addition of 5 μM heme to the titrations of all of the three peptides with Rev-erbα LBD shifted the curves significantly to higher protein concentration, confirming a strong antagonism between heme binding and Rev-erbα LBD/corepressor interactions in vitro (Fig. 7; Table2). A concentration of 5 μM heme led to a very large loss in affinity between the NCoR5-Atto peptide and Rev-erbα, over 130-fold with the Kd increasing from 0.35 to 46 μM. The heme-induced loss in apparent affinity was smaller for the B-2 peptides, 15-20-fold for NCoRID1B-2-Fluo and 20–30-fold for the SmrtID1B-2-Rho peptides, depending on the heme concentration. A greater sensitivity of the NCoR5-Atto interactions with Rev-erbα, compared to the NCorID1B-2-Fluo and SmrtID1B-2-Rho peptides, is not surprising, since the NCoR5 peptide contains the totality of the residues of the β-strand and would hence be more sensitive to the steric incompatibility between the heme-bound and corepressor bound structures than the B-2 peptide constructs.
Figure 7.
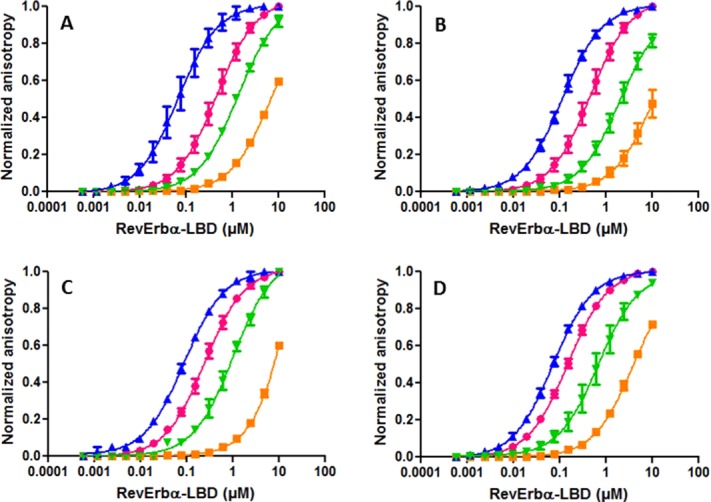
Effect of ligand on the interaction of Rev-erbα LBD with labeled peptides. Normalized anisotropy profiles of the titration by Rev-erbα LBD of (A) NCoRID1B-2-fluorescein in the absence of ligand (pink) and in the presence of 20 µM SGN (blue), 20 µM SD7 (green), and 5 µM heme (orange); (B) NCoRID1B-2-fluorescein in the absence of ligand (pink) and in the presence of 100 µM SGN (blue), 100 µM SD7 (green), and 10 µM heme (orange); (C) SMRT ID1B-2-rhodamine in the absence of ligand (pink) and in the presence of 100 µM SGN (blue), 100 µM SD7 (green), and 10 µM heme (orange); (D) NCor5-Atto647N in the absence of ligand (pink) and in the presence of 100 µM SGN (blue), 100 µM SD7 (green), and 10 µM heme (orange).
Table 2.
Effect of Ligands on the Apparent Affinity of the Labeled Peptides Used in This Study
| (Putative) β-strand | α-Helix | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | Kd μM) | −ΔG kcal/mol | ||
| NCoR | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | −L | 0.6 ± 0.2 | 8.5 ± 0.2 | ||
| ID1B-2 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| NCoR | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | Heme 5 µM | 8.3 ± 1.4 | 6.8 ± 0.1 | ||
| ID1B-2 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| NCoR | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | Heme 10 µM | 13.7 ± 4.4 | 6.6 ± 0.2 | ||
| ID1B-2 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| NCoR | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | SGN 100 µM | 0.1 ± 0.04 | 9.3 ± 0.2 | ||
| ID1B-2 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| NCoR | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | SGN 20 µM | 0.08 ± 0.03 | 9.6 ± 0.2 | ||
| ID1B-2 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| NCoR | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | SD7 100 µM | 3.8 ± 2.3 | 7.4 ± 0.3 | ||
| ID1B-2 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| NCoR | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | SD7 20 µM | 1.8 ± 0.5 | 7.7 ± 0.1 | ||
| ID1B-2 | ||||||||||||||||||||||||
| FITC | ||||||||||||||||||||||||
| Smrt | R | V | V | T | L | A | Q | H | I | S | E | V | I | T | Q | D | Y | T | R | -L | 0.2 ± 0.1 | 9.1 ± 0.2 | ||
| ID1B-2 | ||||||||||||||||||||||||
| Rho | ||||||||||||||||||||||||
| Smrt | R | V | V | T | L | A | Q | H | I | S | E | V | I | T | Q | D | Y | T | R | Heme 5 µM | 4.2 ± 0.7 | 7.2 ± 0.1 | ||
| ID1B-2 | ||||||||||||||||||||||||
| Rho | ||||||||||||||||||||||||
| Smrt | R | V | V | T | L | A | Q | H | I | S | E | V | I | T | Q | D | Y | T | R | Heme 10 µM | 6.3 ± 1.02 | 7.01 ± 0.1 | ||
| ID1B-2 | ||||||||||||||||||||||||
| Rho | ||||||||||||||||||||||||
| Smrt | R | V | V | T | L | A | Q | H | I | S | E | V | I | T | Q | D | Y | T | R | SGN 100 µM | 0.08 ± 0.01 | 9.5 ± 0.07 | ||
| ID1B-2 | ||||||||||||||||||||||||
| Rho | ||||||||||||||||||||||||
| Smrt | R | V | V | T | L | A | Q | H | I | S | E | V | I | T | Q | D | Y | T | R | SD7 100 µM | 0.9 ± 0.5 | 8.2 ± 0.35 | ||
| ID1B-2 | ||||||||||||||||||||||||
| Rho | ||||||||||||||||||||||||
| NCoR5- | T | H | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | −L | 0.35 ± 0.1 | 8.7 ± 0.2 |
| Atto | S | F | A | D | P | A | S | N | L | G | L | E | D | I | I | R | K | A | L | M | G | |||
| NCoR5 | T | H | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | −L | 0.5 ± 0 | 8.4 ± 0 |
| S | F | A | D | P | A | S | N | L | G | L | E | D | I | I | R | K | A | L | M | G | ||||
| NCoR5- | T | H | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | Heme 5 µM | 46 ± 30 | 5.9 ± 0.4 |
| Atto | S | F | A | D | P | A | S | N | L | G | L | E | D | I | I | R | K | A | L | M | G | |||
| NCoR5- | T | H | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | SGN 100 µM | 0.1 ± 0 | 9.4 ± 0 |
| Atto | S | F | A | D | P | A | S | N | L | G | L | E | D | I | I | R | K | A | L | M | G | |||
| NCoR5- | T | H | R | L | I | T | L | A | D | H | I | C | Q | I | I | T | Q | D | F | A | R | SD7 100 µM | 1.4 ± 0.4 | 7.9 ± 0.2 |
| Atto | S | F | A | D | P | A | S | N | L | G | L | E | D | I | I | R | K | A | L | M | G | |||
Estimated errors are standard deviations from three or more experiments. Labeled peptides were used at a concentration of 4 nM. Estimated errors are standard deviations from 3 or more experiments. The label –L indicates in the absence of ligand. The sequences ID1 and ID2 motifs of the NCoR5 construct are given here. The complete 266 residue sequence can be found in the Materials and Methods section. The secondary structures of the corresponding residues in the crystal structures are indicated in yellow for the putative β-strand in yellow and the α-helix in green in the top line of the table. Red indicates that the residues at these positions have been deleted for the corresponding peptide. FITC, Rho, and Atto refer to N-terminal labeling of the peptides with either fluorescein, rhodamine 110 and Atto647N.
Like heme, the SD7 ligand,33 originally designed to antagonize ROR/coactivator interactions, showed antagonism for Rev-erbα LBD corepressor interactions in the anisotropy experiments (Fig. 7; Table2). However, the effect was weaker than that of the heme, such that this antagonism was not apparent at the concentrations used in our SPR measurements [Fig. 4(B)]. In contrast, the SGN ligand which enhances repression by Rev-erb in vivo,34 showed strong enhancement of corepressor peptide binding in our in vitro anisotropy (Fig. 7; Table2), SPR [Fig. 4(C,D)] and mass spectrometry assays (Fig. 8). In our anisotropy assays, the antagonism of the Rev-erbα LBD/peptide interactions by 5 μM heme was larger than the agonistic effects of 20 μM SGN. However, since the ligand concentration in our anisotropy assays must be at least equal to the highest concentration of Rev-erbα LBD used in the titration in order to ensure saturation, the relative potency of heme may be underestimated. Indeed five equivalents of SGN were unable to compete two equivalents of heme in our MS assay [Fig. 8(D)]. Heme affinity for Rev-erb LBD has been reported to be 2–3 μM measured directly by ITC,29 although a recent study by the same group reported a Kd of 353 nM,35 citing aggregation of the LBD in the earlier study as a possible cause for the discrepancy. We measured by ITC a Kd of ∼200 nM for the heme (data not shown) in agreement with the most recent published results.
Figure 8.
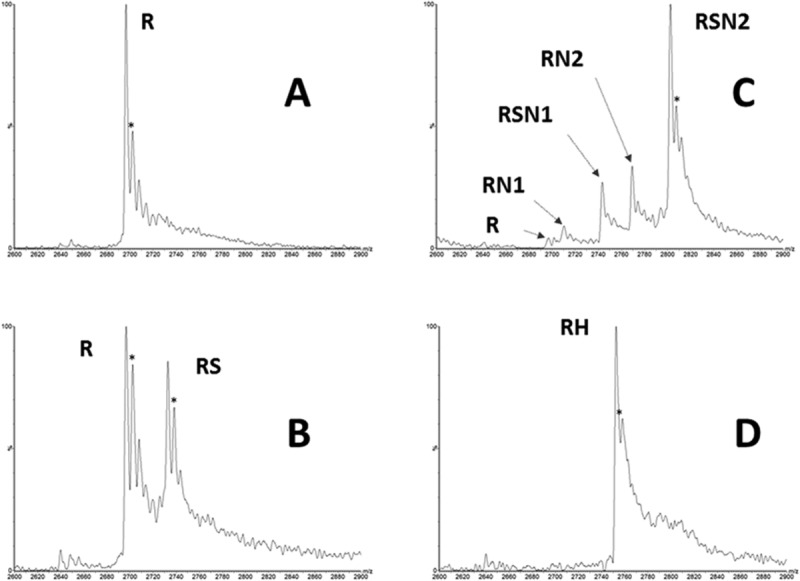
Mass spectrometry measurements of Rev-erbα interactions with ligands and corepressor. Native mass spectrometry experiments were performed using an ESI-MS instrument (see “Materials and Methods” for details). (A) Rev-erbα LBD alone (20 µM); (B) Rev-erbα LBD (20 µM) incubated with 100 µM of SGN complex; (C) Rev-erb αLBD (20 µM) was incubated with 40 µM of NCoRID1 in the presence of 100 µM of SGN; (D) Rev-erb αLBD (20 µM) was incubated with 40 µM hemin (2 equivalents) in the presence of 100 µM of SGN (5 equivalents). R: Rev-erbα LBD. Expected mass: 29651.5 ± 0.7 Da; RS: Rev-erbα LBD-SGN. Expected mass: 30049.5 Da. RN1: Rev-erbα LBD-NCoR(T-S). Expected mass: 32510.5 Da; RSN1: Rev-erbα LBD-NCoR(T-S). Expected mass: 32909.5 Da; RN2: Rev-erbα LBD-NCoR(biot-S). Expected mass: 33218.5 Da; RSN2: Rev-erb αLBD-NCoR(biot-S)-SGN. Expected mass: 30049.5 Da; RH: Rev-erbα LBD–hemin. Expected mass: 30266.5 Da. Symbols: *, gluconoylated HT-Rev-erb adduct; *P, phospho-gluconoylated HT-Rev-erb adduct.
Discussion
Corepressor selectivity
Despite very similar binding modes, Rev-erbα exhibits a preference in vitro and in vivo for the NCoRID1 motif, while RAR recognizes the SmrtID1 motif preferentially. We show here that quantitatively, the affinity of Rev-erbα for the NCoRID1 full-length peptide is 3.5-fold higher than for the SmrtID1 peptide. Comparing the interactions in the Rev-erbα LBD/NCoRID1C complex27 with those of the RAR/SmrtID1B-2 peptide complex26 reveals very few differences that could account for these preferential interactions. We found that deletion of the first two residues of the NCoRID1C peptide led to a 12.5-fold loss in affinity, up to 5 μM, compared to the 1.7 μM Kd for the SmrtID1B-2 peptide. Hence, the structural and energetic determinants of Rev-erbα LBD specificity for NCoRID1 over SmrtID1 appear to lie in the first two residues, Thr2045 and His2046. In fact, the remaining residues in the SmrtID1B-2 peptide have a higher affinity (2 to 3-fold) for Rev-erbα than that of NCoRID1B-2. As pointed out previously26 the two valine residues in the β-strand of SmrtID1 may favor the strand structure more than the corresponding leucine and isoleucine residues in NCoRID1, and the Gln to Glu substitution at position 2056 of NCoRID1 in SmrtID1 would allow for a salt bridge, rather than a hydrogen bond, contributing further stabilization. In contrast, the Cys to Ser substitution at position 2056 of NCoRID1 could mildly destabilize the α-helix in the Smrt peptide. The other substitutions from NCoRID1 to SmrtID1, Asp to Gln at 2053 and Ala to Thr at 2064 should have little effect, since these residues face away from the NCoR/Rev-erbα LBD interface. Interestingly, the relative affinity of the NCoRID1B-2 and SmrtID1B-2 peptides for RAR were similar to those reported here, with the Smrt peptide exhibiting the higher affinity,26 suggesting that at least some determinants for RAR preference for SmrtID1, are found in the helical region.
The two N-terminal residues of the NCoRID1C peptide, Thr2045 and His2046, both make stabilizing contacts to Rev-erbα LBD.27 The carboxyl of Thr2045 makes an H-bond with the backbone amide of Val611. This interaction would not be possible in the full-length NCoR protein, of course, but the Thr2045 backbone carbonyl could still make an H-bond to the backbone amide of Val611. Moreover, the His2046 side chain makes H-bonds to the sidechains of Glu437 and Trp436 of Rev-erbα helix3. The corresponding residues in SmrtID1C are His and Gln, which may not be able to form the appropriate contacts. In mammalian two hybrid assays using a 40-residue peptide containing NCorID1, deletion of residues N-terminal to His2046 of NCorID1 (including Thr2045) led to a ∼3-fold loss in transcriptional activation,21 suggesting with respect to the present results, that the His2046 interactions are at least as important as those involving Thr2045. There was no evidence for a longer β-sheet implicating residues N-terminal to Thr2045 in the reported structure despite use of a longer peptide.27 Hence, we conclude that the NCoRID1C peptide used in the present study encompasses all of the interacting residues, and that the specificity of Rev-erbα LBD for NCoR is defined by Thr2045 and His2046 in NCoRID1. The NCoRID2C and SmrtID2C full-length peptides exhibited similar affinity for Rev-erbα, which was approximately 10-fold lower than the NCoRID1C peptide. While the secondary structural algorithm, JPred,36 predicts an N-terminal β-sheet for NCoRID1C, no secondary structure is predicted for any residues in either NCorID2C or SmrtID2C, regardless of the choice of alignment. Moreover, the helices are predicted to be much shorter for these sequences, providing further support for the notion that the general preference of Rev-erbs for the ID1 motif lies in the intrinsic stability of the peptide's secondary structure, in particular the β-strand.
Ligand binding and corepressor recruitment
Heme is considered to be the natural ligand of Rev-erb.9 The fluctuating heme concentrations linked to circadian rhythms,6,37,38 the strong implication of Rev-erb in entraining the circadian clock,7 and the clear effects of heme addition on the in cellulo activity of Rev-erb,9 all argue strongly for this model. However, Rev-erb in a heme bound form has not been purified from mammalian cells. In contrast to all other nuclear receptors17,18 for which the in vitro binding and structural results explain quite well in vivo functional effects of ligands, the structural information concerning Rev-erb interactions available to date27,30,31,35 (see Fig. 1), along with in vitro binding assays previously reported29,35 and in the present work, indicate that heme binding strongly destabilizes interactions between Rev-erb and the corepressors. As noted in the introduction, this is in strong contradiction with studies in live cells demonstrating increased repression in the presence of heme. Interestingly, spectroscopic studies have shown that the heme iron coordination state is influenced by redox state.30 In Rev-erbβ, when the heme iron is FeIII+, the heme is hexa-coordinated, with the 5th ligand being His 568 (602 in Rev-erbα) located in helix 11 prior to the β-strand that forms upon interaction with the corepressor peptide. The 6th ligand is Cys 384 (418 in Rev-erbα), which is located on helix 3, the conformation of which is key to determining corepressor affinity. As shown in Figure 1, the heme-bound conformation of Rev-erb is incompatible with corepressor binding, particularly given the energetic importance, demonstrated in the present work, of the N-terminal β-strand for stabilizing the interaction. Regions of the protein present in vivo, and not present in the structural and in vitro assays have been invoked to explain this important contradiction in Rev-erb structure–function relationships.27 Disulfide bond formation between Cys384 of Rev-erbβ (which is the 6th heme ligand) and Cys 374 led to a 5-fold reduction in affinity for the heme.39 This modulation of heme affinity by oxidation/reduction of cysteine residues could also explain discrepancies in reported heme affinity, and may play a role in regulating heme binding, and hence Rev-erb function in vivo. It is noteworthy that binding of the SGN ligand to Rev-erbα leads to increased recruitment of corepressor peptides,34 demonstrating that Rev-erbα can exist in a ligand bound state that is compatible with, and even enhances CoR recruitment.
We propose a model which can reconcile the structural and functional data (Fig. 9). The apo-protein is highly malleable and as suggested by the available structural information, it is likely to be highly dynamic, adjusting its conformation to accommodate either ligands and/or the corepressor peptide motif. In vitro, the iron oxidation state of the heme is FeIII+, which results in a conformation that is incompatible with corepressor binding, since in this conformation helix 3 interferes with the N-terminal β-sheet formation. In contrast, binding of the heme in the (usually) reducing environment of the cell would tend to lead to a Rev-erb/heme complex in which the iron is in the reduced FeII+ state, which can be either penta-coordinated or hexa-coordinated, but with His 381, rather than Cys 384 as the 6th ligand, and which, contrary to the FeIII+ state, can bind gas molecules.30 Given the involvement of cysteine residues in helix 3 in heme coordination and the perturbation of helix 11 by heme binding, it is highly possible that heme coordination and ligation state can considerably affect the conformational properties of these regions. Hence the FeII+ heme-bound configuration of Rev-erb could present a structure that favors corepressor binding. Moreover, the interaction of gas molecules such as NO could modulate the conformation and lead to the observed reversal of Rev-erb-mediated repression.30 Such a scheme could explain why addition of heme leads to increased repression in vivo, whereas heme depletion decreases repressor recruitment.9,29 Heme oscillations due to circadian rhythms could modulate Rev-erb activity from mildly to strongly repressing. Intracellular free heme concentrations are quite low ( < 0.1 μM),40 similar to the reported heme affinity of Rev-erb,35 and heme concentration oscillates with circadian rhythms.37 Moreover, the redox state of the cell could lead to switches between oxidized and reduced forms of the disulfide bond between Cys 384 and Cys 374, which modulates heme affinity.39 Rather unexpectedly, cobalt and zinc porphyrins which might be expected to mimic the FeII+ state, were also antagonists of Rev-erb LBD/corepressor interactions in vitro, and consistent with this observation, the crystal structure of Rev-erbβ LBD bound by coporphyrin was almost identical to that of the FeIII+ heme bound form.35 Nonetheless, heme is an extremely subtle biological sensor, and it is probable that these Co and Zn substituted porphyrins do not adequately represent the reduced heme bound form of Rev-erb.
Figure 9.
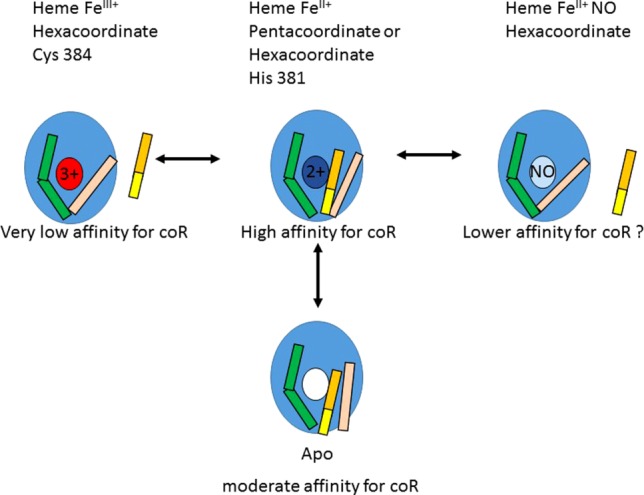
Model of Rev-erb LBD interactions and heme states. The Rev-erb LBD is schematized as a blue ellipse, the ligand binding pocket as the ligand binding site. White is the unbound site, colored small ellipses are heme-bound, either with the iron in the 3 + oxidation state (red), the 2 + oxidation state (dark blue), or bound by NO in the 2 + oxidation state (light blue). Helix 11 is green and helix 3 is in pink. The corepressor is yellow (β-strand) and orange (α-helix).
Confirmation or invalidation of this model for the structure-function relationships in Rev-erb await structures of Rev-erb bound by the reduced heme, NO, CO, and a corepressor peptide. The present results provide quantitative values for corepressor selectivity, the energetic contribution of the N-terminal β-strand of the corepressor motif, and for the effect of heme binding on corepressor recruitment. These values show clearly that reconciliation of the observed effects of heme on corepressor recruitment by Rev-erb in vitro and in vivo must involve considerable structural gymnastics that are controlled by heme binding, heme and/or protein oxidation state and gaseous ligand binding.
Materials and Methods
Proteins, peptides, and ligands
Fluorescence anisotropy experiments used the Rev-erbα LBD: Rev-erbα P281-Q614 (Δ342–413)—6 × histidine. Deletion of the proline rich domain (between α1 and α3 helix containing the α2 helix and the X-domain (342–413)) confers stability to the protein in solution.22 The Rev-erbα-LBD nucleotide sequence was cloned into a pET23 plasmid. Proteins were expressed from E. coli BL21 (DE3). After reaching a 0.8 OD, protein expression was induced (after 1 h at 16°C) by 0.1 mM of IPTG. The bacterial pellet was suspended and lysed at 4°C in phosphate buffer pH 7.4 ; 0.5M NaCl; 1 mM DTT ; 0.5% Triton X-100 ; 0.5 mM PMSF; 1 µM pepstatin. The protein extract was centrifuged and loaded on a His-Trap prep FF (Ni-Sepharose) column equilibrated with 20 mM phosphate buffer pH 7.4; 0.5M NaCl 1 mM DTT; 5% glycerol; 0.5 mM PMSF. Protein was eluted with a linear gradient of imidazole (0–50% of imidazole) in Phosphate buffer pH 7.4; 0.54M NaCl; 0.5M imidazole; 1 mM DTT; 5% glycerol; 0.5 mM PMSF. Purified proteins were conserved at −80°C in the elution buffer.
Corepressor peptides were derived from NCor or SMRT corepressors, chemically synthesized by the EZBiolab Company (Carmel, IN) with a fluorescent label at the N-terminus, either fluorescein or rhodamine 110. Corepressor peptides contain one interaction domain, ID1, 2 or 3, with the exception of NCor5 peptide which contains ID1 and ID2). The NCor5 peptide (below) is derived from the mouse NCoR sequence (GLN 2059 – GLU 2325) and is labeled with a His-Tag and a Tev cleavage site.
QVPRTHRLITLADHICQIITQDFARNQVPSQASTSTFQTSPSALSSTPVRTKTSSRYSPESQSQTVLHPRPGPRVSPENLVDKSRGSRPGKSPERSHIPSEPYEPISPPQGPAVHEKQDSMLLLSQRGVDPAEQRSDSRSPGSISYLPSFFTKLESTSPMVKSKKQEIFRKLNSSGGGDSDMAAAQPGTEIFNLPAVTTSGAVSSRSHSFADPASNLGLEDIIRKALMGSFDDKVEDHGVVMSHPVGIMPGSASTSVVTSSEARRDE
After expression in E. coli BL21 Rosetta bacteria, the peptide was purified on a nickel column then eluted by Tev cleavage. NCor5 peptide was labeled using the NHS ester form of Atto647N (Atto Technology, Amherst NY) at pH 8, which limits labeling to the N-terminus. Three different ligands were tested in this work, heme (Sigma-Aldrich, reference: SLBC4685V) which is the reported natural ligand of Rev-erbα9 and two synthetic ligands SR3335 (ML124) (CID-44237404; SID-85257298)33 (SD7 in this work), and GSK4112 (also known as SR6452) (SGN in this work)34,41 see Scheme 10. Ligands and corepressor peptides were received in powder form and were dissolved and conserved in 100% DMSO, and diluted for experiments at the desired concentration).
Scheme 1.
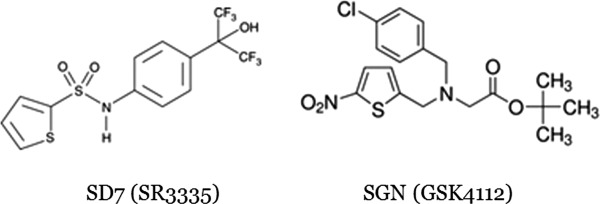
Chemical structures of the synthetic ligands used in this study.
Fluorescence anisotropy assays
All anisotropy experiments were carried out in anisotropy buffer: 200 mM Tris–HCl pH 7.5; 150 mM NaCl; 1 mM EDTA; 5 mM DTT; 10% glycerol; 2.5% DMSO (DMSO concentration in all experiments and wells was maintained at 2.5%). Measurements were made using a Tecan Safire II micro plate reader fluorimeter (Tecan Group AD, Manndorf, Switzerland) and Corning 384 well plates in 60 µL final volumes. Fluorescent labelled peptides were maintained at a fixed concentration of 4 nM throughout titrations of increasing amounts of Rev-erbα LBD (0.0006 to 10 µM, dilutions). When required, ligands (heme, SGN, and SD7) were added at a final concentration of 5, 20, or 100 µM as indicated. Competition experiments evaluate the binding of non labelled corepressor peptide to Rev-erbα-LBD. Fluorescent labelled corepressor peptides, at a final concentration of 4 nM, were titrated by increasing concentrations of Rev-erbα LBD (0.0006–10 µM) in the presence of unlabelled peptides at different concentrations (100, 50, 10, 5, and 2.5 µM).
Anisotropy binding data analysis
Raw anisotropy data are analyzed with BioEqs software42,43 based on numerical calculations of species population in solution and a nonlinear fitting of interaction parameters. This software allows to determine the free energy of complexes (ΔG) as well as their affinity constant (KD) from the adapted binding model considering the mixing of two elements to the solution (protein and fluorescent labelled peptide) and the formation of three possible species at equilibrium (protein alone, protein + fluorescent peptide, and peptide alone) for a classical anisotropy assay. For a competition assay the binding model uses three elements incorporated to the solution (protein, fluorescent labelled peptide and nonlabelled peptide) and five possible species at equilibrium (proteins, fluorescent labelled peptide, nonlabelled peptide, protein + fluorescent peptide, and protein + nonlabelled peptide). In this latter condition two free energy values are obtained: ΔG of the proteins interaction with the fluorescent peptides (already known and fixed in the fit) and ΔG of the protein complex with the nonlabeled peptides. Ligands do not intervene in the affinity constants calculation as we measure the affinity between proteins and fluorescent labelled peptides only, and in the case of the ligands, the proteins are saturated at all concentrations tested. The raw data and the fits obtained were normalized between 0 (anisotropy value of fluorescent labelled peptide alone, no complexes formed) and 1 (anisotropy value when 100% of formed complexes), using the fitted plateau values. Considering the fact that 100% of complexes are not always reached, maximum delta anisotropy cannot be known. For those cases we used the delta anisotropy obtained in the absence of ligand as a reference which is added to the lower anisotropy value in the presence of ligands, and adjusted such that the simple binding rule of 1.908 log units for 10–90% saturation must hold.
Isothermal titration calorimetry
Binding of NCoRID1C and SMRTID1B-2 peptide to Rev-erbα LBD was analyzed by ITC using the ITC200 instrument (MicroCal) at 25°C in ITC buffer: Tris 20 mM, NaCl 200 mM, TCEP 1 mM, 10% glycerol, pH 8.5 + 20% H2O. Prior to titration Rev-erbα LBD was dialyzed against Tris 20mM, NaCl 200 mM, TCEP 1 mM, 10% glycerol, pH 8.5. After dialysis, the protein concentration was determined spectrophotometrically. Rev-erbα LBD concentration in the calorimetric cell was 10 μM, whereas NCoRID1C or SmrtID1B-2 peptide concentrations in the syringe varied from 200 to 300 μM. The heat of dilution was measured by injecting peptide into the protein-free buffer solution or by additional injections of peptide after saturation; the value obtained was subtracted from the heat of reaction to obtain the true effective heat of binding. Data were analyzed using the MicroCal Origin software and were fitted with a “One set of sites” model to obtain the affinity constants (Ka), stoichiometry (N), and enthalpy changes (ΔH). Consequently, the entropy changes (ΔS) were calculated according to the standard equation.
Surface plasmon resonance
The interaction between immobilized NCoR peptide, slightly longer than that used in the anisotropy and ITC assays (Biot-QVPRTHRLITLADHIAQIITQDFARNQVS; Biotinyl-Q-29-S) and Rev-erbα LBD was measured using a Biacore T200 instrument (GE Healthcare) at 25°C. Biotinylated NCoR peptide (10 µM) was tethered on SA sensor chips (GE Healthcare) as described by the supplier (GE healthcare). Typical immobilization levels were around 1000 resonance units (RU). A reference flow cell was prepared by tethering a peptide of similar molecular weight. After the peptide immobilization step, surfaces were saturated by biotin as preconized by the supplier. HBS-EP + containing 1 mM TCEP, 10% glycerol and 5% (v/v) DMSO was used as running buffer. Samples were injected for 120 s followed by a 100 s dissociation phase at a flow rate of 30 μL/min. Results were analyzed by subtracting the signal of the reference flow cell from the signal of the NCoR-bound flow cell. Each experiment series included blanks (running buffer), injection of control and solvent corrections. For competition of NCoR/Rev-erbα LBD binding by selected compounds, Rev-erbα-LBD was injected at 313 nM preincubated without compounds or Smrt in the running buffer containing 5% (v/v) DMSO. This gave a response of around 100 RU. Similar experiments were performed with Rev-erbα pre-incubated with a concentration range of Smrt from 0.1–10 μM [5% (v/v) DMSO]. The responses (in RU) were obtained at the end of the injection time after subtraction of the reference response and DMSO correction. The experiment was repeated two times. An injection of Rev-erbα LBD pre-incubated with running buffer alone between each Smrt or compounds concentration was performed to evaluate the global stability of its conformation. All experiments were performed in duplicate.
ESI-MS measurements
ESI-MS experiments were carried out on an electrospray time-of-flight mass spectrometer (LCT, Waters, Manchester, UK) equipped with an automated chip-based nanoESI device (Triversa Nanomate, Advion Biosciences, Ithaca, NY). External calibration was done in the positive ion mode over the mass range m/z 500–5000 using the multiply charged ions produced by a 0.5 µM horse heart myoglobin solution diluted in a 50/50 water/acetonitrile mixture acidified with 0.5% (v/v) formic acid. Purified Rev-erbα-LBD (P281–Q614 Δ342–413) was buffer exchanged against 200 mM ammonium acetate pH 7.5 using microcentrifuge gel filtration columns (Zeba 0.5 mL, Thermo Scientific, Rockford, IL). The protein concentration was determined spectrophotometrically. The integrity, homogeneity and purity of the Rev-erb alpha was first analyzed under denaturing conditions after the protein was diluted to 1 µM in the acidified 50%/50% water/acetonitrile mixture. The Rev-erbα LBD construct used in the Mass spectrometry analysis was His-Rev-erbα (P281–Q614 Δ342–413)-pET15b with a theoretical mass of 31528.7 Da. The measured mass of the apoprotein (31396 Da) corresponds to the receptor without the N-terminal methionine. Characterization of compound binding to Rev-erbα LBD under nondenaturing conditions was then performed in 200 mM ammonium acetate pH 7.5, 5% (v/v) ethanol. To determine compound interaction, the protein concentration was held constant at 10 µM while the compound was titrated to achieve a final concentration ranging from 20 to 50 µM. Each complex was incubated at room temperature for 30 min and mass spectra were recorded using reduced cone voltage (Vc = 20 V) and elevated interface pressure (Pi = 6 mbar) which corresponds to the fine-tuned instrumental settings previously determined to provide sufficient ion desolvation while preserving the integrity of weak noncovalent complexes in the gas phase. The concentration of free and bound forms of the Rev-erbα LBD ligand was deduced by measuring the peak height of the 12 + charge states of the corresponding species, assuming that compound binding does not alter the protein response factor.
Acknowledgments
The authors would like to thank Valérie Vivat from Novalix, Inc. for the Mass spectrometry experiments.
Glossary
- NR
nuclear receptor
- TF
transcription factor
- ID
interaction domain
- LBD
ligand binding domain
- DBD
DNA binding domain
- AF2
activation function 2
- CoRNR
corepressor nuclear receptor interaction motif
- SPR
surface plasmon resonance
- ITC
isothermal titration calorimetry
- MS
mass spectrometry
- SD7
SR3335
- SGN
GSK4112
References
- Moore JT, Collins JL, Pearce KH. The nuclear receptor superfamily and drug discovery. ChemMedChem. 2006;1:504–523. doi: 10.1002/cmdc.200600006. [DOI] [PubMed] [Google Scholar]
- Everett LJ, Lazar MA. Nuclear receptor Rev-erbalpha: up, down, and all around. Trends Endocrinol Metab. 2014;25:586–592. doi: 10.1016/j.tem.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bois-Joyeux B, Chauvet C, Nacer-Cherif H, Bergeret W, Mazure N, Giguere V, Laudet V, Danan JL. Modulation of the far-upstream enhancer of the rat alpha-fetoprotein gene by members of the ROR alpha, Rev-erb alpha, and Rev-erb beta groups of monomeric orphan nuclear receptors. DNA Cell Biol. 2000;19:589–599. doi: 10.1089/104454900750019344. [DOI] [PubMed] [Google Scholar]
- Harding HP, Lazar MA. The orphan receptor Rev-ErbA alpha activates transcription via a novel response element. Mol Cell Biol. 1993;13:3113–3121. doi: 10.1128/mcb.13.5.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Lazar MA. The monomer-binding orphan receptor Rev-Erb represses transcription as a dimer on a novel direct repeat. Mol Cell Biol. 1995;15:4791–4802. doi: 10.1128/mcb.15.9.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugge A, Feng D, Everett LJ, Briggs ER, Mullican SE, Wang F, Jager J, Lazar MA. Rev-erbalpha and Rev-erbbeta coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012;26:657–667. doi: 10.1101/gad.186858.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng QJ, McMaster A, Beesley S, Lu WQ, Gibbs J, Parks D, Collins J, Farrow S, Donn R, Ray D, Loudon A. Ligand modulation of REV-ERBalpha function resets the peripheral circadian clock in a phasic manner. J Cell Sci. 2008;121:3629–3635. doi: 10.1242/jcs.035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere V, Dufour CR, Eichner LJ, Deblois G, Cermakian N. Estrogen-related receptor alpha, the molecular clock, and transcriptional control of metabolic outputs. Cold Spring Harb Symp Quant Biol. 2011;76:57–61. doi: 10.1101/sqb.2011.76.011031. [DOI] [PubMed] [Google Scholar]
- Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, Waitt GM, Parks DJ, Pearce KH, Wisely GB, Lazar MA. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786–1789. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- Yin L, Wu N, Lazar MA. Nuclear receptor Rev-erbalpha: a heme receptor that coordinates circadian rhythm and metabolism. Nucl Receptor Signal. 2010;8:e001. doi: 10.1621/nrs.08001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Feng D, Emmett MJ, Everett LJ, Loro E, Briggs ER, Bugge A, Hou C, Ferrara C, Seale P, Pryma DA, Khurana TS, Lazar MA. The nuclear receptor Rev-erbalpha controls circadian thermogenic plasticity. Nature. 2013;503:410–413. doi: 10.1038/nature12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste H, Rodriguez JC. Orphan nuclear hormone receptor Rev-erbalphα regulates the human apolipoprotein CIII promoter. J Biol Chem. 2002;277:27120–27129. doi: 10.1074/jbc.M203421200. [DOI] [PubMed] [Google Scholar]
- Raspe E, Duez H, Mansen A, Fontaine C, Fievet C, Fruchart JC, Vennstrom B, Staels B. Identification of Rev-erbalpha as a physiological repressor of apoC-III gene transcription. J Lipid Res. 2002;43:2172–2179. doi: 10.1194/jlr.m200386-jlr200. [DOI] [PubMed] [Google Scholar]
- Vu-Dac N, Chopin-Delannoy S, Gervois P, Bonnelye E, Martin G, Fruchart JC, Laudet V, Staels B. The nuclear receptors peroxisome proliferator-activated receptor alpha and Rev-erbalpha mediate the species-specific regulation of apolipoprotein A-I expression by fibrates. J Biol Chem. 1998;273:25713–25720. doi: 10.1074/jbc.273.40.25713. [DOI] [PubMed] [Google Scholar]
- Delerive P, Monte D, Dubois G, Trottein F, Fruchart-Najib J, Mariani J, Fruchart JC, Staels B. The orphan nuclear receptor ROR alpha is a negative regulator of the inflammatory response. EMBO Rep. 2001;2:42–48. doi: 10.1093/embo-reports/kve007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelivet Y, Rochel N, Moras D. Structural analysis of nuclear receptors: from isolated domains to integral proteins. Mol Cell Endocrinol. 2012;348:466–473. doi: 10.1016/j.mce.2011.08.015. [DOI] [PubMed] [Google Scholar]
- Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–272. doi: 10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osz J, Brelivet Y, Peluso-Iltis C, Cura V, Eiler S, Ruff M, Bourguet W, Rochel N, Moras D. Structural basis for a molecular allosteric control mechanism of cofactor binding to nuclear receptors. Proc Natl Acad Sci USA. 2012;109:E588–E594. doi: 10.1073/pnas.1118192109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–96. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- Hu X, Lazar MA. Transcriptional repression by nuclear hormone receptors. Trends Endocrinol Metab. 2000;11:6–10. doi: 10.1016/s1043-2760(99)00215-5. [DOI] [PubMed] [Google Scholar]
- Hu X, Li Y, Lazar MA. Determinants of CoRNR-dependent repression complex assembly on nuclear hormone receptors. Mol Cell Biol. 2001;21:1747–1758. doi: 10.1128/MCB.21.5.1747-1758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir I, Harding HP, Atkins GB, Horlein A, Glass CK, Rosenfeld MG, Lazar MA. A nuclear hormone receptor corepressor mediates transcriptional silencing by receptors with distinct repression domains. Mol Cell Biol. 1996;16:5458–5465. doi: 10.1128/mcb.16.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir I, Zhang J, Lazar MA. Stoichiometric and steric principles governing repression by nuclear hormone receptors. Genes Dev. 1997;11:835–846. doi: 10.1101/gad.11.7.835. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zamir I, Lazar MA. Differential recognition of liganded and unliganded thyroid hormone receptor by retinoid X receptor regulates transcriptional repression. Mol Cell Biol. 1997;17:6887–6897. doi: 10.1128/mcb.17.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson ML, Jonas BA, Privalsky ML. Alternative mRNA splicing of SMRT creates functional diversity by generating corepressor isoforms with different affinities for different nuclear receptors. J Biol Chem. 2005;280:7493–7503. doi: 10.1074/jbc.M411514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Maire A, Teyssier C, Erb C, Grimaldi M, Alvarez S, de Lera AR, Balaguer P, Gronemeyer H, Royer CA, Germain P, Bourguet W. A unique secondary-structure switch controls constitutive gene repression by retinoic acid receptor. Nat Struct Mol Biol. 2010;17:801–807. doi: 10.1038/nsmb.1855. [DOI] [PubMed] [Google Scholar]
- Phelan CA, Gampe RT, Jr, Lambert MH, Parks DJ, Montana V, Bynum J, Broderick TM, Hu X, Williams SP, Nolte RT, Lazar MA. Structure of Rev-erbalpha bound to N-CoR reveals a unique mechanism of nuclear receptor–co-repressor interaction. Nat Struct Mol Biol. 2010;17:808–814. doi: 10.1038/nsmb.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinking J, Lam MM, Pardee K, Sampson HM, Liu S, Yang P, Williams S, White W, Lajoie G, Edwards A, Krause HM. The Drosophila nuclear receptor e75 contains heme and is gas responsive. Cell. 2005;122:195–207. doi: 10.1016/j.cell.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Raghuram S, Stayrook KR, Huang P, Rogers PM, Nosie AK, McClure DB, Burris LL, Khorasanizadeh S, Burris TP, Rastinejad F. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REV-ERBbeta. Nat Struct Mol Biol. 2007;14:1207–1213. doi: 10.1038/nsmb1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee KI, Xu X, Reinking J, Schuetz A, Dong A, Liu S, Zhang R, Tiefenbach J, Lajoie G, Plotnikov AN, Botchkarev A, Krause HM, Edwards A. The structural basis of gas-responsive transcription by the human nuclear hormone receptor REV-ERBbeta. PLoS Biol. 2009;7:e43. doi: 10.1371/journal.pbio.1000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo EJ, Jeong DG, Lim MY, Jun KS, Kim KJ, Yoon SM, Park BC, Ryu SE. Structural insight into the constitutive repression function of the nuclear receptor Rev-erbbeta. J Mol Biol. 2007;373:735–744. doi: 10.1016/j.jmb.2007.08.037. [DOI] [PubMed] [Google Scholar]
- Yin L, Lazar MA. The orphan nuclear receptor Rev-erbalpha recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol Endocrinol. 2005;19:1452–1459. doi: 10.1210/me.2005-0057. [DOI] [PubMed] [Google Scholar]
- Kumar N, Kojetin DJ, Solt LA, Kumar KG, Nuhant P, Duckett DR, Cameron MD, Butler AA, Roush WR, Griffin PR, Burris TP. Identification of SR3335 (ML-176): a synthetic RORalpha selective inverse agonist. ACS Chem Biol. 2011;6:218–222. doi: 10.1021/cb1002762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant D, Yin L, Collins JL, Parks DJ, Orband-Miller LA, Wisely GB, Joshi S, Lazar MA, Willson TM, Zuercher WJ. GSK4112, a small molecule chemical probe for the cell biology of the nuclear heme receptor Rev-erbalpha. ACS Chem Biol. 2010;5:925–932. doi: 10.1021/cb100141y. [DOI] [PubMed] [Google Scholar]
- Matta-Camacho E, Banerjee S, Hughes TS, Solt LA, Wang Y, Burris TP, Kojetin DJ. Structure of REV-ERB beta ligand-binding domain bound to a porphyrin antagonist. J Biol Chem. 2014;289:20054–20066. doi: 10.1074/jbc.M113.545111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik K, Lee CC. Reciprocal regulation of haem biosynthesis and the circadian clock in mammals. Nature. 2004;430:467–471. doi: 10.1038/nature02724. [DOI] [PubMed] [Google Scholar]
- Rogers PM, Ying L, Burris TP. Relationship between circadian oscillations of Rev-erbalpha expression and intracellular levels of its ligand, heme. Biochem Biophys Res Commun. 2008;368:955–958. doi: 10.1016/j.bbrc.2008.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Ragsdale SW. Thiol-disulfide redox dependence of heme binding and heme ligand switching in nuclear hormone receptor rev-erb{beta} J Biol Chem. 2011;286:4392–4403. doi: 10.1074/jbc.M110.193466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Tyrrell RM. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med. 2000;28:289–309. doi: 10.1016/s0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat Rev. Drug Discov. 2014;13:197–216. doi: 10.1038/nrd4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer CA, Smith WR, Beechem JM. Analysis of binding in macromolecular complexes: a generalized numerical approach. Anal Biochem. 1990;191:287–294. doi: 10.1016/0003-2697(90)90221-t. [DOI] [PubMed] [Google Scholar]
- Royer CA, Beechem JM. Numerical analysis of binding data: advantages, practical aspects, and implications. Methods Enzymol. 1992;210:481–505. doi: 10.1016/0076-6879(92)10025-9. [DOI] [PubMed] [Google Scholar]