

Abstract
Polyglycine hydrolases are secreted fungal proteases that cleave glycine–glycine peptide bonds in the inter-domain linker region of specific plant defense chitinases. Previously, we reported the catalytic activity of polyglycine hydrolases from the phytopathogens Epicoccum sorghi (Es-cmp) and Cochliobolus carbonum (Bz-cmp). Here we report the identity of their encoding genes and the primary amino acid sequences of the proteins responsible for these activities. Peptides from a tryptic digest of Es-cmp were analyzed by LC-MS/MS and the spectra obtained were matched to a draft genome sequence of E. sorghi. From this analysis, a 642 amino acid protein containing a predicted β-lactamase catalytic region of 280 amino acids was identified. Heterologous strains of the yeast Pichia pastoris were created to express this protein and its homolog from C. carbonum from their cDNAs. Both strains produced recombinant proteins with polyglycine hydrolase activity as shown by SDS-PAGE and MALDI-MS based assays. Site directed mutagenesis was used to mutate the predicted catalytic serine of Es-cmp to glycine, resulting in loss of catalytic activity. BLAST searching of publicly available fungal genomes identified full-length homologous proteins in 11 other fungi of the class Dothideomycetes, and in three fungi of the related class Sordariomycetes while significant BLAST hits extended into the phylum Basidiomycota. Multiple sequence alignment led to the identification of a network of seven conserved tryptophans that surround the β-lactamase-like region. This is the first report of a predicted β-lactamase that is an endoprotease.
Keywords: protease, proteinase, family S12, β-lactamase, penicillin-binding protein, SXXK motif, host–pathogen interactions, plant defense, effectors, PAMP-triggered immunity, effector-triggered defense
Introduction
Polyglycine hydrolases are one type of chitinase modifying protein (cmp), secreted fungal proteases that truncate plant class IV chitinases by cleaving near their amino-termini (Fig. 1). Cmp activity is exhibited by at least three types of proteases: fungalysin metalloproteases that cleave a glycine-cysteine bond that is conserved among plant class IV defense chitinases1; alloform-specific proteases that distinguishing a single amino acid difference in maize ChitA alloforms2; and polyglycine hydrolases, proteases that cleave glycine-glycine peptide bonds in the inter-domain linker region of targeted chitinases.3 The proteolytic activity of the polyglycine hydrolase Bz-cmp was first observed in diseased ears of maize that had been inoculated with Cochliobolus carbonum (syn. Bipolaris zeicola).4 A recent mass spectrometry analysis of reaction products showed that both Bz-cmp and Es-cmp—an enzyme from the sorghum pathogen Epicoccum sorghi—can selectively cleave glycine-glycine peptide bonds within a polyglycine linker of targeted chitinases.3
Figure 1.

Chitinase modifying proteins. ChitA and ChitB chitinases from maize are composed of a small, amino-terminal chitin-binding hevein domain and a larger (22 kDa) chitinase domain. These domains are connected by a polyglycine linker. The chitin binding domain is truncated by alloform specific proteases and fungalysin proteases (arrows indicate site of cleavage for each). Polyglycine hydrolases Es-cmp and Bz-cmp have been shown to cleave glycine–glycine peptide bonds within the polyglycine linker by MALDI-TOF/MS analysis of the product peptides released from the N-terminus.
Our initial findings demonstrated that plant class IV chitinases with polyglycine linkers are targeted for truncation by selective polyglycine hydrolases that are secreted by plant pathogenic fungi. Bz-cmp hydrolyzed ChitB predominantly between glycines G3 and G4, whereas the Es-cmp activity was specific for the G1, G3, and G4 residues.3 Site directed mutagenesis of maize ChitB chitinase was used to replace two serine residues in its polyglycine linker region with glycines, resulting in altered substrate specificity for Bz-cmp and Es-cmp such that different glycine-glycine peptide bonds were cleaved. It was also noted that the removal of the N-terminal hevein domain from the ChitB protein led to loss of Es-cmp activity, indicating that interactions outside of the active site are involved in recognition of the polyglycine region. This novel proteolysis of polyglycine motifs was previously unknown, but it was noted that the specificity is similar to that of bacterial lysostaphin proteases, which cleave pentaglycine crosslinks from peptidoglycan.5 We hypothesized that polyglycine hydrolase specificity resulted from two factors: recognition of the amino-terminal chitin binding domain of targeted chitinases through exosite interactions, and selective cleavage of Gly-Gly bonds in the linker by a narrow active site that occludes amino acids with side chains.3
In this manuscript we show that polyglycine hydrolases are serine-type proteases from family S12,6 a protease family comprised of penicillin binding proteins involved in bacterial cell wall biosynthesis,7 β-lactamases that cleave β-lactam antibiotics,8,9 and amino-peptidase cleavage of D-amino acids.10,11 More than half of the protein's sequence is novel, possibly encoding a target recognition domain. Our results show that S12 proteases can function as endoproteases, an activity that has not previously been reported. Moreover their proteolytic specificity to glycine–glycine bonds is remarkable in terms of the more general D-amino acid selectivity of the known bacterial S12 proteases.
Results
Identification of Es-Cmp and Bz-Cmp by heterologous expression of recombinant proteins
We previously reported the activity of two polyglycine hydrolases, Es-cmp and Bz-cmp. Of the two proteases, Es-cmp is produced in greater abundance in culture and we were able to purify this protein and assign its activity to a band on an SDS-PAGE gel.3 Our first attempt to identify the encoding genes for these proteins, in the current study, involved liquid chromatography and MS/MS data collection of tryptic peptides produced from purified Es-cmp. These spectra were first searched against the publicly available genome sequence for C. carbonum,12 which produces Bz-cmp, but few spectra were matched. This surrogate approach, using identified peptide sequences from the protein of one organism to identify a protein with similar function in another, did not work. Next generation sequencing technology was then used to produce a draft genome sequence for E. sorghi. When the spectra from LC-MS/MS analysis were searched against the draft genome, a predicted protein of 642 amino acids was identified as a candidate for polyglycine hydrolase activity. Of the 619 amino acids expected to be present in the mature native protein, 420 residues (68% sequence coverage) were found by mass spectrometry [Fig. 2(A)]. Analysis of deglycosylated protein contributed another five peptides, (76 residues, to give 80% sequence coverage), which are potentially N-glycosylated by virtue of the fact that each now contains an ASP residue instead of the ASN predicted by the sequence. Though these could occur by simple deamidation, if this were the case, we would also expect to find these in the glycosylated sample. Analysis of the amino acid sequence of this protein by the PFAM database13 resulted in identification of a 280 amino acid region [Fig. 2(B), grey] as a predicted β-lactamase region. The SignalP algorithm predicts that the first 23 amino acids [Fig. (2B), underlined] are a secretion signal sequence that is removed during protein maturation.14 Function could not be predicted for the remainder of the protein. The encoding DNA sequence has been submitted (GenBank KM492932). A homolog was identified by BLAST searching the C. carbonum genome (Cocca1_88813). The amino acid identity of the two proteins is <60%. The low amino acid identity between the two proteases may explain why the initial surrogate approach failed.
Figure 2.
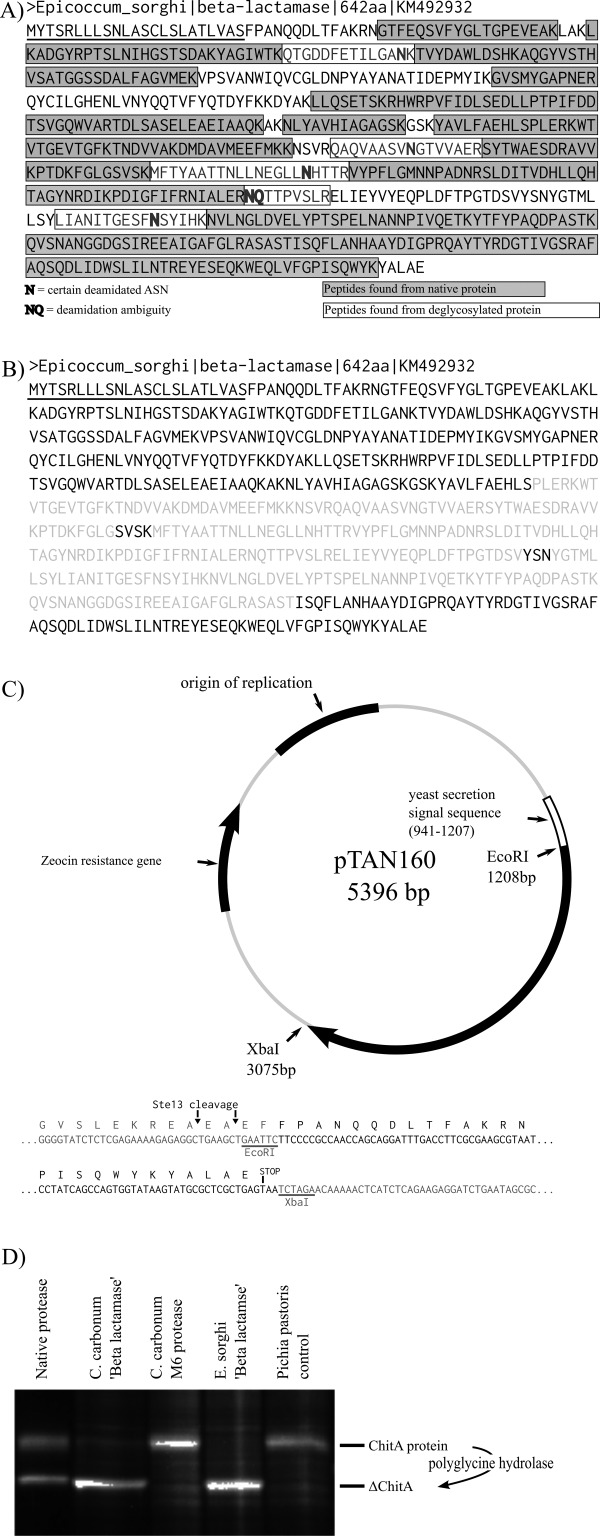
Identification of Es-cmp and Bz-cmp. (A) Peptide mapping. The identified Es-cmp candidate protein is shown. Identified peptides are shown as grey boxes (68% sequence coverage); five additional peptides identified from a PNGaseF-treated sample (deglysosylated) are shown enclosed in black boxes (80% sequence coverage total). (B) Primary sequence analysis. The identified 642 amino acid protein contains a predicted secretion signal sequence (underlined) and a predicted β-lactamase catalytic region (grey); catalytic motifs are black within the grey sequence. (C) Expression vector cloning. The coding DNA for the identified protein and its closest homolog from C. carbonum were cloned into an expression vector and integrated into the genome of the yeast Pichia pastoris. (D) Activity assays. Heterologous strains of P. pastoris were induced by addition of methanol to secrete recombinant proteins. After 4 h, 1 μL of each culture was added to 10 μL in vitro protease reactions containing ChitA protein and incubated for 1 h. Reactions were analyzed by SDS-PAGE followed by protein staining for total protein. Heterologous strains producing either the candidate protein from E. sorghi or its homolog from C. carbonum truncated ChitA into a smaller form that matches that produced by native protein. Heterologous strains producing either a predicted M6 protease from C. carbonum or a control strain that does not produce recombinant protein did not have activity.
To confirm that these identified proteins were polyglycine hydrolases, recombinant proteins were produced from their cDNAs and tested for activity [Fig. 2(C,D)]. Four heterologous strains of the yeast Pichia pastoris were grown. Two strains encoded secreted expression of either the identified candidate protein from E. sorghi or its homolog from C. carbonum. One control strain encoded secreted expression of an M6 protease from C. carbonum. A second contained an integrated expression plasmid that did not have a cloned gene. Cells were grown under non-inducing conditions, pelleted, and re-suspended in induction media. After 4 h of growth in induction media, a sample from each culture was incubated with purified ChitA chitinase to test for polyglycine hydrolase activity. After incubation the reactions were assessed by SDS-PAGE followed by protein staining [Fig. 2(D)]. Activity was observed in reactions containing either the identified E. sorghi protein or the C. carbonum homolog (lanes 2 and 4). In these reactions most of the substrate ChitA protein was converted to a truncated product that matched that produced by purified, native polyglycine hydrolase protein (lane 1). Control reactions (lanes 3 and 5) did not produce this product, confirming that the truncation was due to expression of protein from the cloned cDNAs.
Purification of recombinant polyglycine hydrolases
A method was developed for expression and purification of recombinant Es-cmp. Protein expression conditions were optimized by testing the amount of activity present in the media at different time points and when expressed at various temperatures. The highest polyglycine hydrolase activity occurred in cultures when expressed for 2 days at 20°C (not shown). After expression, cells were precipitated by centrifugation. Ammonium sulfate was gradually added to the supernatant. Insoluble materials were then precipitated by centrifugation, re-suspended in buffer, and dialyzed. Following dialysis, Es-cmp was further purified by mixed-mode cation exchange chromatography [Fig. 3(A)]. Two elution peaks were observed. Elution fractions from the first peak were found to contain Es-cmp activity [Fig. 3(A), 65.6–77.6 ml]. These elution fractions were combined (E) and Es-cmp was further purified by reprecipitation with acetone followed by re-suspension in storage buffer (re-ppt) [Fig. 3(B)]. While gel analysis of protein content shows a high level of protein purity in all steps, it was observed that the appearance of the protein solution changed from dark black (D-RP) to yellow (E) to clear (re-ppt), indicating removal of non-protein contaminants. The final solution, which resulted from a 1 L culture, contained 10 mg of protein in a volume of 0.6 ml. The same method was also applied to purification of recombinant Bz-cmp, with similar results [Fig. 3(C)]. Protease activity in the cell-free media was lower in Bz-cmp expressing cultures (not shown), and final yield was 3.5 mg of protein.
Figure 3.

Purification of recombinant polyglycine hydrolases. Proteins were purified from liquid cultures in four steps: ammonium sulfate precipitation, dialysis, mixed-mode cation exchange chromatography, and acetone precipitation. (A) Mixed-mode cation exchange chromatography of Es-cmp. After ammonium sulfate precipitation, the dialyzed, resuspended pellets (D-RP) were injected onto the column. Unbound proteins were then removed by washing with two column volumes of buffer, followed by a linear elution with increasing salt and pH. The majority of recombinant protein eluted between 65.6 and 77.6 mL (E). Es-cmp was further purified by acetone precipitation. (B) SDS-PAGE analysis of purification. Only the recombinant protein is visible at all steps: prior to chromatography (D-RP), after elution (E), and after re-precipitation (re-ppt). The three solutions went from black to light yellow to clear, indicating removal of contaminating molecules from the expression media. (C) Bz-cmp purification. Recombinant Bz-cmp was expressed and purified by the same method, resulting in pure protein, albeit with lower yield. (D) Gel filtration. The structural integrity of purified proteases were confirmed by gel filtration. Integration analysis indicates that 99.4 and 97.8% of purified Es-cmp and Bz-cmp are correctly folded. Analysis of catalytic mutant Es-cmp(S370G) indicates that this protein retains its structural integrity.
To check the structural integrity of purified proteins, Es-cmp and Bz-cmp were subjected to analysis by gel filtration [Fig. 3(D)]. In each case, chromatographs were dominated by a single large peak with a retention near 12.5 mL. The preps also had small peaks that eluted earlier, indicating presence of a minor amount of unfolded protein. Integration of these peaks demonstrated that Es-cmp and Bz-cmp preps contained 99.4 and 97.8% intact protein. Analysis of Es-cmp(S370G), which contains a mutation in the predicted catalytic serine, showed only a single peak, indicating that this amino acid change does not disrupt the structural integrity of the protease.
In vitro polyglycine hydrolase activity of recombinant Es-Cmp and Bz-Cmp
The ability of purified, recombinant polyglycine hydrolases Es-cmp and Bz-cmp to cleave ChitA and ChitB substrates was assayed in vitro. For each of the four combinations of proteases and substrates, the concentration of protease was varied to determine how much enzyme was necessary to convert half of the substrate to products under constant conditions of 1 μM ChitA substrate at 30°C for 1 h. The results were analyzed by SDS-PAGE followed by total protein staining and densitometry [Fig. 4(A), insets]. For three of the reactions, the E1/2 was similar, at less than 100 pM [Fig. 4(A), first 3 insets]. For the combination of Bz-cmp and ChitB chitinase, it required 10-fold more protein to observe the same amount of cleavage [Fig. 4(A), bottom inset]. This is consistent with observations made previously in reactions with native Bz-cmp, where increased incubation times were necessary to truncate the same amount of ChitB compared to ChitA when the same amount of Bz-cmp was added.3
Figure 4.

In vitro activity of recombinant polyglycine hydrolases on plant chitinase substrates. (A) Reactions containing 1 μM ChitA or ChitB were incubated with Es-cmp or Bz-cmp for 1 h. Cleavage of peptide bonds was mapped by MALDI-TOF/MS analysis of released peptides; the amount of activity was determined by SDS-PAGE analysis and densitometry of truncated chitinases (inset of each panel). The amount of protease necessary to convert 50% of the substrate to products under these conditions is indicated (E1/2). The identity of the final amino acid in the product peptides is indicated above each peptide peak. Unassigned peaks are indicated by asterisks. (B) Catalytic activity summary for native and recombinant polyglycine hydrolases. Deduced cleavage sites within the polyglycine linker regions of ChitA and ChitB as deduced from MALDI-TOF/MS analysis of N-terminal peptide products. For each chitinase, the results from recombinant enzymes (black, pointing up) are compared with cleavage results previously reported for native enzymes (gray, pointing down).
To observe the proteolytic activity in more detail, each of the four combinations of protease and substrate were incubated, using the amount of protease necessary to produce 50% product, and the released amino-terminal peptides were assayed by MALDI-TOF MS [Fig. 4(A)]. As observed previously with the native proteases, the product peptides contain varying numbers of glycine residues, indicating that the polyglycine hydrolase enzymes are selective for the number and position of gly-gly bonds within the polyglycine sequence. The cleavage sites observed and their abundance varied with both the enzyme—Es-cmp or Bz-cmp—and the substrate used. The pattern observed for each combination of protease and chitinase was similar to that reported for native proteases. The largest change was that recombinant Bz-cmp preferentially cleaves after G8 of ChitA rather than G7. A summary of protease products and a comparison to those reported for native Es-cmp and Bz-cmp is presented in Figure 4(B).
Further characterization of Es-Cmp
As polyglycine hydrolases contain a catalytic region that is similar to β-lactamases, we tried to observe β-lactamase or S12 protease-like behavior in Es-cmp and Bz-cmp by testing inhibitors and substrates associated with those proteins. To test inhibitors, they were added to polyglycine hydrolase reactions with Es-cmp or Bz-cmp and ChitA. Reaction products were observed by SDS-PAGE followed by protein staining (Fig. 5). Addition of PMSF, an inhibitor of serine peptidases of class S1, a large family of serine endopeptidases that includes trypsin and chymotrypsin, did not inhibit activity (lanes 3 and 4). Addition of clavulanic acid (a β-lactamase inhibitor, lanes 5 and 6) or ampicillin (PBP inhibitor, lanes 7 and 8) did not inhibit the truncation of ChitA. Additionally, Es-cmp and Bz-cmp were unable to hydrolyze nitrocefin or glycine p-nitroanilide, colorimetric substrates of β-lactamases and amino peptidases (not shown). On the basis of this lack of chemical evidence to link Es-cmp and Bz-cmp to S12 proteases, we attempted to confirm their evolutionary relationship by demonstrating the predicted catalytic serine of Es-cmp is in the SXXK motif. Site-directed mutagenesis was used to mutate this amino acid to glycine. In reactions containing either 3 nM (100-fold increase, lane 9) or 30 nM (1,000-fold increase, lane 10) of purified Es-cmp(S370G) no reaction products were observed, indicating that this serine amino acid is necessary for catalysis. Taken together, these results suggest that the catalytic ability of Es-cmp and Bz-cmp has changed significantly—as indicated by its lack of interaction with known inhibitors and substrates of S12 proteases—but still retains the catalytic mechanism of other members of the group by utilizing the serine amino acid of the SXXK motif to catalyze the endopeptidase reaction.
Figure 5.

Analysis of recombinant proteases. The proteolytic activity of Es-cmp (top) and Bz-cmp (bottom) was not inhibited by addition of the serine protease inhibitor PMSF (lane 3, 0.1 mM; lane 4, 1 mM), the β-lactamase inhibitor clavulanic acid (lane 5, 0.1 mM; lane 6, 1 mM), or the transpeptidase inhibitor ampicillin (lane 7, 0.1 mM; lane 8, 1 mM). In Es-cmp, mutation of the predicted catalytic serine to glycine resulted in apparent loss of all catalytic activity, even at elevated protein concentrations (lane 9, 7 nM, ×100; lane 10, 70 nM, ×1,000). Positive controls did not contain inhibitor, negative controls did not contain protease or inhibitor.
Identification of polyglycine hydrolase homologs
To assess the phylogenetic distribution of polyglycine hydrolases, we searched for Es-cmp homologs from publicly available fungal genome sequences using its amino acid sequence and the BLAST server at the MycoCosm portal15 (Supporting Information S1). In addition to Bz-cmp from C. carbonum, 66 fungal enzymes were found that had significant blast hits. These hits were found beyond the Dothideomycetes, extending to the classes Sordariomycetes, Leotiomycetes, and Eurotiomycetes. Also, hits were found in the phylum Basidiomycota. Fourteen out of the 66 hits were full-length matches to Es-cmp and Bz-cmp. The amino acid sequence of each of these protein homologs was compared pair-wise to Es-cmp and Bz-cmp (Fig. 6). The majority of these homologs, like Es-cmp and Bz-cmp, were identified from fungi in the class Dothidoemycetes while the remaining three were found in the class Sordariomycetes (Fig. 6, grey background).
Figure 6.

Identification of polyglycine hydrolase homologs. (A) Map of primary amino acid sequence of polyglycine hydrolases. (B) Blast search. A blast server at the MycoCosm portal was used to search fungal genomes for predicted proteins that were full-length homologs of Es-cmp. Out of 66 fungal enzymes identified with significant homology, 14 proteins were identified as full-length homologs. Eleven were from fungal plant pathogens in the Dothidiomycetes (top) while three were from fungi in the related class Sordariomycetes (grey background). Each predicted protein was aligned pair-wise with Es-cmp and Bz-cmp; summaries of the alignments are listed. A multiple sequence alignment of these 14 sequences with Es-cmp and Bz-cmp identified a network of seven conserved tryptophan residues (shown in A).
To search for regions of conserved sequence—which could indicate functional importance—the 16 homologous sequences were compared by multiple sequence alignment using the ClustalW algorithm16 (Supporting Information S2). No regions with extensive homology were found within this set of proteins. The longest stretches of absolutely conserved sequence were only four amino acids long. Despite the lack of overall sequence conservation, all of the proteins identified have identical sequences in both the SXXK and YXN motifs—SVSK and YSN. Additionally, there is a network of seven absolutely conserved tryptophan residues that surround the predicted catalytic domain. The first six occur before the catalytic region. The number of amino acids between each tryptophan varies slightly, and fits the regular expression pattern:
The seventh conserved tryptophan occurs near the carboxy terminus, beyond the catalytic region (Fig. 6).
Discussion
In the current manuscript we identify Es-cmp and Bz-cmp, two polyglycine hydrolases. These proteases were identified after LC-MS/MS sequencing of tryptic peptides from purified Es-cmp combined with DNA sequencing of the E. sorghi genome allowed for comparison back to the available C. carbonum genome. Sequence analysis of the identified proteins predicted that they contained β-lactamase domains (PF00144). Nevertheless, we demonstrated that they are endoproteases by expressing recombinant proteins with polyglycine hydrolase activity from their encoding cDNAs. Both recombinant proteins were purified and shown to have high activity for the glycine-rich linker region of maize chitinases ChitA and ChitB [Fig. 4(A)]. The recombinant enzymes cleaved glycine-glycine peptide bonds to produce a defined mixture of product peptides that were similar to those previously reported for native proteins [Fig. 4(B)]. Despite their sequence homology to β-lactamases, PBPs, and other S12 proteases, the polyglycine hydrolases did not interact with substrates or inhibitors of these proteins (Fig. 5). Mutation of serine to glycine in the SXXK motif—the predicted catalytic serine for an S12 protease—resulted in complete loss of protease activity, confirming that these endoproteases belong to class S12 (Fig. 5). To assess the distribution of polyglycine hydrolases within the fungi, we searched for Es-cmp homologs among the genomes represented in the MycoCosm database. The search identified 67 enzymes with significant homology through four classes of the phylum Ascomycota and one class of the Basidiomycota. Full-length hits to both Es-cmp and Bz-cmp were distributed through the Dothideomycetes and Sordariomycetes (Fig. 6).
Sequence analysis identified the catalytic region of polyglycine hydrolases, and by experimentation we have confirmed the identity of the catalytic serine (Fig. 5, lanes 9 and 10). The remainder of the protein is novel. As polyglycine hydrolases show specificity for plant chitinase substrates, and the specificity does not appear to be entirely encoded in the catalytic domain, we speculate that this remaining sequence functions to recognize chitinase and orient the polyglycine linker into the active site for cleavage. An initial clue into the nature of this recognition domain was observed by multiple sequence alignment of Es-cmp with Bz-cmp and 14 additional homologous proteins predicted from genome sequences of fungi in the Dothidiomycetes and Sordariomycetes. These sequences lack regions of extended conserved sequence, but contain a network of seven conserved tryptophans (Fig. 6). Generation of structural information by crystallography combined with functional assay of tryptophan mutant proteins will enable further understanding of this tryptophan network and of how the glycine hydrolases recognize target chitinase protein motifs that lack peptidyl side chains.
C. carbonum and E. sorghi are biotrophic plant pathogens, depending on living host tissues. Many biotrophic pathogens invade and grow within plant tissues by colonizing the intercellular space, termed the apoplast. In the apoplast, biotrophic pathogens secrete effector proteins that combat apoplastic plant defense proteins and facilitate invasion by suppressing plant defenses.17,18 One aspect of plant defenses against fungi is detection of chitin. Successful fungal pathogens secrete diverse proteins to overcome chitin detection. This has been most elegantly demonstrated for Cladosporium fulvum, a fungal pathogen of tomatoes. This fungus secretes two pathogenesis promoting proteins, Avr4 and Ecp6, to interfere with chitin detection by protecting the fungal cell wall from plant chitinases and binding chitin oligosaccharides to prevent their detection.19–22 In comparison, polyglycine hydrolases, and all chitinase modifying proteins (Fig. 1), directly attack plant chitinases to disrupt their activity. Despite also being a member of the fungal class Dothidiomycetes, C. fulvum does not contain a gene encoding for polyglycine hydrolases, and has developed different mechanisms to interfere with the same plant defense target, a common theme in plant pathogenesis.23 A greater understanding of the mechanisms that plant pathogens use to promote disease promises to lead to improved methods for their treatment and control.
Materials and Methods
Purification of Es-cmp
Polyglycine hydrolase Es-cmp was purified as described.3 Briefly, E. sorghi NRRL 54204 was grown on agar slants, and used as inoculum for 1 kg of autoclaved maize seed. Cultures were grown for 14 days at 25°C. Secreted proteins were extracted from the culture by mixing with 1 L of protein extract buffer (10 mM sodium acetate, pH 4.7, 100 mM NaCl). Particulate material was separated from the protein extract by a combination of filtration and centrifugation. The clarified extract was injected onto a Capto MMC XK 26/20 column (GE Healthcare, Waukesha WI) resulting in capture of Es-cmp. Es-cmp was eluted from the column using a linear gradient towards elution buffer (100 mM MES, pH 6.0, 1.0 M NaCl). Eluted Es-cmp was further purified and concentrated by lectin affinity chromatography (HiTrap ConA 4B, 1 ml, GE Healthcare). Eluted proteins were precipitated with cold acetone, and resuspended in gel filtration buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl). The final purification step was gel filtration (Superdex 200 10/300 GL, GE Healthcare). The most active fraction, as determined by cmp assay, was concentrated by acetone precipitation.
LC-MS/MS analysis of purified Es-cmp
About 10 µL of purified Es-cmp was loaded onto an SDS-PAGE gel, and proteins were separated at 200 V for 1 h. The gel was stained with Bio-Safe Coomassie stain (Bio-rad, Hercules CA). The stained band was removed with a scalpel, placed in a microcentrifuge tube, destained and digested overnight with trypsin. Recovered peptides were analyzed on an LTQ-Orbitrap Velos Pro (Thermo Fisher, Waltham, MA) with an online Dionex 3000 RSLCnano HPLC system (Thermo Fisher), using an Acclaim® PepMap RSLC column (2 µm, 100 Å, 50 × 0.075 mm2) employing a 20 min run time from sample to sample at a flow rate of 300 nL min−1 from 2 to 45% B (A = 100% water, 0.1% formic acid; B = 80% acetonitrile, 0.1% formic acid) over 9.1 min at 35°C. Peptides were trapped on a C18 PepMap100 pre-column (5µm, 100Å, 5 × 0.3 mm2) and washed for 3 min at 5 µL min−1 before switching the trap pre-column inline. Mass spectra were acquired using collision-induced dissociation (CID) and the raw data files processed into mgf files using Proteome Discoverer (Thermofisher; version 1.4) and searched using Mascot (Matrix Science, London, UK; version 2.5.0), first against the C. carbonum database12 with limited success, and subsequently against a FASTA protein sequence database, derived from genome sequencing of E. sorghi and gene annotation using GeneMark-ES.24
Genome sequencing of E. sorghi
Eppicoccum sorghi, NRRL 54204, was inoculated from an agar slant into a 20 mL liquid culture of YM Broth (20 g dextrose, 5 g peptone, 3 g yeast extract, 3 g malt extract per L) and grown at 25°C with shaking for 4 days. Fungus was collected from the mature liquid culture by filtration and freeze-dried. Genomic DNA was purified from freeze-dried mycelia by the CTAB method.25 A sequencing library was prepared using 500 ng of genomic DNA and the NEBNext Fast DNA Fragmentation & Library Prep Set (New England Biolabs, Ipswich, MA). Size selection was performed on a BluePippin using a 2% agarose gel (Sage Science, Beverley, MA). The size selected library was purified with Agencourt AMPure XP (Beckman Coulter, Brea, CA) according to manufacturer's instructions and the final library concentration was determined on an Agilent 2200 TapeStation (Agilent Technologies, Santa Clara, CA). Emulsion PCR was carried out on an Ion One Touch 2 instrument using an Ion PGM Template OT2 400 Kit (Life Technologies, Grand Island, NY). Sequencing was carried out on an Ion Torrent Personal Genome Machine using an Ion 318 Chip v2 and the Ion PGM 400 Sequencing Kit (Life Technologies, Grand Island, NY). De novo assembly was performed using 5,157,511 quality trimmed reads (1.8 Gbp of quality trimmed sequence) using the CLC Genomics Workbench (CLC bio, Boston, MA) with default parameters and a minimum contig size of 1000 bp. The resulting assembly consisted of 936 contigs totaling 31.9 Mbp at an average coverage of 54×.
Expression of recombinant Es-cmp and Bz-cmp
The cDNA sequences for the identified polyglycine hydrolases were cloned into a commercial expression plasmid, pPICZ-α-A (Invitrogen, Carlsbad CA). Strains of heterologous Pichia pastoris were produced following the guidelines in the EasySelect Pichia expression kit manual (Invitrogen, manual K1740-01). The cDNAs were cloned into the expression plasmid following the method of Gibson,26 utilizing a commercial kit (New England Biolabs, Ipswich MA). Oligonucleotide primers used are listed in Table1. The same methods were used to produce a strain that expresses Es-cmp(S370G).
Table 1.
Oligonucleotide Sequences Used in This Study
| Name | Sequence 5′–3′ | Description |
|---|---|---|
| Es-lactamase_Fwd | GAGAGGCTGAAGCTGAATTCTTCCCCGCCAACCAGCAGGATTTGA | Forward oligo with vector overlap |
| Es-lactamase_Rev | AGATGAGTTTTTGTTCTAGATTACTCAGCGAGCGCATACTTATAC | Reverse oligo with vector overlap |
| Bz-lactamase_Fwd | GAGAGGCTGAAGCTGAATTCGAGAGGCTGAAGCTGAATTCTCTCCCACCACTAACCAGAATTTGC | Forward oligo with vector overlap |
| Bz-lactamase_Rev | AGATGAGTTTTTGTTCTAGATTATAAGGGAAGCGCCGAATCGAGG | Reverse oligo with vector overlap |
| S370G_Fwd | CGGTCTCGGCGGCGTCTCGAAGATGTTCACGTACG | Fwd mutagenic oligo with nucleotide change |
| S370G_Rev | TCGAGACGCCGCCGAGACCGAATTTGTCAGTAGGC | Reverse mutagenic oligo with nucleotide change |
Purification of recombinant Es-cmp and Bz-cmp
Heterologous Pichia strains were grown in a shake incubator in non-inducing media (1 L of buffered glycerol-complex medium, 1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% yeast nitrogen base, 4 × 10−5% biotin, 1% glycerol) at 25°C until OD600 = 10. Cells were harvested by centrifugation and re-suspended in induction media (1 L of buffered methanol-complex medium, 1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% yeast nitrogen base, 4 × 10−5% biotin, 0.5% methanol). Proteins were expressed in baffled flasks with shaking at 20°C for 48 h.
After expression, cells were removed from the media by centrifugation. Proteins were then precipitated from the cell-free media by addition of ammonium sulfate (608 g L−1) followed by centrifugation. The precipitated pellet was re-suspended in 10 ml cation exchange buffer (50 mM sodium acetate, pH 4.7, 100 mM NaCl) and dialyzed to remove residual ammonium sulfate. The dialyzed, re-suspended pellet was then injected onto a HiScreen Capto MMC column (GE Healthcare, Waukesha WI) that had been equilibrated with cation exchange buffer. The column was washed with 10 mL buffer. Bound proteins were then eluted with a linear gradient ending in 100% elution buffer (100 mM MES, pH 6.0, 1.0M NaCl). Fractions containing recombinant Es-cmp (or Bz-cmp) were combined, and protein was further purified and concentrated by acetone precipitation (50% v/v acetone). The purified proteins were pelleted by centrifugation, and re-suspended in 600 μL buffer (20 mM Tris-Cl, pH 7.5) and were stored at −80°C in 1-μL aliquots. For assays, proteins were thawed and diluted.
To ensure accurate concentration measurements of polyglycine hydrolases, their extinction coefficients were determined experimentally.27,28 For recombinant Es-cmp and Bz-cmp the extinction coefficients at 280 nM were determined to be 110,740 and 108,538 M−1 cm−1.
Gel filtration analysis
Purified proteases were analyzed by gel filtration. Proteins were diluted 10-fold in gel filtration buffer (20 mM Tris-Cl, pH 7.5, 0.15M NaCl) and 20 µL were injected onto a superdex 200 10/300 GL column (GE Healthcare), with a flow rate of 0.5 mL min−1. Protein retention was detected by measuring absorbance at 280 nM. Peak integration was performed using UNICORN 5.31.
Expression and purification of recombinant plant chitinases
Maize plant class IV chitinases ChitA (LH82 alloform) and ChitB were expressed as recombinant proteins and purified as described previously.1,29 To improve accuracy of protein concentration measurements, their extinction coefficients were determined experimentally.27,28 For ChitA and ChitB the extinction coefficients at 280 nM were determined to be 43,191 and 55,337 M−1 cm−1.
Enzyme assays
Protease (chitinase modifying protein, cmp) assays contained 1 μM chitinase substrate (ChitA or ChitB) in 10 mM sodium acetate buffer (pH 5.2). The assays were initiated by addition of 1 μl of protease to 10 μL substrate and were incubated at 30°C for 1 h. The reactions were stopped by either addition of SDS-PAGE loading buffer followed by heating in boiling water for 1 min (for the gel analysis) or addition of an equal volume of matrix followed by spotting onto a MS target (for MALDI-TOF MS). The choice of assay conditions and determination of E1/2 was patterned after Timmer et al.30
Purified Es-cmp and Bz-cmp were tested for aminopeptidase and β-lactamase activities with the colorimetric substrates nitrocefin or glycine p-nitroanilide, respectively, as described previously.31,32 No activity was observed with either substrate, even after incubation for 24 h at 30°C with 200 nM enzyme.
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS)
The peptides released from the N-terminus of the ChitA or ChitB chitinases were assayed by MALDI-TOF MS, as described previously.3 The instrument was a Bruker-Daltonics Microflex LRF (Bruker-Daltonics, Billerica, MA), with a pulsed N2 laser (3000 shots, 337 Hz, 60 Hz pulse), and reflectron acquisition. The calibrant was Peptide Calibration Standard II mono with insulin (Bruker-Daltonics), and the matrix used was 2,5-dihydrobenzoic acid (2,5-DHB).
Acknowledgments
The authors thank Kurt Sollenberger, Jacob Brown, Trina Hartman, and Nathane Orwig for technical assistance. The mention of firm names or trade products does not imply that they are endorsed or recommended by the US Department of agriculture over other firms or similar products not mentioned. The USDA is an equal opportunity provider and employer.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information
References
- Naumann TA, Wicklow DT, Price NPJ. Identification of a Chitinase-modifying protein from fusarium verticillioides: truncation of a host resistance protein by a fungalysin metalloprotease. J Biol Chem. 2011;286:35358–35366. doi: 10.1074/jbc.M111.279646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann TA, Wicklow DT. Allozyme-specific modification of a maize seed chitinase by a protein secreted by the fungal pathogen stenocarpella maydis. Phytopathology. 2010;100:645–654. doi: 10.1094/PHYTO-100-7-0645. [DOI] [PubMed] [Google Scholar]
- Naumann TA, Wicklow DT, Price NPJ. Polyglycine hydrolases secreted by pleosporineae fungi that target the linker region of plant class IV chitinases. Biochem. J. 2014;460:187–198. doi: 10.1042/BJ20140268. [DOI] [PubMed] [Google Scholar]
- Naumann TA, Wicklow DT, Kendra DF. Maize seed chitinase is modified by a protein secreted by bipolaris zeicola. Physiol Mol Plant Pathol. 2009;74:134–141. [Google Scholar]
- Firczuk M, Mucha A, Bochtler M. Crystal structures of active LytM. J Mol Biol. 2005;354:578–590. doi: 10.1016/j.jmb.2005.09.082. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Morton FR, Kok CY, Kong J, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2008;36:D320–D325. doi: 10.1093/nar/gkm954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JA, Kuzin AP. The refined crystallographic structure of a DD-peptidase penicillin-target enzyme at 1.6 a resolution. J Mol Biol. 1995;254:223–236. doi: 10.1006/jmbi.1995.0613. [DOI] [PubMed] [Google Scholar]
- Jelsch C, Mourey L, Masson JM, Samama JP. Crystal structure of Escherichia coli tem1 beta-lactamase at 1.8 a resolution. Proteins. 1993;16:364–383. doi: 10.1002/prot.340160406. [DOI] [PubMed] [Google Scholar]
- Jacoby GA. AmpC β-lactamases. Clin Microbiol Rev. 2009;22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano Y, Kato Y, Yamada A, Kondo K. Structural similarity of D-aminopeptidase to carboxypeptidase DD and β-lactamases. Biochemistry. 1992;31:2316–2328. doi: 10.1021/bi00123a016. [DOI] [PubMed] [Google Scholar]
- Bompard-Gilles C, Remaut H, Villeret V, Prange T, Fanuel L, Delmarcelle M, Joris B, Frere JM, Beeumen JV. Crystal structure of a D-aminopeptidase from Ochrobactrum anthropi, a new member of the “penicillin-recognizing enzyme” family. Structure. 2000;8:971–980. doi: 10.1016/s0969-2126(00)00188-x. [DOI] [PubMed] [Google Scholar]
- Condon BJ, Leng Y, Wu D, Bushley KE, Ohm RA, Otillar R, Martin J, Schackwitz W, Grimwood J, MohdZainudin N, Xue C, Wang R, Manning VA, Dhillon B, Tu ZJ, Steffenson BJ, Salamov A, Sun H, Lowry S, LaButti K, Han J, Copeland A, Lindquist E, Barry K, Schmutz J, Baker SE, Ciuffetti LM, Grigoriev IV, Zhong S, Turgeon BG. Comparative genome structure, secondary metabolite, and effector coding capacity across cochliobolus pathogens. PLoS Genet. 2013;9:e1003233. doi: 10.1371/journal.pgen.1003233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coffill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. The pfam protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0:discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42:D699–704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Balentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. ClustalW and ClustalX version 2. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Okmen B, Doehlemann G. Inside plant: biotrophic strategies to modulate host immunity and metabolism. Curr Opin Plant Biol. 2014;20:19–25. doi: 10.1016/j.pbi.2014.03.011. [DOI] [PubMed] [Google Scholar]
- Stotz HU, Mitrousia GK, De Wit PJGM, Fitt BDL. Effector-triggered defence against apoplastic fungal pathogens. Trends Plant Sci. 2014;19:491–500. doi: 10.1016/j.tplants.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten MHAJ, Cozijnsen TJ, De Wit PJGM. Host resistance to a fungal tomato pathogen lost by a single base-pair change in an avirulence gene. Nature. 1994;367:384–386. doi: 10.1038/367384a0. [DOI] [PubMed] [Google Scholar]
- Van Den Burg HA, Harrison SJ, Joosten MHAJ, Vervoort J, De Wit PJGM. Cladosporium fulvum avr4 protects fungal cell walls against hydrolysis by plant chitinases accumulating during infection. Mol Plant-Microbe Int. 2006;19:1420–1430. doi: 10.1094/MPMI-19-1420. [DOI] [PubMed] [Google Scholar]
- De Jonge R, Van Esse HP, Kombrink A, Shunya T, Desaki Y, Bours R, Van Der Krol S, Shibuya N, Joosten MHAJ, Thomma BPHJ. Conserved fungal LysM effector ecp6 prevents chitin-triggered immunity in plants. Science. 2010;329:953–955. doi: 10.1126/science.1190859. [DOI] [PubMed] [Google Scholar]
- Bolton MD, Van Esse HP, Vossen JH, De Jonge R, Stergiopoulos I, Stulemeijer IJE, Van Den Berg GCM, Borras-Hidalgo O, Dekker HL, De Koster CG, De Wit PJGM, Joosten MHAJ, Thomma BPHJ. The novel Cladosporium fulvum lysine motif effector ecp6 is a virulence factor with orthologues in other fungal species. Mol Microbiol. 2008;69:119–136. doi: 10.1111/j.1365-2958.2008.06270.x. [DOI] [PubMed] [Google Scholar]
- Muktar MS, Carvunis AR, Dreze M, Epple P, Steinbrenner J, Moore J, Tasan M, Galli M, Hao T, Nishimura MT, SJ Pevzner, SE Donovan, L Ghamsari, B Santhanam, V Romero, MM Poulin, F Gebreab, BJ Gutierrez, S Tam, D Monachello, M Boxem, CJ Harbort, N McDonald, L Gai, H Chen, Y He European Union Effectoromics Consortium. J Vandenhaute, FP Roth, DE Hill, JR Ecker, M Vidal, J Beynon, P Braun, JL Dangl. Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science. 2011;333:596–601. doi: 10.1126/science.1203659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA. Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. III. [DOI] [PubMed] [Google Scholar]
- Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsley GR, Pace CN. Spectrophotometric determination of protein concentration. Curr Protoc Protein Sci. 2003:3.1.1–3.1.9. doi: 10.1002/0471140864.ps0301s33. [DOI] [PubMed] [Google Scholar]
- Naumann TA. Modification of recombinant maize ChitA chitinase by fungal chitinase-modifying proteins. Mol Plant Pathol. 2010;12:365–372. doi: 10.1111/j.1364-3703.2010.00677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmer JC, Zhu W, Pop C, Regan T, Snipas SJ, Eroshkin AM, Riedl SJ, Salvesen GS. Structural and kinetic determinants of protease substrates. Nat Struct Biol. 2009;16:1101–1108. doi: 10.1038/nsmb.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Callaghan CA, Morris A, Kirby SM, Shingler AH. Novel method for detection of β-lactamases by using a chromogenic cephalosporin substrate. Antimicrob Agents Chemother. 1972;1:283–288. doi: 10.1128/aac.1.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasafirek E, Fric P, Slaby J, Malis F. Nitroanilides of 3-carboxypropionyl-peptides. Their cleavage by elastase, trypsin, and chymotrpysin. Eur J Biochem. 1976;69:1–13. doi: 10.1111/j.1432-1033.1976.tb10852.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information