

Abstract
Aims
The aims of the study were to compare [14C]-paracetamol ([14C]-PARA) paediatric pharmacokinetics (PK) after administration mixed in a therapeutic dose or an isolated microdose and to develop further and validate accelerator mass spectrometry (AMS) bioanalysis in the 0–2 year old age group.
Methods
[14C]-PARA concentrations in 10–15 µl plasma samples were measured after enteral or i.v. administration of a single [14C]-PARA microdose or mixed in with therapeutic dose in infants receiving PARA as part of their therapeutic regimen.
Results
Thirty-four infants were included in the PARA PK analysis for this study: oral microdose (n = 4), i.v. microdose (n = 6), oral therapeutic (n = 6) and i.v. therapeutic (n = 18). The respective mean clearance (CL) values (SDs in parentheses) for these dosed groups were 1.46 (1.00) l h–1, 1.76 (1.07) l h–1, 2.93 (2.08) l h–1 and 2.72 (3.10) l h–1, t1/2 values 2.65 h, 2.55 h, 8.36 h and 7.16 h and dose normalized AUC(0-t) (mg l–1 h) values were 0.90 (0.43), 0.84 (0.57), 0.7 (0.79) and 0.54 (0.26).
Conclusions
All necessary ethical, scientific, clinical and regulatory procedures were put in place to conduct PK studies using enteral and systemic microdosing in two European centres. The pharmacokinetics of a therapeutic dose (mg kg–1) and a microdose (ng kg–1) in babies between 35 to 127 weeks post-menstrual age. [14C]-PARA pharmacokinetic parameters were within a two-fold range after a therapeutic dose or a microdose. Exploratory studies using doses significantly less than therapeutic doses may offer ethical and safety advantages with increased bionalytical sensitivity in selected exploratory paediatric pharmacokinetic studies.
Keywords: accelerator mass spectrometry, exploratory clinical study, microdosing, paediatric pharmacokinetics, paracetamol
What is Already Known about this Subject
The paediatric pharmacokinetics of paracetamol are well described as are the pharmacokinetics of a paracetamol microdose in the adult population. The use of [14C]-labelled drugs to study adult pharmacokinetics with accelerator mass spectrometry bioanalysis has also been documented. The current observational study was conducted to establish if it was feasible to conduct a paediatric (0–2 years of age) isolated microdose study using [14C]-paracetamol and accelerator mass spectrometry bioanalysis and to compare the pharmacokinetic data obtained with that after a therapeutic dose.
What this Study Adds
This study reports that it is possible to put in place an operational plan to permit paediatric microtracer and microdose [14C]-paracetamol administration in the 0–2 year old age group. The paracetamol microdose used of 6 ng kg–1 is lower than any other isolated microdose reported in the literature and yet the pharmacokinetics were still approximately dose proportional with therapeutic doses for both enteral and intravenous administrations. The methods reported here need to be extended to other drugs to establish the general utility of the microdose approach.
Introduction
Paediatric drug treatment is frequently based on ‘best-guess’ modifications of adult dosage regimens without detailed knowledge of pharmacokinetics (PK) in children 1. PK data are pivotal in understanding a drug's safety and efficacy and are essential to determine therapeutic dosages in specific age groups 2,3. The ontogeny of many enzymes and physiological processes involved in PK are reflected in age-specific effects on drug disposition 4,5. US and European regulations require the separate investigation of new medicines in children as well as investigations into already existing medicines used in paediatric care 6. As a consequence of these regulations, new medicines are now being more regularly evaluated in children as part of clinical development plans.
When there is no prior knowledge of the appropriate dosage in children, an empiric choice is often made based on allometry and/or paediatric physiologically based pharmacokinetic modelling (PBPK) 7. This approach may lead to over- or under-dosage which does not provide a complete picture of the PK situation while running the risk of exposure to toxic concentrations of parent drug or metabolites 8. This problem can arise because extrapolation from adults or animals does not take into account gaps in our knowledge of the ontogeny of drug disposition (for example the activity of drug metabolizing enzymes in the liver) 9,10.
An alternative approach is to obtain exploratory PK data using doses that are substantially below the anticipated therapeutic dose, see for example ICH M3 R2 11. Microdosing or human phase 0 studies have been widely used to obtain adult PK data 12–14 but infrequently in infants. Microdose studies assess drug metabolism and PK/PD parameters as a basis for future therapeutic dose determination. These studies involve limited human exposure to the drug, have no therapeutic intent and are not designed to examine clinical tolerability 11. In children this approach could overcome the issues with dose selection by providing directly relevant PK data, as long as the PK are approximately dose-proportional between a microdose and a therapeutic dose.
Microdosing can be conducted with a microtracer [14C] label as is required for AMS bioanalysis 15 or with a trace of cold drug and LC/MS analysis 16. For definitions of microtracer and microdose see reference 15. A comparison of PK between a microtracer [14C]-labelled therapeutic dose and a microdose can be used to establish whether or not PK are dose-proportional across the doses used. In adult microdose validation studies, dose proportionality was assumed if the PK parameters between a microdose and a therapeutic dose were within a two-fold margin 17,18, a margin routinely used when making allometry comparisons to establish PK equivalency between animal models and humans.
GC/MS or LC/MS are currently the main methods of bioanalysis to study paediatric PK 19,20 but insensitivity (low pg ml–1) in analyzing drug concentrations in small volume blood samples after microdose administration could be a significant barrier to use in paediatric PK studies. Furthermore because only small blood volumes can be withdrawn from babies, sparse sampling methods are generally used 21. An alternative methodology is accelerator mass spectrometry (AMS) capable of measuring a drug's plasma concentration in the atto- to zeptogram ml–1 range (10−18 to 10−21 g ml–1) 22,23. This provides up to a million-fold increase in sensitivity over LC/MS. Furthermore AMS can detect [14C]-labelled drug concentrations when the drug is labelled at the level of background radiation exposures. AMS has been extensively demonstrated to be an effective methodology to study drug metabolism and PK in the adult population 24,25. However, only two examples in limited numbers of children have been reported for the paediatric population 26,27. Gordi et al. demonstrated the utility of microdosing and microtracing research in paediatric research using 14C-ursodiol 26. The microdose varied between 3–30 ng kg–1 and in total eight patients were included. Mooij et al., in a preliminary communication, reported on a fundamentally different study design to that described here (see Discussion section). They focused on intestinal and hepatic drug disposition of 14C-PARA using an absolute bioavailability study design of an oral paracetamol microdose administered together with a concomitant i.v. therapeutic paracetamol dose. The Mooij et al. study dosed approximately 3 ng kg–1 and nine patients were included, up to the age of 6 years. No group PK data were presented in this short communication.
This paper reports the results of a European-wide collaborative research programme known as the Paediatric Accelerator Mass Spectrometry Evaluation Research Study (PAMPER). The collaborators put together an operational plan in two European countries viz. the United Kingdom and Estonia to address the necessary legal/ethical, regulatory, clinical and scientific procedures required to permit the application of AMS in paediatric microdose studies. The study involved a well-characterized and regularly used drug in paediatric medicine, paracetamol (PARA) (paracetamol) whose PK have been reported in this population (paediatric PK studies summarized in 28. Here we aimed to examine the feasibility of giving an isolated microdose in young children through an assessment of whether PK parameters of PARA in infants and neonates following a therapeutic dose (using [14C]-PARA mixed in a therapeutic dose as a microtracer) are similar to PK parameters for the single isolated microdose of [14C]-PARA not administered at the same time as a therapeutic dose, to determine dose-linearity of the approach. The results highlight the potential to use an AMS microdosing approach also for (new) less characterized compounds than PARA in the paediatric population.
The objectives of this proof of concept exploratory study were:
To prepare all the necessary ethical, regulatory and scientific documentation to permit a [14C]-microtracer and an isolated microdose paediatric study using PARA as a model drug, in two European countries.
To conduct a microtracer/isolated microdose comparison study in children up to the age of 2 years.
To establish the PK of a microtracer of [14C]-PARA ncorporated in a therapeutic dose using non-compartmental analysis (NCA) and extant data.
To compare NCA PARA PK parameters for an isolated microdose not administered at the same time as a therapeutic dose.
Methods
Test substances and reagents
Oral PARA syrup (Efferalgan, 30 mg ml–1; Bristol Myers Squibb or Pharmacopoiea grade equivalent) and PARA (Perfalgan, Bristol Myers Squibb or European Pharmacopoiea grade) for intravenous administration were used for this study. [14C]-PARA (Moravek Biochemicals Inc, Brea, USA), specific radioactivity 2.85 GBq mmol–1 was repurified and certificated by the Pharmaceutical Research Institute, Warsaw, Poland to a purity of 99.9% w/w, (1.1 ml ethanol solution contained approximately 5.55 MBq [14C]-PARA concentration 0.27 mg ml–1, 5.032 MBq ml–1) and shipped to Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK for GMP i.v. formulation. Stability testing of the ethanol stock solution of [14C]-PARA when stored at −20°C showed no degradation over a 12 month period. PARA standards for HPLC were USP grade or equivalent. All other chemicals and reagents used were pharmacopoeia grade or equivalent.
For UPLC method development and AMS validation [14C]-PARA purchased from American Radiolabeled Chemicals Inc (ARC Inc, United Kingdom) was used. 12C-PARA was purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands). Blank human EDTA-plasma was obtained from Bioreclamation Inc (USA). All plasma samples were screened for background PARA concentrations. Only blank plasma samples, negative for PARA, were included in the study. A pool of blank plasma was prepared by mixing equal volumes from six individuals.
Dose formulation and administration
An intravenous sterile formulation of [14C]-PARA in 5% w/v glucose solution (0.22 ml containing 111Bq [14C]-PARA, specific radioactivity 2.85 GBq mmol–1 equivalent to 5.91 ng PARA) was prepared in the MHRA GMP accredited Radiopharmacy Department, Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK. This sterile formulation was used both for enteral or intravenous administration to paediatric patients. The sterile formulation was stored at 2–8°C and showed no degradation over the study period nor was there any evidence of non-specific binding to the filtration apparatus or storage vials.
Administration of [14C]-PARA was either enterally or intravenously (111 Bq kg–1) in one of two scenarios. Scenario 1 (microtracer dose) involved administration of the sterile [14C]-PARA formulation alongside either an enteral or i.v. therapeutic PARA dose. The latter dose was determined by the baby's body weight and is documented in Table1. Scenario 2 involved administration of the sterile [14C]-PARA formulation (111 Bq kg–1, 5.91 ng kg–1) either enterally or intravenously alone (microdose). Scenario 1 dosing was part of normal clinical practice with [14C]-label administration occurring alongside a scheduled therapeutic dose of PARA. Scenario 2 dosing was only in infants not given PARA, providing information on dose linearity. All details of the dosing procedures can be found in Table1.
Table 1.
Detailed patient information with individual pharmacokinetic parameters (AUC(0,t) and CL) for APAP
| Patient number | Route of administration | *Dose kg–1 | Post-menstrual age (weeks) | Body weight (kg) | Gender | †AUC(0-t) (ng ml–1 h) | †‡CL (l h–1) |
|---|---|---|---|---|---|---|---|
| AH15 | i.v. | 6 ng | 44.9 | 3.0 | M | 0.0261 | 0.6848 |
| AH17 | i.v. | 6 ng | 73.7 | 8.0 | F | 0.0234 | 2.0815 |
| AH19 | i.v. | 6 ng | 36.3 | 2.6 | F | 0.0255 | 0.6166 |
| AH21 | i.v. | 6 ng | 127.0 | 9.8 | F | 0.0200 | 2.8451 |
| AH23 | i.v. | 6 ng | 42.9 | 5.1 | F | 0.0234 | 1.2721 |
| AH35 | i.v. | 6 ng | 35.6 | 3.0 | M | 0.0058 | 3.0660 |
| AH14 | Oral | 6 ng | 39.1 | 2.3 | M | 0.0174 | 0.7941 |
| AH16 | Oral | 6 ng | 75.4 | 5.5 | F | 0.0111 | 2.9355 |
| AH18 | Oral | 6 ng | 44.1 | 2.9 | M | 0.0211 | 0.8228 |
| AH20 | Oral | 6 ng | 77.0 | 6.3 | F | 0.0294 | 1.2887 |
| AH01 | i.v. | 7.4mg | 41.0 | 3.4 | F | 18964.88 | 1.3182 |
| AH03 | i.v. | 14.9mg | 57.7 | 6.7 | F | 46125.36 | 2.1680 |
| AH05 | i.v. | 14.1mg | 75.4 | 7.1 | M | 53037.72 | 1.8855 |
| AH08 | i.v. | 15.0mg | 76.3 | 8 | M | 63645.26 | 1.8855 |
| AH09 | i.v. | 15.0mg | 86.0 | 10 | M | 50845.19 | 2.9501 |
| AH10 | i.v. | 15.0mg | 79.3 | 5.9 | M | 55117.83 | 1.6329 |
| AH12 | i.v. | 20.0mg | 38.6 | 2.6 | F | 15527.7 | 1.2880 |
| AH13 | i.v. | 7.8mg | 39.9 | 3.2 | M | 30322.2 | 0.8245 |
| AH24 | i.v. | 14.1mg | 66.9 | 6.4 | F | 43440.96 | 2.0718 |
| AH25 | i.v. | 14.8mg | 52.0 | 4.1 | F | 21867.59 | 2.7438 |
| AH26 | i.v. | 7.1mg | 40.1 | 3.5 | M | 13069.82 | 1.9128 |
| AH27 | i.v. | 7.0mg | 41.3 | 3.6 | M | 8119.676 | 3.0789 |
| AH28 | i.v. | 7.4mg | 38.6 | 3.2 | M | 11855.91 | 2.0243 |
| AH29 | i.v. | 15.2mg | 49.1 | 3.3 | M | 19569.3 | 2.5550 |
| AH31 | i.v. | 7.3mg | 41.9 | 3.7 | F | 11848.15 | 2.2788 |
| TP09 | i.v. | 8.0mg | 36.0 | 3.9 | F | 2069.503 | 14.4962 |
| TP13 | i.v. | 9.0mg | 36.0 | 2.3 | F | 18410.7 | 1.0863 |
| AH02 | Oral | 7.2mg | 38 | 2.5 | M | 134629.9 | 0.4457 |
| AH04 | Oral | 15.4mg | 79.4 | 7.8 | F | 41954.4 | 2.8602 |
| AH06 | Oral | 15.0mg | 68.7 | 6.0 | M | 40109.02 | 2.2439 |
| AH11 | Oral | 13.6mg | 52.1 | 4.4 | M | 11495.91 | 1.5658 |
| AH32 | Oral | 15.0mg | 50.9 | 5.0 | F | 15655.72 | 4.7906 |
| AH33 | Oral | 15.0mg | 43.6 | 4.0 | M | 12051.86 | 4.9785 |
*Infants were administered either a) 111 Bq kg–1 [14C]-PARA as a microdose (6 ng kg–1) or b) a microtracer dose alongside the therapeutic dose listed in the table.
†Individual AUC(0-t) and CL values based on dose of PARA administered.
‡Apparent clearance (CL/F) was obtained for oral route.
Paediatric patients
Children were eligible to be included in this study if they were preterm neonates (32–36 gestational weeks at birth) up to 2 years of age and had intravenous lines in place (for i.v. administration) or were able to tolerate enteral administration of [14C]-labelled PARA and had suitable vascular access for blood sampling. Exclusion criteria were a history of allergy or hypersensitivity to PARA, serious hepatic or renal impairment, haemofiltration, peritoneal/haemodialysis or ECMO (extracorporeal membrane oxygenation). Ethical approval was obtained from the relevant Research Ethics Committees for the hospitals where patient enrolment occurred and all parents or an adult who carried parental responsibility provided fully informed consent for their child to be included as defined in the Declaration of Helsinki 29. No radioactive substance administration approval was required as the administered radioactive dose was below 1 μSievert, the UK Administration of Radioactive Substances Advisory Committee (ARSAC) exemption level. The clinics participating in this study were Paediatric Intensive Care Unit (PICU), Alder Hey Children's NHS Foundation Trust, Liverpool, UK and Tartu University Hospital, Tartu, Estonia.
All i.v. microdosed children received i.v. therapeutic PARA prior to the microdose at varying preceding time intervals. Two out of the four enterally microdosed babies had no preceding PARA.
Blood collection
Blood samples were obtained from an arterial line, central venous line or capillary sample. A predose sample was obtained before administration of the [14C]-PARA and subsequently up to five post dose samples at selected time points (typical sampling times: enteral dosing 0.5, 1, 2, 3 and 8 h; i.v. dosing 0.1, 0.25, 5, 5.5 and 6 h) which had previously been determined from PARA PK modelling using literature data as likely to provide the most informative PK data. Blood 100–250 µl was collected in Microtainers (Becton Dickenson) containing EDTA and centrifuged shortly after collection to obtain plasma. Aliquots (10 or 15 l) of plasma were saved into small cryotubes (usually 2–3 per sample) and immediately frozen for storage at a minimum of −20°C. The total volume of blood obtained from each baby did not exceed 1.1 ml, a volume well within the European Medicines Agency recommended limits for blood sampling for all age groups included in the study 30. Samples were shipped on dry ice to TNO Zeist, The Netherlands for UPLC and AMS bioanalysis.
Plasma [14C]-PARA measurement by UPLC-AMS
Plasma 10 or 15 µl was diluted with 0.9% w/v NaCl to 45 µl and subsequently extracted using 175 µl 100% v/v methanol containing 6.6 µg ml–1 PARA in 96-well protein precipitation plates. The pellet was washed with 100 µl 0.9% NaCl : 100% methanol (1 : 4 v/v). Resulting filtrates were evaporated to dryness and redissolved in 30 µl 10 m m ammonium phosphate pH 3.4 (Eluent A) of which 25 µl was used for UPLC analysis. A PARA solution, specific radioactivity = 3700 Bq [14C]-PARA 100 µg–1 PARA in blank pooled plasma was used to prepare eight calibrator levels and three quality control sample levels from 0.4 to 180 mBq ml–1, and from 1.7 to 131 mBq ml–1, respectively.
Calibrators (duplicate), QCs (triplicate) and sample extracts were injected onto a UPLC coupled to a PDA. Chromatograpic conditions can be found in Table2. PARA in 100% methanol was added to each collected fraction to increase the [12C] carbon content to 25 µg. Fractions were transferred to a tin foil cup and evaporated to dryness.
Table 2.
UPLC conditions for recovery of [14C]-PARA from plasma extracts
| Eluent A | 10 m m ammonium phosphate pH 3.4 |
|---|---|
| Eluent B | 100 % v/v methanol |
| UPLC column | Aquity UPLC (Waters), BEH C18 1.7 µm 2.1 × 100 mm column |
| Flow rate | 0.3 ml min–1 |
| Column temperature | 30°C |
| Chromatography conditions | 0–1 min 100% A and 0% B |
| 1–10 min linear gradient from 100% A and 0% B to 95% A and 5% B | |
| 10–12 min 95% A and 5% B | |
| 12–15 min linear gradient from 95% A and 5% B to 0% A and 100% B | |
| 15–20 min 0% A and 100% B | |
| 20–20.10 min linear gradient from 0% A and 100% B to 100% A and 0% B | |
| 20.10–20.50 min 100% A and 0% B | |
| 20.50–28 min 100% A and 0% B at a flow rate of 0.4 ml min–1 | |
| 28–29 min 100% A and 0% B |
A novel AMS sample introduction method was used in this study 31. Briefly, a tin foil cup was combusted using an elemental analyzer (Vario Micro, Elementar, Germany), and the resulting CO2 was captured on a zeolyte trap. CO2 was released by heating of the trap and transferred to a vacuum syringe using helium. The resulting 6% v/v gas mixture of CO2 with helium was infused at a pressure of 1 bar at 60 µl min–1 into the titanium target in the SO110 ion source of a 1 MV Tandetron AMS (High Voltage Engineering Europe B.V., The Netherlands).
The scientific method of validation for the LC/AMS analysis was based on the recommendation of the European Bioanalytical Forum 32. Details of the validation results are presented in the supporting information.
AMS data processing
The combustion-CO2-AMS method uses a calibration line (CAL) procedure in which the individual baby's PARA plasma concentration is determined by extrapolating from the CAL line. Results are expressed as mBq ml–1 plasma which is then converted to the PARA concentration from the specific radioactivity of the dose administered to each baby. For the microdose arm of the study (Scenario 2) a dose value of 6 ng kg–1 paracetamol was assumed. Concentrations of PARA in plasma were calculated from the concentration in the UPLC fraction, the volume of plasma extracted and the volume of extract analyzed.
Pharmacokinetics
PK parameters were calculated using a non-compartmental model with WinNonLin software version 6.3 (Certara, St Louis, Missouri, USA). The input data were the plasma concentration values of PARA in ng ml–1, sampling times (h) and doses administered. Output parameters were time to Cmax, terminal half-life, clearance, apparent volume of distribution and area under the curve (AUC). AUCs were calculated by use of a combination of linear and log trapezoidal approximations.
Results
Study setup
The UK Medicines and Healthcare products Regulatory Agency (MHRA) agreed that this was a physiological study of a medicine used within the terms of its marketing authorization to validate a new methodology and so was not a Clinical Trial of an Investigational Product (CTIMP). Radiation dose calculations were undertaken using ICRP's recommended model 33 where the body is regarded as a single compartment with a biological half-life of 40 days. Assuming an administered dose of 1387 Bq (111 Bq kg–1 to a 12.5 kg infant) this gave a whole body effective dose value of 0.8 μSieverts or 0.3 μSieverts using an alternative model 34. Based on the ICRP conservative estimate this radioactive dose equates to less than 10% of background exposure. 50% of parents approached about the trial agreed to the participation of their child.
The study recruited at two paediatric clinics between January 2013 and December 2013 in Liverpool, United Kingdom (Alder Hey Children's NHS Foundation Trust) and Tartu, Estonia (Tartu University Hospital). The youngest baby recruited was 35.6 weeks and the oldest 127 weeks post-menstrual age (see Tables1 and 3). In general it was not possible to administer an isolated microdose of PARA to PARA naïve infants because PARA therapy is a normal part of treatment for sick babies in intensive care. PARA microdose administration was separated in time from the pre- and post-therapeutic PARA dose administration.
Summary of patient details and dosing information for infants dosed with [14C]-PARA
| Dose and dosing route | Number of infants and gender (Male/Female) | Median post-menstrual age (weeks)(range) | Median body weight (kg) (range) | ¶Age classification – ICH E11 |
|---|---|---|---|---|
| i.v. microdose* | 6 (2M/4F) | 43.9 (35.6–127) | 4.1 (3–9.8) | Preterm – 2 |
| Term – 1 | ||||
| Infants – 3 | ||||
| i.v. therapeutic dose† | 18 (10M/8F) | 41.9 (36–86) | 3.7 (2.3–10) | Preterm – 2 |
| Term – 7 | ||||
| Infants – 9 | ||||
| Oral microdose‡ | 4 (2M/2F) | 59.7 (39.1–77) | 4.2 (2.3–6.3) | Preterm – 1 |
| Term – 1 | ||||
| Infants – 2 | ||||
| Oral therapeutic dose§ | 6 (4M/2F) | 51.5 (38–79.4) | 4.7 (2.5–7.8) | Preterm – 0 |
| Term – 2 | ||||
| Infants – 4 | ||||
6ng kg–1 PARA and 111 Bq [14C] kg–1 PARA i.v.
i.v. therapeutic PARA dose as appropriate for age and weight of infant plus 111 Bq [14C] PARA kg–1, 6 ng PARA kg–1.
Oral 6ng kg–1 PARA and 111 Bq [14C] PARA kg–1 administered by enteral tube.
Oral therapeutic PARA dose as appropriate for age and weight of infant plus 111 Bq [14C] PARA kg–1, 6 ng kg–1 PARA by enteral tube.
Based on post-menstrual age where term is 40 weeks.
Ten babies in total received either an enteral or i.v. microdose of [14C]-PARA alone whilst the remainder received [14C]-PARA mixed with a therapeutic PARA dose. The microdose route of administration was selected on the basis of whether or not the babies might have required therapeutic PARA by this route at some stage during their treatment. Only babies who had a) blood collected from in-dwelling cannulae, b) a minimum of a predose sample and three time points after [14C]-PARA and c) valid AMS bioanalysis measurements were included in the non-compartmental analysis making a total of 33 babies for PK analysis. This dataset was substantially larger with more children of a younger age group than the previously published PARA microtracer study 27
Optimal blood sampling time windows were estimated using PopDes software 35, where a structural PK model (values of the PK parameters, the between-subject variability of the PK parameters, the residual variability and an initial sampling scheme) was specified using literature data.
PK analysis
A summary of the PK analysis is presented in Table4 which presents the PARA PK parameters. The Cmax and AUC(0,t) values are presented non-normalized and dose-normalized. Table4 shows that for both enteral and i.v. routes the microdose tmax, clearance and half-life was approximately half the values found with the microtracer incorporated into a therapeutic dose. The results of the PK NCA conducted with the i.v. microtracer mixed with a therapeutic dose were similar, but not identical, to the results from a similar NCA conducted with unlabelled PARA 31. Time to Cmax was slightly longer in this study and t1/2 was slightly shorter in this study. The differences were within the two-fold variation we used as an a priori threshold for discrepant parameters.
Table 4.
PARA pharmacokinetic parameters in neonates and infants
| *Dose and route | tmax (h) | †Cmax (mg l–1) | †AUC(0,t) (mg l–1 h) | ‡AUC(0,t) (mg l–1 h) | t1/2 (h) | §¶CL (l h–1) | **††Vss (l) |
|---|---|---|---|---|---|---|---|
| i.v. therapeutic | 0.93 (1.84) | 0.16 (0.06) | 0.54 (0.26) | 28.46 (19.32) | 3.78 (3.09) | 2.72 (3.10) | 7.16 |
| i.v. microdose | 0.47 (0.72) | 0.30 (0.19) | 0.84 (0.57) | 2.071 x 10−5 (0.760 x 10−6) | 1.69 (0.88) | 1.76 (1.07) | 2.55 |
| Oral therapeutic | 1.05 (0.74) | 0.14 (0.12) | 0.70 (0.79) | 21.326 (47.122) | 2.62 (3.05) | 2.93 (2.08) | 8.36 |
| Oral microdose | 0.65 (0.36) | 0.24 (0.1) | 0.90 ((0.43) | 1.975 x 10−5 (0.766 x 10−6) | 1.64 (1.02) | 1.46 (1.00) | 2.65 |
Data are presented as mean and (SD).
tmax, time to maximum concentration;
t1/2, half-life;
Cmax, maximum concentration;
AUC(0,t), Area under the curve from 0 h to last time point t (last time point was ∼ 6 h in i.v. and ∼8 h in oral PARA);
Vss, apparent volume of distribution calculated from dose/ AUC(0,t) assuming 100% bioavailability.
all plasma concentrations from subjects listed in Table1 were used in the PK calculations.
Dose normalized.
Values not normalized.
Systemic clearance (CL) was calculated by Dose/AUC(0,t).
Apparent clearance CL/F.
Vss was calculated by CL × mean residence time (MRT).
Apparent volume of distribution V/F.
Figure1 is a semi-logarithmic plot of the PARA clearance curve after i.v. administration of either a therapeutic or a microdose (6 ng kg–1). The data are presented as a scatter plot with the line of best fit drawn since the blood collection times after PARA dosing were not identical between babies or between doses. Figure2 presents clearance data from the enteral therapeutic and microdosing arm of this study also with dose-normalization.
Figure 1.

Semilog plots of the dose-normalized PARA plasma concentration–time profiles after administration of either i.v. single therapeutic doses or a single 6 ng kg–1 microdose. Results are presented as mean ± 1 SD.  i.v. therapeutic dose;
i.v. therapeutic dose;  i.v. microdose
i.v. microdose
Figure 2.
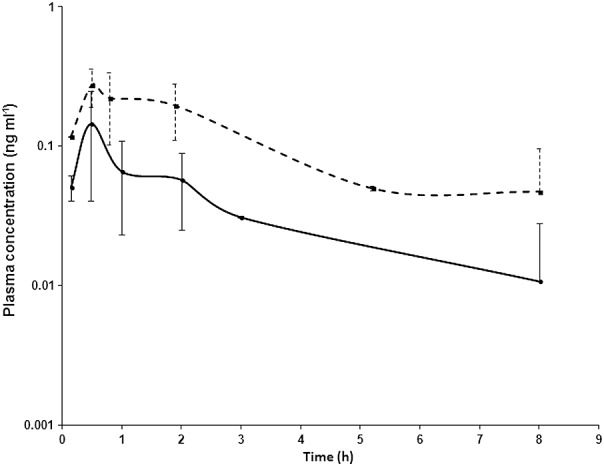
Semilog plots of the dose-normalized mean PARA plasma concentration–time profiles after administration of either oral single therapeutic doses or a single 6 ng kg–1 microdose. Results are presented as mean ± 1 SD.  therapeutic dose;
therapeutic dose;  microdose
microdose
Discussion
This European collaborative project, known as PAMPER, was set up to develop methodology using [14C]-PARA as a model drug to conduct a paediatric exploratory clinical microdose study in the 0–2 year old age group and to examine the relationship between PK for a microdose given at a different time to a therapeutic dose and a microtracer mixed in with a PARA therapeutic dose. PARA is a widely prescribed drug whose paediatric PK have been well-documented 36. The feasibility of the methodology has been demonstrated using a well-characterized probe molecule. The data illustrate that the systemic PARA PK parameters between the two doses are comparable when dose-normalized.
Preparation and formulation of the labelled medicines did not pose any practical problems. We encountered no significant barriers to the setup of this study from ethical or radiological perspectives. This study was conducted in the United Kingdom and Estonia and so met the regulatory requirements for a clinical study in two EU Member States. In other jurisdictions the regulatory authorities may have different requirements. Whilst this study did not offer any prospect of direct benefit to the participants, the additional [14C]-labelled PARA dose of 6 ng kg–1 was trivial in comparison with the therapeutic dose and for the microdose represented less than permitted API impurity levels for the glucose vehicle. The radiological exposure was calculated using a worst case scenario to be well below background exposures. Accordingly the participants will have experienced no harm due to participation in the study. On the other hand, the results from the study could be of general utility for the development of drugs for the paediatric population and hence the study can be ethically justified.
The 50% acceptance rate among parents approached about the study suggests that any concerns relating to the radiation exposure for a number of families were overcome. This acceptance rate was similar to that for PK studies conducted on these units which do not use labelled probes 37. The parents who gave a reason for not participating most commonly said that they were concerned that their child was too sick to be in any studies and/or that they were concerned about the volume of blood that would be sampled. None of the parents expressed concerns about the radioactivity after the explanations had been given. Our participants were in-patients in a paediatric intensive care unit and all had intra-arterial access. Review of the blood biochemistry indicated that there were no clinically relevant abnormalities in liver function or renal function among the participants.
The results of the PK NCA conducted with the i.v. microtracer mixed with a therapeutic dose were similar, but not identical, to the results from a similar NCA conducted with unlabelled PARA 38. Time to Cmax was longer and the clearance was slower. The changes were within the two-fold variation we used as an a priori threshold for discrepant parameters. Encouragingly the values for CL (apparent clearance for enteral route) obtained in our study for therapeutic and microdoses (enteral and i.v.) of PARA were broadly in agreement with those reported by others of approximately 2.0–3.0 l h–1 36,38,39 accepting that our study had a relatively small numbers of babies. In contrast the type of care received by the participants in the Zuppa et al. study 38 was not stated although all participants had intravenous access. It is possible that comorbidities in our group prolonged distribution and elimination without leading to changes in biochemical measures of renal or hepatic function.
The values for tmax and t1/2 following the administration of a microdose appear to be similar to what would be seen following the administration of a therapeutic dose to a PARA naïve infant. In the paper by Zuppa et al., tmax for i.v. administration to infants was median 0.29 h, range 0.3–1.4 h which is similar to the mean 0.47 (SD 0.75) observed here for the microdose not administered at the same time as a therapeutic dose 38. In the light of clinical circumstances in an ICU where PARA is routinely used and where there is little possibility of recruiting a PARA naïve baby, the microdoses were administered a median of 4 h (range 1.5–17 h) after a previous therapeutic dose and a median of 9 h (range 0.5–15.3 h) before the next therapeutic dose. Given an elimination half-life for PARA of circa 2 h the microdose time separation from a therapeutic dose is a reasonable approximation to a true microdose in a PARA-naïve participant.
Most of the parameters in the non-compartmental analyses for microtracer and microdose were within the a priori threshold of a two-fold difference. This threshold provides a frame of reference for comparison between different PK studies (40–42 but is not intended to provide a criterion for the acceptance or rejection of this technique. Comparisons also need to take into account other aspects of the similarities and differences between dosing regimens. The shape of the plasma clearance curve for either an enteral or i.v. dose was similar for a microdose not administered at the time of a therapeutic dose and a microtracer mixed with therapeutic dose administration.
The microdose used here was administered at a different time from a therapeutic dose but the patients were not PARA naïve. A true microdose would have required the recruitment of PARA naïve infants. This was a consequence of conducting this study in infants with appropriate vascular access and a medicine that parents are familiar with. Since the microdose was so small (ng kg–1), it is possible in microdose babies, that a residual pool of PARA was present from prior therapeutic dose PARA administration and that the [14C]-PARA microdose pulse labelled this pool. In other words, the PK data obtained were that associated with pool turnover rather than the isolated microdose per se. This does not detract from our demonstration of proof-of-concept for exploratory PK studies involving enteral and systemic microdoses in young children.
The patient numbers in this study are still relatively small, particularly when broken down into the separate dosing groups. For example only four babies were administered an enteral microdose. This reflected the opportunities available in clinical practice. Nevertheless despite the small group size, PK parameters between a microdose and a therapeutic dose were similar and compared with literature values.
Paediatric microdosing with AMS bioanalysis may offer some advantages over current LC/MS methods to gather exploratory PK data including a) the use of 10–15 microliter sample plasma volumes, which is even two to three times lower than used in both other microdosing/tracing articles in paediatric patients 26,27, b) the ability to take multiple blood samples at time points after drug administration, c) high analytical sensitivity (attograms to zeptograms) permitting trace drug doses to be administered and d) reduction or elimination of safety and pharmacology issues since there is no chance that drug targets become saturated. Conversely routine AMS bioanalysis after microdose administration requires a) the administered drug to be [14C]-labelled albeit at background levels of radioactivity, b) that there is wide availability of AMS instruments for bioanalysis and c) that there is dose-proportionality between a microdose and a therapeutic dose. A preliminary report of the PAMPER study after five babies were recruited was presented at the European Society for Developmental, Perinatal and Paediatric Pharmacology in Salzburg, Austria in June 2013. A short communication, published by Mooij et al. 27, appeared whilst our manuscript was under review and differs substantially from this study. Our study reports on the PK of an isolated microdose given either orally or i.v. whereas the Mooij et al. communication reported on an oral microdose given at the same time as an i.v. therapeutic dose 43.
This observational study has demonstrated that an operational plan can be put in place to meet the scientific, legal, regulatory and ethical challenges in order to conduct paediatric microdose studies with AMS bioanalysis. The procedures developed here, when extended to other drugs, could be used to establish if microdosing has general utility for routine paediatric development of appropriate drugs, such as those which are not transporter dependent and which are metabolized before elimination. This could permit new drugs to enter into the neonate and infant population earlier than currently practised. Microdose PK data, obtained after intensive blood sampling, could be used to compare PK parameters calculated from paediatric PBPK modelling studies in order to determine more accurately the therapeutic dose for clinical efficacy studies. Furthermore microdose PK data alongside PBPK modelling studies 44 should give greater confidence that modelling data truly reflects a drug's metabolism in this sensitive population. Finally we suggest that further microdose/therapeutic dose paediatric PK studies should be undertaken with other model drug substrates to establish when these methods could be used in selected Paediatric Investigation Plans.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
This research programme was funded under the ERA-Net PRIOMEDCHILD programme (Proposal No 40-41800-98-022). Individual country contributions were as follows; United Kingdom, Medical Research Council grant number G1100801 as part of the Medical Research Council Drug Safety Science Centre (grant number G0700654) awarded to the University of Liverpool; The Netherlands, ZonMW grant 113205022; Estonia, Estonian Science Foundation, grant no 18/2011; Poland, Narodowym Centrum Bada I Rozwojugrant.
We would like to thank Kishor Solanki, Radiopharmacy Department, Addenbrookes Hospital, Cambridge, United Kingdom for GMP PARA formulation, Babs Fabriek for management of the AMS activities, Hugo Sandman, Hans Mocking and Dimitri Grossouw, TNO the Netherlands for laboratory execution of the LC+AMS analysis, Lenne-Triin Kõrgvee, University of Tartu for preparation of the study protocol and ethics application, Elaine Scott, Alder Hey Children's NHS Foundation Trust for supporting the clinical research team and Mark Gannon, Nuclear Medicine, Cambridge University Hospitals, Cambridge, United Kingdom for dosimetry calculations.
Contributors
Professor Colin Garner managed the PAMPER project on behalf of the Consortium. He wrote the manuscript and designed part of the research programme.
Dr Mark Turner contributed to all clinical aspects of this study as Principal Investigator. He also part wrote the manuscript and analyzed data.
Professor Kevin Park was the PAMPER Project Co-ordinator and assisted in designing and implementing the research programme as well as being involved in manuscript preparation.
Dr Neil French assisted in the co-ordination of the PAMPER programme including study design and implementation.
Dr Caroline Earnshaw contributed to developing new analytical assays.
Dr Alessandro Schipani conducted all pharmacokinetic analysis of the data including modelling.
Dr Andrew Selby was the main Clinical Investigator at Alder Hey Children's NHS Foundation Trust and contributed to the design of the study and writing of the manuscript.
Ms Lindsay Byrne and Ms Sarah Siner were responsible for recruitment, dose administration and blood collection at Alder Hey Children's NHS Foundation Trust.
Mr Francis Crawley provided expertise on the ethical aspects of the study and contributed to the writing and reviewing of the manuscript.
Dr Wouter Vaes was in overall charge of AMS bioanalysis.
Dr Esther vanDuijn and Dr Rianne deLigt were responsible for day to day supervision of the AMS bioanalysis programme and data calculation.
Dr Heili Varendi participated in the design of research, study protocol and writing of the manuscript; performed clinical research.
Dr Jana Lass participated in the design of research and study protocol.
Professor Grzegorz Grynkiewicz was responsible for supervising the purification and certification of [14C]-PARA.
Dr Wioleta Maruszak was responsible for purification land certification of [14C]-PARA.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1 Mean deviations for calibration standards.
Table S2 Accuracy and precision details for the quality control samples.
Supporting info item
References
- 1.Barker CI, Standing JF, Turner MA, McElnay JC, Sharland M. Antibiotic dosing in children in Europe: can we grade the evidence from pharmacokinetic/pharmacodynamic studies–and when is enough data enough? Curr Opin Infect Dis. 2012;25:235–42. doi: 10.1097/QCO.0b013e328353105c. [DOI] [PubMed] [Google Scholar]
- 2.FDA, U.S. 2013. Guidance for industry, pediatric study plans:content of and process for submitting initial pediatric study plans and amended pediatric study plans. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM360507.pdf.
- 3.Bartelink IH, Rademaker CM, Schobben AF, van den Anker JN. Guidelines on paediatric dosing on the basis of developmental physiology and pharmacokinetic considerations. Clin Pharmacokinet. 2006;45:1077–97. doi: 10.2165/00003088-200645110-00003. [DOI] [PubMed] [Google Scholar]
- 4.Leeder JS, Kearns GL, Spielberg SP, van den Anker J. Understanding the relative roles of pharmacogenetics and ontogeny in pediatric drug development and regulatory science. J Clin Pharmacol. 2010;50:1377–87. doi: 10.1177/0091270009360533. [DOI] [PubMed] [Google Scholar]
- 5.Hines RN. Developmental expression of drug metabolizing enzymes: impact on disposition in neonates and young children. Int J Pharm. 2013;452:3–7. doi: 10.1016/j.ijpharm.2012.05.079. [DOI] [PubMed] [Google Scholar]
- 6.Turner MA, Catapano M, Hirschfeld S, Giaquinto C Global Research in Paediatrics. Paediatric drug development: the impact of evolving regulations. Adv Drug Deliv Rev. 2014;73:2–13. doi: 10.1016/j.addr.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Leong R, Vieira ML, Zhao P, Mulugeta Y, Lee CS, Huang SM, Burckart GJ. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin Pharmacol Ther. 2012;91:926–31. doi: 10.1038/clpt.2012.19. [DOI] [PubMed] [Google Scholar]
- 8.Johnson TN. The problems in scaling adult drug doses to children. Arch Dis Child. 2008;93:207–11. doi: 10.1136/adc.2006.114835. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez W, Selen A, Avant D, Chaurasia C, Crescenzi T, Gieser G, Di Giacinto J, Huang S-M, Lee P, Mathis L. Improving pediatric dosing through pediatric initiatives: what we have learned. Pediatrics. 2008;121:530–9. doi: 10.1542/peds.2007-1529. [DOI] [PubMed] [Google Scholar]
- 10.Hines RN. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther. 2008;118:250–67. doi: 10.1016/j.pharmthera.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Guideline IM. 2008. Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. ICH M3 (R2). International Conference on Harmonization.
- 12.Lappin G, Garner RC. Big physics, small doses: the use of AMS and PET in human microdosing of development drugs. Nat Rev Drug Discov. 2003;2:233–40. doi: 10.1038/nrd1037. [DOI] [PubMed] [Google Scholar]
- 13.Garner RC, Lappin G. The phase 0 microdosing concept. Br J Clin Pharmacol. 2006;61:367–70. doi: 10.1111/j.1365-2125.2006.02575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garner RC. Practical experience of using human microdosing with AMS analysis to obtain early human drug metabolism and PK data. Bioanalysis. 2010;2:429–40. doi: 10.4155/bio.10.6. [DOI] [PubMed] [Google Scholar]
- 15.Lappin G, Noveck R, Burt T. Microdosing and drug development: past, present and future. Expert Opin Drug Metab Toxicol. 2013;9:817–34. doi: 10.1517/17425255.2013.786042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamane N, Tozuka Z, Sugiyama Y, Tanimoto T, Yamazaki A, Kumagai Y. Microdose clinical trial: quantitative determination of fexofenadine in human plasma using liquid chromatography/electrospray ionization tandem mass spectrometry. J Chromatogr B. 2007;858:118–28. doi: 10.1016/j.jchromb.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 17.Lappin G, Kuhnz W, Jochemsen R, Kneer J, Chaudhary A, Oosterhuis B, Drijfhout WJ, Rowland M, Garner RC. Use of microdosing to predict pharmacokinetics at the therapeutic dose: experience with 5 drugs. Clin Pharmacol Ther. 2006;80:203–15. doi: 10.1016/j.clpt.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 18.Lappin G, Shishikura Y, Jochemsen R, Weaver RJ, Gesson C, Brian Houston J, Oosterhuis B, Bjerrum OJ, Grynkiewicz G, Alder J, Rowland M, Garner C. Comparative pharmacokinetics between a microdose and therapeutic dose for clarithromycin, sumatriptan, propafenone, paracetamol (acetaminophen), and phenobarbital in human volunteers. Eur J Pharmaceut Sci. 2011;43:141–50. doi: 10.1016/j.ejps.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Patel P, Mulla H, Tanna S, Pandya H. Facilitating pharmacokinetic studies in children: a new use of dried blood spots. Arch Dis Child. 2010;95:484–7. doi: 10.1136/adc.2009.177592. [DOI] [PubMed] [Google Scholar]
- 20.Suyagh MF, Kole PL, Millership J, Collier P, Halliday H, McElnay JC. Development and validation of a dried blood spot-LC-APCI-MS assay for estimation of canrenone in paediatric samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:769–76. doi: 10.1016/j.jchromb.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 21.Abdel-Rahman S, Reed M, Wells T, Kearns G. Considerations in the rational design and conduct of phase I/II pediatric clinical trials: avoiding the problems and pitfalls. Clin Pharmacol Ther. 2007;81:483–94. doi: 10.1038/sj.clpt.6100134. [DOI] [PubMed] [Google Scholar]
- 22.Kaye B, Garner RC, Mauthe RJ, Freeman SP, Turteltaub KW. A preliminary evaluation of accelerator mass spectrometry in the biomedical field. J Pharm Biomed Anal. 1997;16:541–3. doi: 10.1016/s0731-7085(97)00104-0. [DOI] [PubMed] [Google Scholar]
- 23.Garner R. Accelerator mass spectrometry in pharmaceutical research and development a new ultrasensitive analytical method for isotope measurement. Curr Drug Metab. 2000;1:205–13. doi: 10.2174/1389200003339054. [DOI] [PubMed] [Google Scholar]
- 24.Boulton DW, Kasichayanula S, Keung CF, Arnold ME, Christopher LJ, Xu XS, Lacreta F. Simultaneous oral therapeutic and intravenous (14C)-microdoses to determine the absolute oral bioavailability of saxagliptin and dapagliflozin. Br J Clin Pharmacol. 2013;75:763–8. doi: 10.1111/j.1365-2125.2012.04391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham RA, Lum BL, Morrison G, Chang I, Jorga K, Dean B, Shin YG, Yue Q, Mulder T, Malhi V, Xie M, Low JA, Hop CE. A single dose mass balance study of the Hedgehog pathway inhibitor vismodegib (GDC-0449) in humans using accelerator mass spectrometry. Drug Metab Dispos. 2011;39:1460–7. doi: 10.1124/dmd.111.039339. [DOI] [PubMed] [Google Scholar]
- 26.Gordi T, Baillie R, Vuong LT, Abidi S, Dueker S, Vasquez H, Pegis P, Hopper AO, Power GG, Blood AB. Pharmacokinetic analysis of 14C-ursodiol in newborn infants using accelerator mass spectrometry. J Clin Pharmacol. 2014;54:1031–1037. doi: 10.1002/jcph.327. [DOI] [PubMed] [Google Scholar]
- 27.Mooij MG, van Duijn E, Knibbe CA, Windhorst AD, Hendrikse NH, Vaes WH, Spaans E, Fabriek BO, Sandman H, Grossouw D. Pediatric microdose study of [14C] paracetamol to study drug metabolism using accelerated mass spectrometry: Proof of concept. Clin Pharmacokinet. 2014;53:1045–51. doi: 10.1007/s40262-014-0176-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C, Allegaert K, Tibboel D, Danhof M, van der Marel CD, Mathot RA, Knibbe CA. Population pharmacokinetics of paracetamol across the human age-range from (pre)term neonates, infants, children to adults. J Clin Pharmacol. 2014;54:619–29. doi: 10.1002/jcph.259. [DOI] [PubMed] [Google Scholar]
- 29.Association, WM. 2008. Declaration of Helsinki. Ethical principles for medical research involving human subjects. Available at http://www wma net/e/policy/b3 htm (last accessed 24 February 2015)
- 30. Available at ftp://ftp.cordis.europa.eu/pub/fp7/docs/ethical-considerations-paediatrics_en.pdf. Final 2008 (last accessed 24 February 2015)
- 31.van Duijn E, Sandman H, Grossouw D, Mocking JA, Coulier L, Vaes WH. Automated combustion accelerator mass spectrometry for the analysis of biomedical samples in the low attomole range. Anal Chem. 2014;86:7635–41. doi: 10.1021/ac5015035. [DOI] [PubMed] [Google Scholar]
- 32.Higton D, Young G, Timmerman P, Abbott R, Knutsson M, Svensson LD. European Bioanalysis Forum recommendation: scientific validation of quantification by accelerator mass spectrometry. Bioanalysis. 2012;4:2669–79. doi: 10.4155/bio.12.242. [DOI] [PubMed] [Google Scholar]
- 33.(ICRP), I. C. o. R. P. Limits for Intakes of Radionuclides by Workers ICRP Publication 30 (Part 1) Ann. ICRP. 1979;2:3–4. doi: 10.1016/0146-6453(79)90122-2. [DOI] [PubMed] [Google Scholar]
- 34.Manger RP. A generic biokinetic model for Carbon-14. Radiat Prot Dosimetry. 2011;143:42–51. doi: 10.1093/rpd/ncq332. [DOI] [PubMed] [Google Scholar]
- 35.Gueorguieva I, Ogungbenro K, Graham G, Glatt S, Aarons L. A program for individual and population optimal design for univariate and multivariate response pharmacokinetic–pharmacodynamic models. Comput Methods Programs Biomed. 2007;86:51–61. doi: 10.1016/j.cmpb.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 36.Wang C, Allegaert K, Tibboel D, Danhof M, van der Marel CD, Mathot RA, Knibbe CA. J Clin Pharmacol. 2013 doi: 10.1002/jcph.259. Population pharmacokinetics of paracetamol across the human age-range from (pre)term neonates, infants, children to adults. doi: 10.1002/jcph.259 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 37.Zhao W, Hill H, Le Guellec C, Neal T, Mahoney S, Paulus S, Castellan C, Kassai B, van den Anker JN, Kearns GL. Population pharmacokinetics of ciprofloxacin in neonates and young infants less than 3 months of age. Antimicrobial Agents Chemother. 2014:6572–80. doi: 10.1128/AAC.03568-14. 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuppa AF, Hammer GB, Barrett JS, Kenney BF, Kassir N, Mouksassi S, Royal MA. Safety and population pharmacokinetic analysis of intravenous acetaminophen in neonates, infants, children, and adolescents with pain or Fever. J Pediatr Pharmacol Therapeut. 2011;16:246–61. doi: 10.5863/1551-6776-16.4.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson BJ, Pons G, Autret-Leca E, Allegaert K, Boccard E. Pediatric intravenous paracetamol (propacetamol) pharmacokinetics: a population analysis1. Pediatr Anesth. 2005;15:282–92. doi: 10.1111/j.1460-9592.2005.01455.x. [DOI] [PubMed] [Google Scholar]
- 40.Rowland M. Microdosing: a critical assessment of human data. J Pharm Sci. 2012;101:4067–74. doi: 10.1002/jps.23290. [DOI] [PubMed] [Google Scholar]
- 41.Madan A, O'Brien Z, Wen J, O'Brien C, Farber RH, Beaton G, Crowe P, Oosterhuis B, Garner RC, Lappin G, Bozigian HP. A pharmacokinetic evaluation of five H1-receptor antagonists after an oral and intravenous microdose to human subjects. Br J Clin Pharmacol. 2009;67:288–98. doi: 10.1111/j.1365-2125.2008.03351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lappin G, Shishikura Y, Jochemsen R, Weaver RJ, Gesson C, Houston B, Oosterhuis B, Bjerrum OJ, Rowland M, Garner C. Pharmacokinetics of fexofenadine: evaluation of a microdose and assessment of absolute oral bioavailability. Eur J Phramaceut Sci. 2010;40:125–31. doi: 10.1016/j.ejps.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 43.Lappin G, Rowland M, Garner RC. The use of isotopes in the determination of absolute bioavailability of drugs in humans. Expert Opin Drug Metab Toxicol. 2006;2:419–27. doi: 10.1517/17425255.2.3.419. [DOI] [PubMed] [Google Scholar]
- 44.Jiang X, Zhao P, Barrett J, Lesko L, Schmidt S. Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. Clin Pharmacol Ther: pharmacometrics & systems pharmacology. 2013;2:e80. doi: 10.1038/psp.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item